

Abstract
Background
The healthy microbiome protects against the development of Clostridium difficile infection (CDI), which typically develops following antibiotics. The microbiome metabolizes primary to secondary bile acids, a process if disrupted by antibiotics, may be critical to initiation of CDI.
Aim
To assess the levels of primary and secondary bile acids associated with CDI and associated microbial changes.
Methods
Stool and serum were collected from patients with (1) first CDI (fCDI), (2) recurrent CDI (rCDI) and (3) healthy controls. 16S rRNA sequencing and bile salt metabolomics were performed. Random forest regression models were constructed predict disease status. PICRUSt analyses were used to test for associations between predicted bacterial bile salt hydrolase (BSH) gene abundances and bile acid levels.
Results
60 patients (20 fCDI, 19 rCDI, and 21 controls) were enrolled. Secondary bile acids in stool were significantly elevated in controls compared to rCDI and fCDI (p<0.0001 and p=0.0007 respectively). Primary bile acids in stool were significantly elevated in rCDI compared to controls (p<0.0001) and in rCDI compared to fCDI (p=0.02). Using random forest regression, we distinguished rCDI and fCDI patients 84.2% of the time using bile acid ratios. Stool deoxycholate to glycoursodeoxycholate was the single best predictor. PICRUSt analyses found significant differences in predicted abundances of bacterial BSH genes in stool samples across the groups.
Conclusion
Primary and secondary bile acid composition in stool was different in those with rCDI, fCDI, and controls. The ratio of stool deoxycholate to glycoursodeoxycholate was the single best predictor of disease state and may be a potential biomarker for recurrence.
Keywords: Clostridium difficile, bile salts, microbiome
Background
Clostridium difficile is a spore-forming, potentially pathogenic organism that colonizes the human gut. Recurrent disease complicates 20–30% of cases and represents an important source of morbidity and mortality.1–3 The intestinal microbiota protects against the development of Clostridium difficile infection (CDI), and antibiotic-induced changes in the microbiota likely permit initiation of the disease. While toxins associated with this disease have been well characterized, the host microbial and metabolic alterations, which facilitate CDI, as well as their role in the disease and its recurrences, are not well understood.
The immediate cause of CDI is evident: the disease is communicated by ingestion of spores.4 Spores are resistant to heat and antibiotics and are able to survive outside of the colon. Spores germinate in the GI tract and become vegetative cells which can produce toxin.5 While the organism itself is noninvasive6, diarrhea is mediated by the release of toxin.7 However, the underlying pathogen-commensal interactions that gives rise to the resulting pseudomembranous colitis remains poorly defined.
Germination of spores is critical to initiate CDI. Bile acids are vital to the germination process. Primary bile acids are produced in the liver and released into the small intestine to assist in digesting fat5. A majority of bile acids are reabsorbed in the small intestine, however a portion are passed into the colon where they undergo biotransformations by intestinal bacteria.5 The normal intestinal microbiota metabolizes primary bile acids through 7α-dehydroxylation and a series of other biochemical transformations.8 This process may be disrupted with antibiotics, producing an increase in primary bile acids and a reduction of secondary bile acids.9
The relationship between Clostridium difficile and bile acids is complex. In vitro studies suggest that the primary bile acids cholate, taurocholate and glycocholate can stimulate germination of C. difficile spores, however the primary bile acid chenodeoxyholate can inhibit germination.10 Additionally the secondary bile acid deoxycholate can also stimulate germination but more importantly deoxycholate inhibits growth of the vegetative form.11,12 Antibiotic therapy is thought to ablate critical members of the commensal microbiota that generate inhibitory (protective) secondary bile acids. Stool extracts from antibiotic treated mice have been found higher concentrations of primary bile acids, whereas stool extracts from untreated mice have relatively higher secondary bile acids.9
While the interaction between bile acids and CDI is illustrated in in vitro studies, the relationship between the host microbiome and bile salt metabolism is less well defined in humans with CDI.13 An understanding of this interaction is essential to identify those at greatest risk for recurrence, to develop better strategies to treat the disease and to prevent recurrences. We aim to delineate the relationship between bile acid concentration, host microbiome, and CDI status.
Methods
This is a cross sectional study of three well-defined groups: individuals experiencing their first episode of CDI (fCDI), individuals with recurrent CDI (rCDI) defined as 3 or more episodes, and healthy controls.
The rCDI group and healthy controls were recruited from patients and rigorously screened healthy donors who had been referred for fecal transplantation for recurrent CDI. Donor selection, screening for relevant communicable diseases, and stool processing were performed as outlined by the Fecal Microbiota Transplantation Working Group.14 The patients experiencing their first episode of CDI were recruited from the inpatient service at Brigham and Women’s Hospital informed by daily reports of patients with positive tests for C. difficile provided by the BWH Clinical Microbiology Laboratory.15,16 CDI testing was performed with an Elisa for Tox A/B and this was confirmed with PCR. After informed consent, stool and blood samples were collected from each patient. Controls were not exposed to antibiotics for a minimum of 3 months. Samples from fCDI patients were collected prior to initiation of antibiotics to treat CDI. For rCDI patients, samples were collected on stable doses of chronic oral vancomycin prior to fecal transplantation. Stool samples were flash frozen at −80° C within 4 hours. Clinical data was extracted from medical records and patient interview. Blood plasma and stool samples were analyzed for bile salt metabolomic profiles; and stool was sent for high-throughput 16S rRNA amplicon sequencing to determine the composition of the microbiota. Any patients on antibiotics for other reasons or those taking bile salt binding agents were excluded from the study. This protocol was approved by the institutional review board (IRB) at Brigham and Women’s Hospital.
Preparation of DNA and sequencing protocols
A multiplexed amplicon library covering the 16S rDNA gene V4 region was generated from human stool sample DNA. Bacterial genomic DNA was extracted using the Mo Bio Power Fecal DNA Isolation kit (Mo Bio Laboratories, Carlsbad, CA) according to the manufacturer’s instructions for high yields of DNA with the following modifications to increase yields from difficult to lyse microbes. An additional bead beater step using the Faster Prep FP120 (Thermo) at 6 meters/second for 1 min was used instead of vortex agitation. Incubation with buffers C2 and C3 was increased to 10 min at 4°C.
Starting nucleic acid concentrations were determined by a Qubit Fluromoter (Life Technologies, Carlsbad, CA). A multiplexed amplicon library covering the 16S rDNA gene V4 region was prepared using the protocol of with dual-index barcodes.17 The aggregated library pool was size selected from 300–500 bp on a pippin prep 1.5% agarose cassette (Sage Science, Beverly, MA) according to the manufacturer’s instructions. The concentration of the pool was measured by qPCR (Kapa Biosystems, Willmington, MA) and loaded onto the MiSeq Illumina instrument (300 bp kit) at 6–9pM with 40% phiX spike-in to compensate for low base diversity according to Illumina’s standard loading protocol.
16S rRNA data analysis
Sequencing of the stool samples on Illumina MiSeq instrument generated 5,244,155 total raw sequencing reads. Raw reads were processed using the mothur software package (v.1.34.2) and custom Python scripts, which perform denoising, quality filtering, alignment against the ARB Silva reference database of 16S rDNA gene sequences, and clustering into Operational Taxonomic Units (OTUs) at 97% identity.18 In total, 8,330 OTUs were generated. After filtering OTUs that failed to have a mean number of reads per sample ≥ 1 in at least one cohort, 597 OTUs were available for further analysis. To identify the OTUs with increased taxonomic resolution, the Pplacer software package was used to perform phylogenetic placement.19 Pplacer uses a likelihood-based methodology to place short sequencing reads of 16S rRNA amplicons on a reference tree, and also generates taxonomic classifications of the short sequencing reads using a least common ancestor-based algorithm. The reference tree required for phylogenetic placement was generated using full-length or near full-length (>1,200 nt) 16S rDNA sequences of type strains from the Ribosomal Database Project (RDP).20
A measure of overall microbial community diversity, the Shannon entropy18,21,22, was calculated for the samples, and the nonparametric Wilcoxon rank-sum test was applied for hypothesis testing.23 To visualize differences in overall microbial community structure, the unweighted Unifrac measure24 was calculated between all pairs of samples, and Principal Coordinates Analysis (PCoA) plots were generated using custom R scripts. Analysis of Molecular Variance (AMOVA) was used for statistical hypothesis testing18,25,26 of differences in overall microbial community structure between cohorts as assessed with the Unifrac measures. DESeq2 within the phyloseq package was applied to test for differential abundance of phyla and genera in different cohort groups.27 For all statistical testing for 16S rDNA data analysis, p-values were adjusted for multiple hypothesis testing using the method of Benjamini and Hochberg.28
To predict bile salt hydrolase gene content in the samples, 486 of the OTUs defined as described above, accounting for more than 97% of all reads, were mapped to OTUs defined in the 13_5 release of the Greengenes database whose representative sequences were at least 99% identical to representative sequences of the OTUs.29 Relative abundances of those Greengenes OTUs were used to predict the abundances of genes corresponding to KEGG orthology K01442, cholylglycine hydrolase, with PICRUSt 1.0.0.30 OTUs contributing to changes in BSH abundance between groups were identified using the Pplacer results described above, and changes in abundances of those OTUs between groups were individually tested for significance using the Wilcoxon rank-sum test.
Bile Salt Metabolomics
Bile acids were measured using liquid chromatography tandem mass spectrometry (LC-MS) method operated on a Nexera X2 U-HPLC (Shimadzu Scientific Instruments; Marlborough, MA) coupled to a Q Exactive hybrid quadrupole orbitrap mass spectrometer (Thermo Fisher Scientific; Waltham, MA) methods. Stool samples were homogenized in a weight proportional volume of water (4μL/mg of stool) using a TissueLyser II (Qiagen). Plasma or stool homogenates (30 μL) were extracted using 90 μL of methanol containing PGE2-d4 as an internal standard (Cayman Chemical Co.; Ann Arbor, MI) and centrifuged (10 min, 10,000 × g, 4°C). The supernatants (10 μL) were injected onto a 150 × 2.1 mm ACQUITY UPLC BEH C18 column (Waters; Milford, MA). The column was eluted isocratically at a flow rate: 450μL/min with 20% mobile phase A (0.1% formic acid in water) for 3 minutes followed by a linear gradient to 100% mobile phase B (acetonitrile with 0.1% formic acid) over 12 minutes. MS analyses were carried out using electrospray ionization in the negative ion mode using full scan analysis over m/z 70–850 at 70,000 resolution and 3 Hz data acquisition rate. Additional MS settings were: ion spray voltage, −3.5 kV; capillary temperature, 320°C; probe heater temperature, 300 °C; sheath gas, 45; auxiliary gas, 10; and S-lens RF level 60. Targeted processing of a bile acid profiles was conducted using TraceFinder software (Thermo Fisher Scientific; Waltham, MA) and visually inspected for quality.
Additional statistical analyses
Baseline patient demographics differences were evaluated as mean ± SD for continuous variables. Bile acid concentrations were not normally distributed according to the Shapiro–Wilk test; thus, the Wilcoxon rank-sum test with a bonferroni correction was used. The total of primary and secondary bile acids were calculated by averaging the concentration of all unconjugated and conjugated species. The relative proportion of a given acid corresponds to its concentrations divided by the total of bile acids. These statistical analyses were performed using SAS v.9.4.
Random forest regression models were constructed using the random forest R package with 100,000 trees and two variables per branch. To construct each tree, a random selection of 2/3 of the samples were used to train the model, and the remaining 1/3 used to test the model (3-fold cross-validation). Study cohort groups were the dependent variables and either OTUs (597 total) or log-transformed bile acids (27 measured in blood and stool) and/or part-to-whole ratios of bile acids (bile acid x/(bile acid x + bile acid y)) (351 total) were used as predictors. For samples with bile acid abundance below the limit of detection, the missing data was imputed using the MICE package in R. To assess the random forest model constructed, study groups were shuffled randomly and 100 random forest regressions were computed. The out-of-bag error estimate was compared to the un-shuffled dataset using a one sample Wilcoxon one sample signed rank test to assess the performance of the regression model.31
Results
A total of 60 subjects were enrolled in the study: 21 controls, 19 rCDI subjects and 20 fCDI subjects. The rCDI group had an average of 3.6 (range 3–6) confirmed recurrences and these patients were all being screened for fecal transplantation. All rCDI patients were on chronic vancomycin at the time of collection. All but one of the fCDI patients were inpatients at Brigham and Women’s Hospital. The reasons for hospitalization varied and included rectal bleeding, abdominal pain, diarrhea, elective surgery, shortness of breath and alcohol withdrawal. Eleven of the 20 fCDI patients had not received antibiotics in the 8 weeks prior to diagnosis. Of the patients that did, the antibiotics given included clindamycin, ciprofloxacin, levofloxacin and cefpodoxime. None of the fCDI patients met criteria for severe or severe/complicated disease. Controls were all healthy patients who passed the fecal transplant donor screening clinical and laboratory assessment14. Groups did not differ greatly in their demographic characteristics, however controls were significantly younger then fCDI patients (47 ± 16.8 vs 66.6 ± 17.2, p=0.0007). Additionally, both fCDI and rCDI patients were more likely to be taking proton pump inhibitors compared to controls (9 vs 1, p=0.0025 and 12 vs 1 p <.0001 respectively) (Table 1). There were no significant relationships between proton pump inhibitor use and either bile salts or OTUs using spearmon correlation and random regression analysis.
Table 1.
Patient Characteristics
| Recurrent CDI N=19 |
First Time CDI N=20 |
Controls N=21 |
P-Value | |
|---|---|---|---|---|
| Age -mean +/− SD | 56.8 +/− 16.2 | 66.6 +/− 17.2 | 47 +/− 16.8 | 0.002 |
|
| ||||
| White n(%) | 17 (89.5) | 14 (73.7) | 19 (85.3) | 0.51 |
|
| ||||
| Male n(%) | 10 (45.5) | 8 (42.1) | 5 (26.3) | 0.42 |
|
| ||||
| BMI mean +/− SD | 24.1 +/− 5.3 | 28.2 +/− 1.1 | 28.6 +/− 1.4 | 0.05 |
|
| ||||
| Cholecystectomy n(%) | 0 (0) | 1 (5.2) | 1 (4.5) | 0.61 |
|
| ||||
| Dietary | 0.27 | |||
| Restrictions %(n) | ||||
| Gluten Free | 1 (5.26) | 0 (0) | 1 (4.55) | |
| Lactose Free | 2 (10.5) | 0 (0) | 0 (0) | |
| Vegetarian | 0 (0) | 1 (5.2) | 0 (0) | |
|
| ||||
| On PPI n(%) | 63.2 (12) | 47.4 (9) | 4.5 (1) | <0.001 |
|
| ||||
| Current Smoker n(%) | 15.8 (3) | 10.5 (2) | 9.1 (2) | 0.40 |
Bile salt analysis
Stool bile acid analysis
The median secondary bile acids lithocholate and deoxycholate in stool were significantly elevated in controls compared to both fCDI (1.54E+07 vs 2.26E+06, p=<0.0001 and 4.60E+07 vs 1.20E+07, p=0.0001 respectively) and to rCDI (1.54E+07 vs 2.71E+04, p=<0.0001 and 4.60E+07 vs 1.32E+05 p=<0.0001 respectively). Additionally they were significantly elevated in fCDI vs rCDI (2.26E+06 vs 2.71E+04, p=0.02 and 1.20E+07 vs 1.32E+05, p=<0.05 respectively). The primary bile acids cholate and chenodeoxycholate were significantly elevated in rCDI compared to controls (3.91E+07 vs 8.74E+06, p=0.001 and 1.07E+07 vs 7.18E+05, p=0.02 respectively) (Figure 1a). Patients with rCDI overall had a significantly higher abundance of primary bile salts then both fCDI and controls. Similarly, rCDI patients had significantly lower secondary bile salts then controls and fCDI(Figure 1b).
Figure 1.
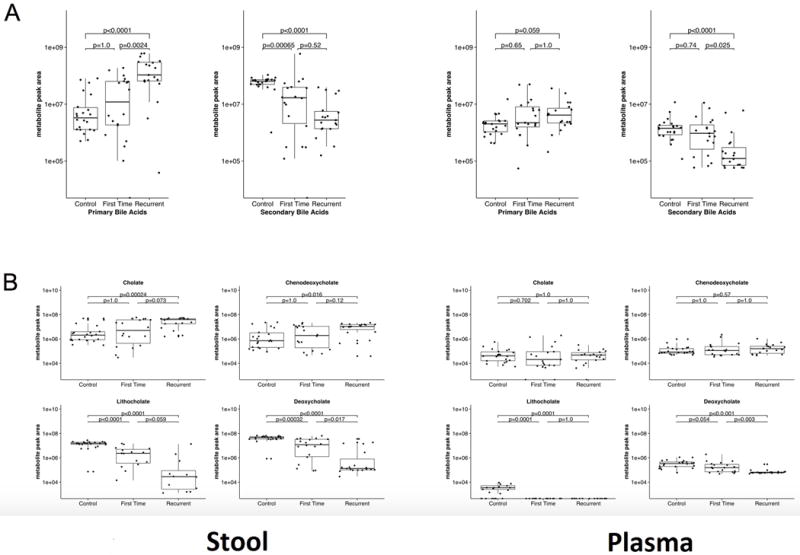
Plasma bile salt analysis
Individual primary bile acids from blood plasma were not significantly different between the groups (Figure 1a and 1b). Secondary bile acids were present in the blood but at lower abundances than in the stool. Median deoxycholic acid in the blood was significantly higher in fCDI than rCDI (1.54E+05 vs 6.12E+04, p=0.003) as well as significantly higher in controls than in both fCDI and rCDI patients respectively (3.41E+05 vs 1.54E+05, p=0.05 and 3.41E05 vs 6.12E+04, p<.0001).
Microbiome analyses
16S rRNA gene analyses revealed significant differences in ecological (alpha) diversity as measured by Shannon entropy (Figure 2). These differences were most pronounced with rCDI vs controls (adjusted p <0.001), however significant differences were also seen when comparing fCDI and rCDI (adjusted p <0.05) and controls and fCDI (adjusted p <0.05). While variation occurred among the subjects, clear differences were seen in the phyla present in each cohort (Figure 3) with multiple significant differences between groups (Table 2). Overall microbial community structure (beta diversity) differed significantly between all groups as measured by unweighted Unifrac analysis (Figure 4, p-values = 0.001).
Figure 2.
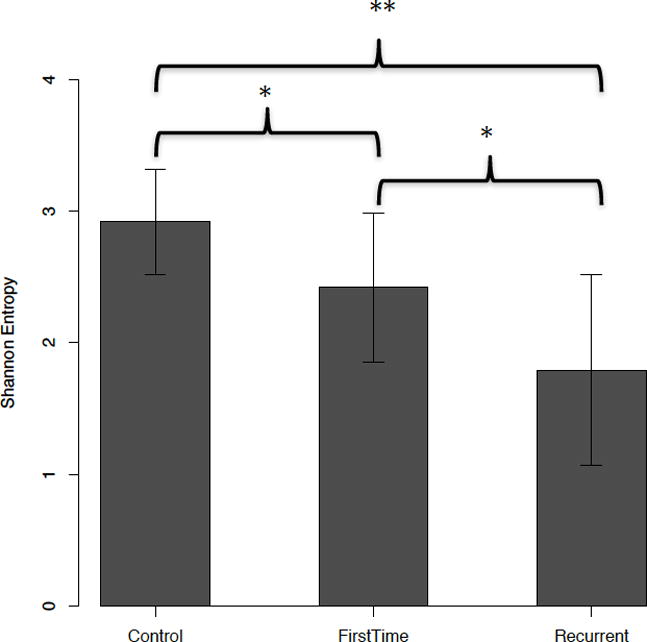
Shannon Entropy Analysis of Microbial Ecological Diversity. P-values from Wilcoxon-Man-Whitney test. Adjusted p-values collected for multiple hypothesis testing using the method of Benjamini-Hochberg (22).
*Denotes adjusted-p-value <0.05
**Denotes adjusted-p-value < 0.001
Figure 3.
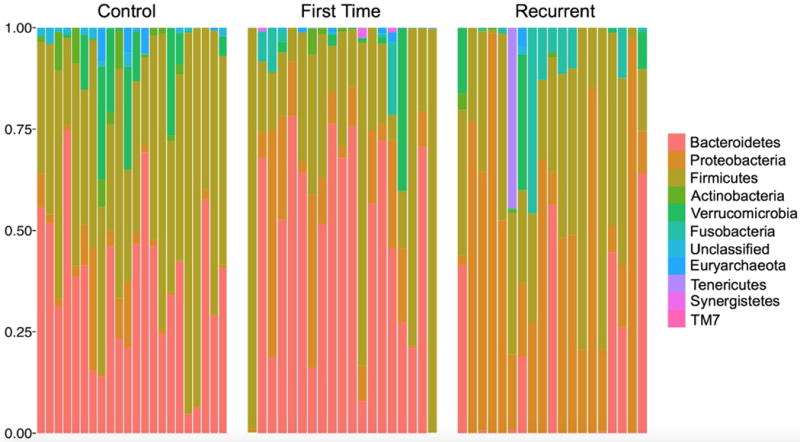
Relative abundances of microbial phyla in each subject (Control subjects n=21, First time subjects n=20, Recurrent subjects n=19).
Table 2.
Phyla showing significant differences among the cohort groups using DESeq2. Stated p-values were adjusted for multiple hypothesis testing using the method of Benjamini-Hochberg (22).
| Cohort Groups | Phylum Name | log2FoldChange | Adjusted p-values |
|---|---|---|---|
| Controls Compared to fCDI | |||
| Higher in Controls | Actinobacteria | −2.408 | 0.0004 |
| Higher in Controls | Verrucomicrobia | −1.533 | 0.0042 |
| Higher in fCDI | Fusobacteria | 1.644 | 0.0342 |
| Higher in fCDI | Synergistetes | 2.195 | 0.0004 |
| Higher in fCDI | TM7 | 1.186 | 0.0381 |
| Controls Compared to rCDI | |||
| Higher in Controls | Euryarchaeota | −2.000 | 0.0076 |
| Higher in Controls | Verrucomicrobia | −1.031 | 0.063 |
| Higher in Controls | Actinobacteria | −2.624 | 0.0001 |
| Higher in Controls | Bacteroidetes | −2.381 | 0.0004 |
| Higher in rCDI | TM7 | 1.466 | 0.011 |
| Higher in rCDI | Fusobacteria | 7.300 | 0 |
| Higher in rCDI | Proteobacteria | 3.870 | 0 |
| rCDI compared fCDI | |||
| Higher in fCDI | Bacteroidetes | −2.249 | 0.0008 |
| Higher in fCDI | Euryarchaeota | −2.848 | 0.0001 |
| Higher in fCDI | Synergistetes | −2.293 | 0.0005 |
| Higher in rCDI | Proteobacteria | 2.986 | 0 |
| Higher in rCDI | Fusobacteria | 5.656 | 0 |
Figure 4.
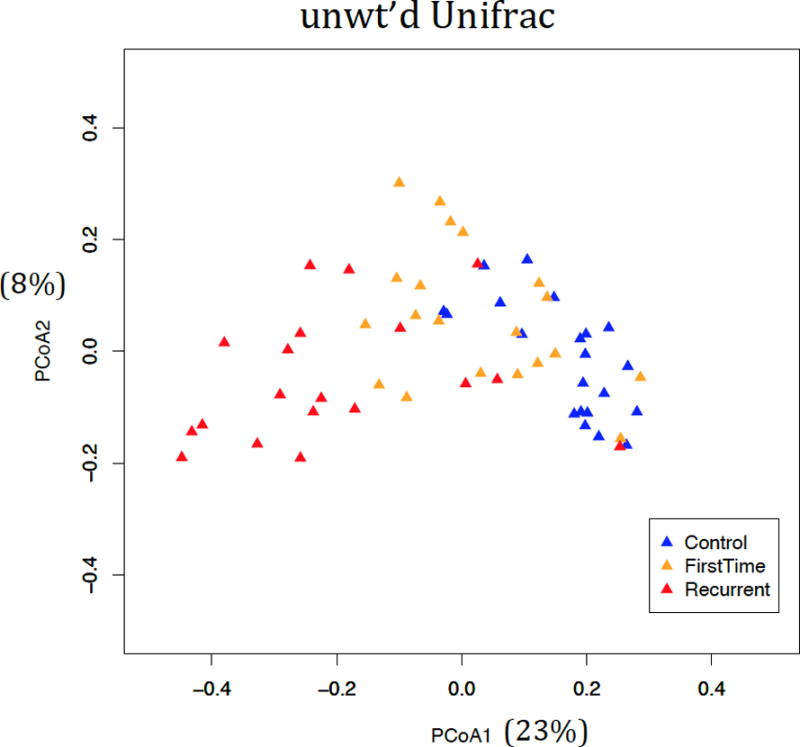
Beta diversity of gut microbiome in all subjects. Beta-diversity values were calculated using the unweighted Unifrac dissimilarity measure, to assess differences in overall microbial community structure. Overall community structure differed significantly between all groups, considered pairwise (adjusted p-values = 0.001), using Analysis of Molecular Variance for statistical hypothesis testing.
Significant differences in relative abundances at the level of individual taxa were also observed. These differences were seen between all groups, notably between control and fCDI and even more profoundly between control and rCDI (Supplementary Table 1). In particular, statistically significant increases in abundances of multiple species in the Order Clostridiales in controls compared to both CDI groups were observed. Collinsella species (a member of phylum Actinobacteria) were also statistically significantly decreased in both fCDI and rCDI subjects compared to controls, members of which carry 7α-dehydrogenase enzymes with activity on bile acids, suggesting that the loss of these members may be associated with decreased abundance of secondary bile acids.32,33
To determine whether bile salts, OTUs or combinations thereof could serve as biomarkers distinguishing between fCDI and rCDI, we constructed random forest regressions of these variables against patient status (control, fCDI, or rCDI). The model including only OTUs had a 23.7% error rate, misclassifying 3/20 fCDI patients as rCDI and 6/19 rCDI patients as fCDI. This model, which takes into account all OTU abundances, ranks Enterobacteriaceae and Lactobacillus OTUs (OTUs resolved at different levels) as the most important variables for predicting patient status, both of which are increased in relative abundance in rCDI relative to fCDI patients.
When using bile salts only as predictors, the best indicators of disease status are the secondary serum bile acids taurodeoxycholate and glyodeoxycholate, with an error rate of 21.05%. However, when part-to-whole ratios of bile salts are added in the model, the error rate improves to 15.8%, with the ratio of stool deoxycholate (a secondary bile acid) to stool glycoursodeoxycholate (a conjugated secondary bile acid) being the best predictor of rCDI versus fCDI, with this ratio significantly decreased in most rCDI patients. We evaluated the performance of this classification model by constructing an ROC curve, which suggests that measured bile acid levels perform better than chance in discerning rCDI from fCDI patients (Figure 5a). Using this ratio alone, it is possible to classify 84% of cases correctly, as only 3/20 fCDI and 3/19 rCDI patients are misclassified (Figure 5b). The top two OTUs predicting patient status, Enterobacteriaceae and Lactobacillus OTUs, are the only OTUs strongly negatively correlated with this ratio, suggesting that either the deoxycholate/glycoursodeoxycholate ratio or Enterobacteriaceae and Lactobacillus OTUs could be used as potential biomarkers for disease status.
Figure 5.
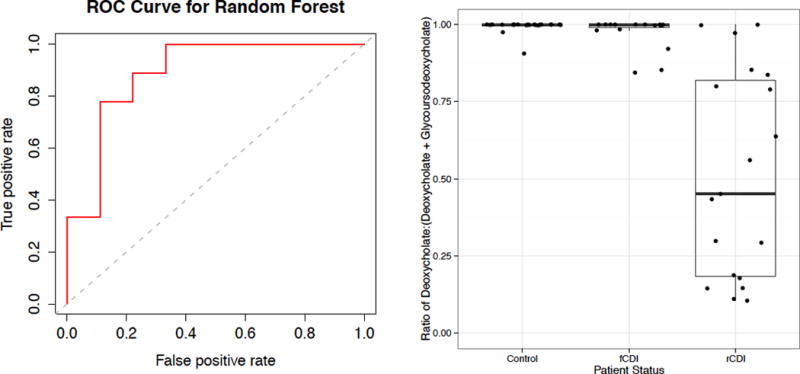
Random forest model accurately assigns patients to groups based on bile acid part-to-whole ratios, (a) ROC curve for the random forest regression using bile acid part-to-whole ratios as independent variables where false positive rate is the incidence of assigning rCDI as fCDI and true positive rate is the incidence of assigning rCDI as rCDI. (b) ratio of deoxycholate to glycoursodeoxycholate and deoxycholate, which was found to be the most important predictor variable in the random forest model.
To assess the levels of bacterial salt hydrolase genes present in samples, we applied PICRUSt30, a tool that predicts the abundances of bacterial genes using 16S rRNA sequencing data. The median predicted BSH gene abundance in the rCDI group was 19% of the median value in the control group (p<0.001), and was also significantly lower than the fCDI group (p<0.001). In contrast, there were no significant differences between predicted bile salt hydroloase gene abundances in control and fCDI groups. The difference in predicted bile salt hydrolase gene abundance between the control group and the rCDI group was found to be largely the result of changes in the abundances of ten bacterial taxa (Table 3). Taxonomic classifications for these OTUs are listed in Supplementary Table 2. Predicted bile salt hydrolase abundances were significantly correlated with bile acid levels. Specifically, predicted bile salt hydrolase abundances were negatively correlated with the conjugated and unconjugated forms of the primary acids, but positively correlated with the secondary acids (Table 4).
Table 3.
Operational taxonomic units with largest contributions to the decrease in average predicted bile salt hydrolase gene levels among recurrent CDI patients as compared to controls. Collectively, these 10 OTUs account for 67.3% of the decrease in predicted BSH abundance. Except for Otu00134, the difference in abundance of each individual OTU between control and rCDI groups is significant
| OTU | BSH abundance | Percent of total decrease | Identification | ||
|---|---|---|---|---|---|
| Control | rCDI | Decrease | |||
| Otu00002 | 1474 | 345 | 1129 | 16.5 | Bacteroides dorei |
| Otu00008 | 932 | 96 | 836 | 12.2 | Blautia wexlerae |
| Otu00007 | 843 | 298 | 545 | 7.9 | Eubacterium rectale |
| Otu00018 | 520 | 48 | 472 | 6.9 | Ruminococcus bromii |
| Otu00017 | 775 | 376 | 399 | 5.8 | Akkermansia muciniphila |
| Otu00021 | 368 | 4 | 364 | 5.3 | family Lachnospiraceae |
| Otu00134* | 254 | 0 | 254 | 3.7 | Eubacterium coprostanoligenes |
| Otu00016 | 287 | 64 | 223 | 3.3 | Parabacteroides distasonis |
| Otu00035 | 213 | 3 | 210 | 3.1 | Blautia luti |
| Otu00011 | 209 | 26 | 183 | 2.7 | genus Bifidobacterium |
Table 4.
Spearman correlation coefficients and p-values for predicted BSH genes and bile acid abundances in patient stool samples.
| Correlation Coefficient | P-value | Bile Acid |
|---|---|---|
| Primary Bile Acids | ||
| −0.39 | 0.0029 | chenodeoxycholate |
| −0.52 | 4.50E-05 | taurochenodeoxycholate |
| −0.58 | 3.80E-06 | Glycochenodeoxycholate |
| −0.42 | 0.0014 | Cholate |
| −0.48 | 0.00017 | Taurocholate |
| −0.58 | 3.90E-06 | Glycocholate |
| Secondary Bile Acids | ||
| 0.46 | 0.00064 | lithocholate |
| 0.43 | 0.0011 | Deoxycholate |
Discussion
In this study, we demonstrate that significant differences in bile salt profiles are associated with the first episode of CDI as well as recurrent CDI. Microbiome analyses further identified underlying alterations of the microbiota associated with CDI, and an association with reduced predicted bacterial bile salt hydrolase gene abundances that may be associated with a diminished capacity to metabolize bile acids.
A healthy intestinal microbiome prevents the development of CDI, though the mechanism of this colonization resistance has been uncertain. This study provides support for the role of secondary and primary bile salts in CDI. The protective effect of the healthy microbiome against the development of CDI may be through its metabolic effects on bile acids. In health, secondary bile acids predominate in stool, which are inhibitory to the germination of C difficile spores. It has been hypothesized that antibiotics, by reducing a subset of bacteria with enzymatic capability of producing secondary bile acids, diminishes the metabolic capacity of the microbiome. A reduction in bile salt metabolizing enzymes leads to an increase in primary bile salts, which are permissive to the germination of C difficile spores, culminating in CDI.34,35
It has been previously demonstrated by Weingarden et al. that FMT done for rCDI induced dramatic changes in fecal bile acid composition.36 Specifically two primary bile acids, cholic acid and chenodoxycholic acid, were significantly elevated in patients prior to FMT but were absent post-FMT and were also absent in the donor stool samples. Additionally, secondary bile acids that were absent pre-FMT were identified in the post-FMT samples. In this study we were able to build on this previous data and target a group of particular interest, those experiencing their first episode of CDI. Additionally, we were able to capture data from these patients prior to the initiation of antibiotics for treatment of C. difficle. Understanding the gut microbial and metabolic changes that occur even at the time of diagnosis of CDI may allow us to identify those at highest risk for recurrence and act earlier with fecal transplantation or other more targeted therapies in the future. This would prevent the significant morbidity of recurrent CDI but also reduce the cost these patients generate with multiple courses of vancomycin and hospitalizations.37
The relationship between bile salts and C. difficile spore germination has been well documented in vitro. Sorg et al reported that C. difficile spores can germinate in the presence of taurocholic acid.10,38 While a wide variety of bacterial species contain bile salt hydrolases, a smaller subset of organisms possess 7α-dehydroxylase and it’s associated enzymatic reactions which lead to the complete conversion of primary bile salts to secondary bile salts.39,40 Recently, Buffie et al identified a microbial cluster felt to be protective against CDI.13 Specifically, they identified Clostridium scindens, a species with known 7α dehydroxylase activity41,42, as being protective against CDI. An OTU identified as C. scindens was present in only a minority of subjects in this study, at very low abundances, making it difficult to assess its relationship to bile salt levels; it is likely only one of several microbial species that perform this function in vivo. Additionally, we identified 10 OTUs that accounted for 67.3% of the decrease in predicted bile salt hydrolase abundance in rCDI patients compared to controls (table 2). This suggests that a core group of bacterial species may possess the majority of bile salt metabolizing capabilities and pending how much of this functional capacity is knocked out with the use of broad spectrum antibiotics, is a key determining factor in who will clear CDI and who will go on to recur.
We evaluated the utility of bile acid profiles to predict the clinical phenotype of patients, i.e., control, fCDI, or rCDI. Specifically, the secondary bile acids taurodeoxycholate and glycodeoxycholate in the plasma were the best predictors of disease status when considering all bile acids as factors in the model. In addition, when ratios were included in the model, the part-to-whole ratio of deoxycholic acid and glycoursodeoxycholic acid in the stool were the most effective predictors of cohort membership. Therefore this ratio may serve as potential biomarkers for recurrence as this ratio was able to distinguish between fCDI and rCDI with higher accuracy than serum taurodeoxycholate and glycodeoxycholate as individual factors. We cannot confirm the validity of these potential biomarkers in this cross sectional study and recognize that a error rate of 15% remains significant. Prospective studies are needed to identify the sensitivity and specificity of these biomarkers in predicting recurrence at the time of first CDI which would allow clinicians to identity those at high risk much earlier in their course.
Additionally, these individual bile acids and ratios provide further mechanistic insight into the roles bile acids may be playing in rCDI, given the significant reduction of secondary bile acids in the rCDI cohort. These finding raise additional questions about the potential for modifying bile acids to be utilized as a therapeutic agent to treat CDI, especially rCDI. Further studies testing the therapeutic potential of secondary bile acids as a novel therapeutic agent would help elucidate whether they have a direct protective effect in treating and also preventing rCDI.
It is important to note the limitations of this study. The rCDI samples were all collected while patients were on chronic antibiotic therapy. Chronic vancomycin therapy may explain the decreased microbial diversity and shift in bile salts. However, patients experiencing recurrent CDI are almost always on chronic antibiotic therapy, so our study may well reflect the metabolic state of the colon underlying CDI recurrence. In addition, the fCDI samples in our study were all collected prior to antibiotic therapy, and these samples demonstrate similar trends to those of rCDI patients. Another limitation of our results is the cross-sectional nature of the study; we do not have follow up data to conclude which, if any, of the fCDI patients experienced a recurrence. We were also unable to control for diet. We did collect dietary information, additionally all donors were household contacts of the rCDI patients with similar diets. We were not able to control for fCDI patient diets however. Further directed studies will be necessary to determine if the observed metabolomic derangements represent a cause or effect of the CDI.
In conclusion, these results re-confirm our prior knowledge of the mechanistic role gut microbiota play in converting primary to secondary bile salts, which in health protect against CDI. When antibiotics kill commensal bacteria, which carry the ability to perform 7α-dehydroxylation, an abundance of primary bile salts likely is a permissive factor for recurrent disease. This is the first study in humans to investigate the role of bile salt profiles in patients experiencing their first episode of CDI who are antibiotic naïve in order to better understand the genesis of this process. We have also suggested the role of secondary bile salts in stool as possible novel biomarkers of recurrence. These insights may help identify those at highest risk for recurrent disease and support the development of new treatments that target bile salt metabolism.
Supplementary Material
Acknowledgments
Declaration of Funding:
This study was funded in full by the American College of Gastroenterology Clinical Research Award. Award ACG-JR-017-2015
Footnotes
Financial disclosures:
no conflicts of interest to report for any authors in regards to the work presented in this manuscript.
Guarantor of the article: Jessica R. Allegretti
Author Contribution:
JRA contributed to study design, acquisition of data, analysis and interpretation of data, and drafting of the manuscript
SK, NL, EB, KB and CC contributed to analysis and interpretation of data, statistical analysis and critical review of the manuscript
JK contributed to study design, interpretation of data and critical review of the manuscript
GG EA and LB contributed to statistical analysis, interpretation of data and critical review of the manuscript.
All authors approved the final version of this manuscript
References
- 1.Bartlett JG. Narrative review: the new epidemic of Clostridium difficile-associated enteric disease. Ann Intern Med. 2006;145:758–764. doi: 10.7326/0003-4819-145-10-200611210-00008. [DOI] [PubMed] [Google Scholar]
- 2.O’Connor JR, Johnson S, Gerding DN. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology. 2009;136:1913–1924. doi: 10.1053/j.gastro.2009.02.073. [DOI] [PubMed] [Google Scholar]
- 3.Kassam Z, Cribb Fabersunne C, Smith MB, et al. Clostridium difficile associated risk of death score (CARDS): a novel severity score to predict mortality among hospitalised patients with C. difficile infection. Aliment Pharmacol Ther. 2016;43:725–733. doi: 10.1111/apt.13546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shivashankar R, Khanna S, Kammer PP, et al. Clinical predictors of recurrent Clostridium difficile infection in out-patients. Aliment Pharmacol Ther. 2014;40:518–522. doi: 10.1111/apt.12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol. 2008;190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rupnik M. Is Clostridium difficile-associated infection a potentially zoonotic and foodborne disease? Clin Microbiol Infect. 2007;13:457–459. doi: 10.1111/j.1469-0691.2007.01687.x. [DOI] [PubMed] [Google Scholar]
- 7.Leffler DA, Lamont JT. Clostridium difficile Infection. N Engl J Med. 2015;373:287–288. doi: 10.1056/NEJMc1506004. [DOI] [PubMed] [Google Scholar]
- 8.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto S, Igimi H, Uchida K, Satoh T, Benno Y, Takeuchi N. Effects of beta-lactam antibiotics on intestinal microflora and bile acid metabolism in rats. Lipids. 1996;31:601–609. doi: 10.1007/BF02523830. [DOI] [PubMed] [Google Scholar]
- 10.Sorg JA, Sonenshein AL. Chenodeoxycholate is an inhibitor of Clostridium difficile spore germination. J Bacteriol. 2009;191:1115–1117. doi: 10.1128/JB.01260-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorg JA, Sonenshein AL. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol. 2010;192:4983–4990. doi: 10.1128/JB.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giel JL, Sorg JA, Sonenshein AL, Zhu J. Metabolism of bile salts in mice influences spore germination in Clostridium difficile. PLoS One. 2010;5:e8740. doi: 10.1371/journal.pone.0008740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buffie CG, Bucci V, Stein RR, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakken JS, Borody T, Brandt LJ, et al. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin Gastroenterol Hepatol. 2011;9:1044–1049. doi: 10.1016/j.cgh.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy S, Churchill S, Bry L, et al. Instrumenting the health care enterprise for discovery research in the genomic era. Genome Res. 2009;19:1675–1681. doi: 10.1101/gr.094615.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pecora ND, Li N, Allard M, et al. Genomically Informed Surveillance for Carbapenem-Resistant Enterobacteriaceae in a Health Care System. MBio. 2015;6:e01030–15. doi: 10.1128/mBio.01030-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsen FA, Kodner RB, Armbrust EV. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics. 2010;11 doi: 10.1186/1471-2105-11-538. 538-2105-11-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole JR, Wang Q, Fish JA, et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–42. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shannon CE. The mathematical theory of communication. 1963. MD Comput. 1997;14:306–317. [PubMed] [Google Scholar]
- 23.Bauer DF. Circular triads when not all paired comparisons are made. Biometrics. 1978;34:458–461. [PubMed] [Google Scholar]
- 24.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baxter NT, Zackular JP, Chen GY, Schloss PD. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome. 2014;2 doi: 10.1186/2049-2618-2-20. 20-2618-2-20. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9:811–818. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- 29.DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langille MG, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cutler DR, Edwards TC, Jr, Beard KH, et al. Random forests for classification in ecology. Ecology. 2007;88:2783–2792. doi: 10.1890/07-0539.1. [DOI] [PubMed] [Google Scholar]
- 32.Hirano S, Masuda N. Epimerization of the 7-hydroxy group of bile acids by the combination of two kinds of microorganisms with 7 alpha- and 7 beta-hydroxysteroid dehydrogenase activity, respectively. J Lipid Res. 1981;22:1060–1068. [PubMed] [Google Scholar]
- 33.Hirano S, Masuda N. Characterization of NADP-dependent 7 beta-hydroxysteroid dehydrogenases from Peptostreptococcus productus and Eubacterium aerofaciens. Appl Environ Microbiol. 1982;43:1057–1063. doi: 10.1128/aem.43.5.1057-1063.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Theriot CM, Koenigsknecht MJ, Carlson PE, Jr, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun. 2014;5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Theriot CM, Young VB. Microbial and metabolic interactions between the gastrointestinal tract and Clostridium difficile infection. Gut Microbes. 2014;5:86–95. doi: 10.4161/gmic.27131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weingarden AR, Chen C, Bobr A, et al. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am J Physiol Gastrointest Liver Physiol. 2014;306:G310–9. doi: 10.1152/ajpgi.00282.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varier RU, Biltaji E, Smith KJ, et al. Cost-effectiveness analysis of treatment strategies for initial Clostridium difficile infection. Clin Microbiol Infect. 2014;20:1343–1351. doi: 10.1111/1469-0691.12805. [DOI] [PubMed] [Google Scholar]
- 38.Giel JL, Sorg JA, Sonenshein AL, Zhu J. Metabolism of bile salts in mice influences spore germination in Clostridium difficile. PLoS One. 2010;5:e8740. doi: 10.1371/journal.pone.0008740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Sorg JA. Microbial bile acid metabolic clusters: the bouncers at the bar. Cell Host Microbe. 2014;16:551–552. doi: 10.1016/j.chom.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doerner KC, Takamine F, LaVoie CP, Mallonee DH, Hylemon PB. Assessment of fecal bacteria with bile acid 7 alpha-dehydroxylating activity for the presence of bai-like genes. Appl Environ Microbiol. 1997;63:1185–1188. doi: 10.1128/aem.63.3.1185-1188.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kitahara M, Takamine F, Imamura T, Benno Y. Assignment of Eubacterium sp. VPI 12708 and related strains with high bile acid 7alpha-dehydroxylating activity to Clostridium scindens and proposal of Clostridium hylemonae sp. nov., isolated from human faeces. Int J Syst Evol Microbiol. 2000;50(Pt 3):971–978. doi: 10.1099/00207713-50-3-971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.