

Abstract
Methanol‐to‐olefin (MTO) catalysis is a very active field of research because there is a wide variety of sometimes conflicting mechanistic proposals. An example is the ongoing discussion on the initial C−C bond formation from methanol during the induction period of the MTO process. By employing a combination of solid‐state NMR spectroscopy with UV/Vis diffuse reflectance spectroscopy and mass spectrometry on an active H‐SAPO‐34 catalyst, we provide spectroscopic evidence for the formation of surface acetate and methyl acetate, as well as dimethoxymethane during the MTO process. As a consequence, new insights in the formation of the first C−C bond are provided, suggesting a direct mechanism may be operative, at least in the early stages of the MTO reaction.
Keywords: methanol-to-olefin reaction, operando spectroscopy, reaction mechanisms, solid-state NMR spectroscopy, zeolites
The growing concern on climate change stimulates both academia and industry to develop more sustainable chemical processes. The methanol‐to‐olefin (MTO) process over H‐SAPO‐34 and H‐ZSM‐5 is one of these promising catalytic routes, which provides a means to indirectly produce important chemical building blocks, such as propylene, from natural gas, biomass and waste.1, 2, 3, 4, 5 Owing to the inherent complex nature of the MTO process, more than 20 different mechanisms have already been proposed in the literature.6 Among them, the most experimentally verified one is the “dual‐cycle” mechanism, which is a modified version of the “hydrocarbon pool” (HCP) mechanism, originally proposed by Dahl and Kolboe.2, 7 However, the exact route for the formation of the first carbon–carbon (C−C) bond in this mechanism still remains elusive.8 For a long time, the commonly acknowledged assumption was that the presence of traces of impurities (e.g. in the used methanol, catalyst, and/or carrier gas) is responsible for the first C−C bond formation. An alternative option, known as the direct mechanism (e.g. coupling of two methanol or dimethyl ether molecules), still lacks clear experimental support.9 Interestingly, Hunger and co‐workers showed already in 2006 that traces of organic impurities neither have any significant influence on product distribution, nor do they govern the formation of HCP species.10 Since then, the research groups of Hunger,10, 11, 12, 13, 14, 15 Kondo,16 Fan,17, 18 Copéret and Sautet,19 and Lercher20 have provided both experimental and theoretical evidences in support of the direct mechanism during the initial stages of the MTO process.
The earlier proposed direct MTO routes include the a) oxonium ylide, b) carbene, and c) methane–formaldehyde mechanisms.2, 6 Although, these mechanisms still have their (limited) experimental evidences in literature, the theoretical probability of such direct coupling is anticipated to be low because of their higher activation energies and unstable reaction intermediates.2, 21 However, a surface methoxy species (SMS), formed upon adsorption of methanol onto a Brønsted acid site, is the most experimentally verified intermediate of the MTO process.14, 22 Moreover, some recent reports identified that several initial hydrocarbon species can be directly generated as a result of coupling between SMS and methanol/dimethyl ether (DME). For instance, with the help of IR and GC‐MS spectroscopy Kondo et al. proposed that propylene can be generated as a result of the coupling between SMS and ethylene.16 Based on both experimental and theoretical evidences, the direct C−C bond‐forming route proposed by Fan et al. involves: i) the formation of a methoxymethyl cation (CH3OCH2 + from SMS and DME) and ii) its subsequent direct C−C coupling with another DME or methanol molecule to form CH3OCH2CH2OR (R=H, CH3) moiety, which is considered to be a precursor for olefins.17 An interesting study was reported by the research groups of Copéret and Sautet, where the formation of the first C−C bond from DME was proposed to be catalyzed by extra‐framework Al atoms in acidic zeolites.19 Their experimental and theoretically verified proposal involves the C−C bond formation over Al2O3 through the C−H activation of methane in an alumino‐oxonium (AlO=CH2 +)/methane adduct, which has conceptual resemblance with the “methane–formaldehyde” mechanism proposed by Hutchings et al.,23 Kubelkova and co‐workers24 and Hirao and co‐workers.25 Independently from each other, the research groups of Copéret and Sautet19 and Lercher et al.20 postulated the involvement of formate and acetate species bound to the zeolite surface during the first C−C bond formation at the early stages of the MTO reaction. This surface formate species could lead to the formation of dimethoxymethane (DMM, CH3OCH2OCH3) during the course of the MTO reaction, as suggested by the research groups of Kubelkova,24 and Chang.26 Within this context it is important to mention that Lercher and co‐workers very recently postulated that methyl acetate (CH3CO2CH3) is the very first C−C bond‐containing intermediate (derived from surface‐bound acetate species) during the MTO reaction over ZSM‐5.20 Despite these encouraging results, no direct evidence has been presented for the existence of DMM as well as surface‐bound acetate and methyl acetate species during the zeolite‐catalyzed MTO reaction.
In this work, we provide spectroscopic evidence for the direct formation of the first C−C bond during the H‐SAPO‐34‐catalyzed MTO reaction, through the identification of surface‐trapped formate and acetate species and methyl acetate as well as DMM. This has been made possible by using a combination of UV/Vis diffuse reflectance spectroscopy (DRS), mass spectrometry (MS), and magic angle spinning (MAS)27 solid‐state nuclear magnetic resonance (ssNMR) studies using a 13C‐methanol reacted catalyst. The multispectroscopic approach, illustrated in Figure S1 in the Supporting Information, allows the investigation of the initial stages of the MTO reaction, consisting of the formation of surface formate/acetate, methyl acetate and DMM from SMS.
In the first stage of our study, operando UV/Vis DRS with online MS was used to identify and differentiate between neutral and carbocationic HCP‐type species, as well as gas‐phase products formed during the MTO reaction over zeolite H‐SAPO‐34 at a reaction temperature of 673 K for 60 minutes. The results of this approach are summarized in Figures S2 and S3. Figure S2a reveals major spectral changes during the first 10 minutes of the MTO reaction, as absorption bands at 297, 350, 387, 414, and 640 nm increase in intensity with increasing time‐on‐stream (Figure S3b). After 7 minutes of reaction, a decrease in intensity was observed only in the case of the 350 and 640 nm bands, implying the existence of intramolecular transformations within the zeolite framework. The observed bands at 297, 350, 479, and 640 nm are attributed to neutral methylated benzenes, dienylic carbocationic/methylbenzenium (up to three methyl groups) ions, trienylic and methylated poly‐arenium ions, respectively.10, 11, 12, 28, 29, 30 Similarly, the 387 nm band is characteristic for a hexamethylbenzenium ion (HMB+) and its bathochromic shift to 414 nm with increasing time‐on‐stream can be explained by the further methylation of HMB+ to, for example, hepta‐methylbenzenium ions.31 We propose that the cracking of polyaromatics into trienylic carbenium species and olefins are responsible for the concomitant increase and decrease of the 479 and 640 nm bands, respectively, after 10 minutes of reaction (Figures S3b). Interestingly, the online MS data presented in Figures S2c,d reveal the presence of methane, DME, lower olefins (i.e., ethylene, propylene, and butylene) and also traces of DMM. The existence of DMM is supported by its characteristic m/z of 75, which exclusively belongs to DMM (Figure S3c). The gradually increasing amount of DMM with increasing time‐on‐stream could be attributed to the formation of a higher quantity of its precursor, the surface formate species, during the course of reaction. This is consistent with the work of Kubelkova et al., who used temperature‐programmed desorption to identify DMM.24
In the second stage of our study, we have performed advanced ssNMR spectroscopy on H‐SAPO‐34 after being exposed to the MTO reaction for 30 minutes at 673 K using 13C‐enriched methanol. Such isotope enrichment not only greatly increased ssNMR signal intensities but also enabled a detailed analysis of two‐dimensional ssNMR correlation experiments discussed below. The 1H‐13C cross‐polarization (CP) and 13C direct excitation (DE) ssNMR spectra at 10 kHz MAS of the MTO‐reacted catalyst are presented in Figure 1. The following features were observed: i) 18–22 ppm aliphatic methyl groups, ii) 50–65 ppm methoxy groups, iii) 90–105 ppm acetal moiety, iv) 120–155 ppm (methylated) aromatic/olefinic groups, and v) 170–185 ppm carbonyl groups.2, 11, 12, 13, 32, 33, 34, 35 Because of the overlap with spinning side bands, we employed higher MAS rates to unambiguously identify weak signals at 200 and 220 ppm, indicative for poly‐methylbenzenium ions (Figure S4 and Table S1). Interestingly, we do not observe strong signals around 40 ppm, which indicates the absence of typical sp3 CH2 groups. The strongest aliphatic signal at 52 ppm was due to the methanol, whereas the peaks at 55, 57, and 62 ppm are due to acetate, methoxy/SMS, and adsorbed (side‐on) DME, respectively (Table S1). Moreover, the strongest peak at 132 ppm suggests the presence of neutral methylated benzenes and HMB+ as the dominating HCP intermediate.
Figure 1.
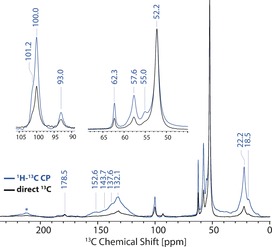
13C DE (black) and 1H‐13C CP (blue) ssNMR spectra of trapped products obtained after the methanol‐to‐olefin (MTO) reaction over H‐SAPO‐34 at 673 K for 30 minutes (*=spinning sideband).
In order to elucidate in more detail the zeolite‐trapped species, additional 2D ssNMR spectra were recorded: i) 13C PDSD (Proton‐Driven Spin‐Diffusion) spectra36 with short (30 ms) and long (150 ms) mixing times for short‐ and long‐range 13C–13C correlations, respectively, and ii) proton‐detected 2D 13C–1H CP‐HETCOR (HETCOR: HETero‐nuclear CORrelation spectroscopy)37 with a short (50 μs) and long (500 μs) CP contact time for the second 13C–1H CP, for short‐ and long‐range 13C–1H correlations (Figure S5). In those spectra we can clearly distinguish between the broad signals of trapped/immobile aromatic species, and sharp signals for mobile non‐aromatic/small molecules. The latter signals remain visible under (dipolar‐based) CP experiments because of their restricted molecular mobility in H‐SAPO‐34. Because of the large difference in NMR signal behavior, the NMR spectra were processed to either focus on the broad signals (using only initial points and applying additional line‐broadening) or on the sharp signals (using full dataset and additional linear prediction). The strong signal at 52.2 (13C) and 3.59 (1H) ppm can easily be assigned to surface adsorbed methanol, while the signal at 57.7 (13C) and 3.54 (1H) ppm is due to surface‐adsorbed methoxy species (Figure 2 a,b).14, 15, 22 Interestingly, a strong cross‐peak between these C signals is observed already at short (13C,13C) mixing times (Figures 2 a and S5), which indicates the close proximity of surface adsorbed methanol and SMS. Such close proximity could be a possible signature of their reaction through the polarization of the C−H bond of SMS by a neighbouring adjacent oxygen, as observed independently before by the groups of Kondo, Anderson, and Senchenya (Figure 2 c).16, 38, 39 As a result, SMS of a carbene/ylide nature are likely to be formed, as it was previously characterized by Kondo et al. and Hunger et al. by IR and NMR spectroscopy, respectively.15, 40 The existence of such carbene‐like SMS was experimentally confirmed as well, by a trapping experiment with cyclohexane as a probe molecule at ≥493 K, where methylcyclohexane was formed through an insertion reaction of carbene/ylide into the sp3 C−H bond of cyclohexane.14, 15 Therefore, we believe the strong cross‐peak at short (13C,13C) mixing times between 57.7 and 52.2 ppm is an indication of a similar type of carbene/ylide insertion into the sp3 C−H of the incoming methanol, as shown in Figure 2 c. It should be noted that SMS is highly capable of forming C−C bonds at higher reaction temperatures, which is well established in zeolite chemistry.1, 3, 14, 16, 22 Only at a long mixing time, an additional cross‐peak with 57.7 was observed at 100.1 ppm, whose corresponding 1H resonates at 4.72 ppm, which can be assigned to dimethoxymethane (DMM).41, 42, 43 Remarkably, a second minor signal at 101.3 ppm is observed for the DMM‐CH2 group, that shows an exchange cross‐peak with 100.1 at longer mixing times (Figure 2 a). This is an indication of exchange between anomeric DMM structures on a sub‐second time‐scale (trans, trans and gauche, gauche, Figure 2 d).42, 44 Although the plausible existence and/or influence of DMM during MTO was already proposed by Kubelkova et al.,24 Chang et al.26 and Lercher et al.,20 these results provide the first spectroscopic evidence that DMM is indeed formed during MTO. The acetal signal at 93 ppm also has a cross‐peak in the CH spectrum at 4.7 ppm, but lacks a correlation with other C signals, hence identified as methanediol, as the hydrolyzed product of DMM (Figure 2 e).41
Figure 2.
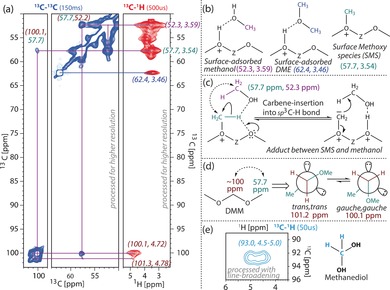
ssNMR spectra of methanol, methoxy, and acetal species in H‐SAPO‐34 after the methanol‐to‐olefin (MTO) reaction for 30 minutes at 673 K. a) Zooms from 2D 13C–13C (blue) and 13C–1H (red) ssNMR spectra with long mixing (150 ms) or CP contact time (500 μs), respectively. b) NMR assignment of surface species. c) Identification of a surface adduct between SMS and methanol (normal arrows: electron flows, dotted arrows: 13C‐13C NMR correlation). d) Chemical exchange of anomeric conformations of DMM observed in 13C–13C spectra. e) Identification of methanediol in 13C–1H spectrum (light blue) with short CP contact time (50 μs).
In the carbonyl region of the 13C–13C spectra, clear cross‐peaks to methyl carbon atoms were observed at both long and short mixing times (Figure 3), indicative for acetate species.20 While the signal at 180.5 ppm shows no additional cross‐peaks, in accordance with surface‐bound acetate, the signal at 178.5 ppm has a clear cross‐peak with a 13C signal at 55.1 ppm at longer mixing times. This 13C signal correlates with a H signal at 3.82 ppm and has an additional very weak signal with the methyl signal at 22.3 ppm at longer C−C mixing times. This cross‐peak pattern is typical for methyl acetate (Figure 3 a). Similarly, in the 2D 1H–13C ssNMR spectra, the C signl at 173 ppm correlates with a H signal at 8–8.5 ppm, which is compatible with surface formate (Figure 3 b).19 Actually, two signals were observed in this region, which could be indicative for different resonance structures for surface formate. Together, these observations support the existence of surface‐bound formate/acetate species‐assisted C−C bond formation route during MTO, as has recently been proposed by the research groups of Copéret and Sautet,19 and Lercher et al.20 Our study provides evidence for the formation of the first C−C bond‐containing intermediates (i.e., methyl acetate and surface acetate) during the MTO reaction over H‐SAPO‐34. Several other correlations between aliphatic (13C/1H: 20–30/2.2–2.4 ppm) and aromatic moieties (13C/1H: 132–154/ 6–10 ppm) in Figure 3 c reveal the presence of various HCP‐type intermediates; that is, conjugated polyenes, methylated neutral benzenes and benzeniums, which are known to be formed in the alkene and arene cycles of the HCP mechanism (Scheme S1 and Table S1).2, 11, 12, 13, 32, 33, 34, 35 However, the lack of signals in the 65–90 ppm region signifies the absence of a CH3OCH2CH2OR (R=H, CH3) moiety, as previously proposed by Fan and co‐workers.17
Figure 3.
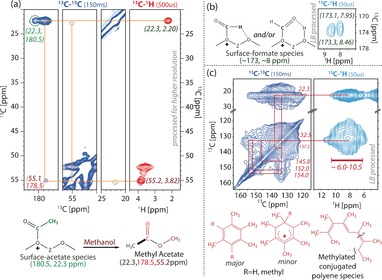
ssNMR correlations of acetate, formate, and methylated polyene/benzene in H‐SAPO‐34 after the methanol‐to‐olefin (MTO) reaction for 30 minutes at 673 K. a) Zooms from 2D 13C–13C (blue) and 13C–1H (red) MAS ssNMR spectra with long mixing (150 ms) or CP contact time (500 μs), respectively, indicating surface acetate and methyl acetate resonances. b) ssNMR signals of surface‐bound formate in the 13C–1H spectra (light blue) with a short CP contact time (50 μs). c) Zoom of aromatic signals from 2D 13C–13C (blue) and 13C–1H (light blue) MAS NMR spectra with long mixing (150 ms) or short CP contact time (50 μs), respectively.
Based on the above‐described results, we propose in Scheme 1 a pathway for the formation of the first C−C bond during the initial stages of the MTO process. First, the CH3 group of SMS, formed by methylation of a Brønsted acid site (denoted as ZeOH, Scheme 1), interacts initially with another molecule of methanol/DME (Species A in path a, Scheme 1). The methyl C−H of SMS is then polarized because of its interaction with a neighboring O and the first C−C bond formation step is assisted by hydrogen bonding with the C−H proton (species A in path a, Scheme 1). As a result, the first C−C bond formation takes place through the typical insertion reaction of carbene/ylide from SMS into the sp3 C−H bond of methanol/DME during MTO (species A/A′ in path a, Scheme 1 and Figure 2 c).14, 38, 39, 40 In addition to our ssNMR observation (Figure 2 a–c), the existence of such neighboring oxygen group‐assisted SMS‐type species has been characterized by Hunger et al.14, 15 and Kondo et al.16, 40 Species A′ eventually led to surface‐ethanolic species (B in path a, Scheme 1), which undergoes dehydration (through protonation of alcohol/ether oxygen atoms by Brønsted acid sites of the zeolite) to form ethylene and regenerates ZeOH.17 In principle, the presence of two adjacent methoxy groups (Figure 2 a–c) could also be consistent with the existence of the oxonium ylide mechanism (Scheme S2). However, considering the reported theoretical calculations based on the oxonium mechanism and the observed carbene/ylide features of SMS in our study, we favor the carbene/ylide insertion pathway.2, 6 Moreover, our NMR data is not fully compatible with the intermediates of oxonium mechanism, as was discussed in Scheme S2. Species B alternatively could also be derived from the direct C−H bond activation of the CH3 group of SMS by methanol, as computationally confirmed by Blaszkowski and van Santen (path b in Scheme 1).45 Here, the additional water or methanol can act as an assisting molecule to allow favorable transition‐state geometry and reduce the energy barrier.45
Scheme 1.
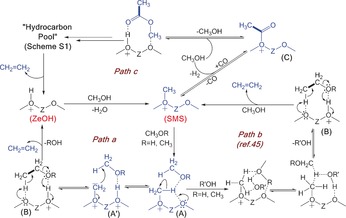
Plausible catalytic cycle of the “direct mechanism” for the first C−C bond formation during the early stages of the MTO reaction. The reaction products and intermediates indicated in blue have been experimentally observed in this work. The first C−C bond in paths a/b is highlighted in bold.
However, Hirao and co‐workers indicated that the spectroscopic detection of surface species B would be very difficult as the final conversion to ethylene is expected to be fast.25 Next, the first C−C bond could also be derived from surface‐bound acetate species (C in path c, Scheme 1), which is a “Koch‐type” carbonylation product of SMS (Figure 3 a).20, 46 Methyl acetate is formed from the surface‐bound acetate species, which initiates the formation of HCP species and hence, produces an olefin (path c, Scheme 1 and Section SIII in the Supporting Information). Therefore, the present work also provides evidence in support of the first C−C bond formation route through methanol carbonylation of SMS (path b, Scheme 1), as also recently proposed by Lercher and co‐workers.20 Indeed, the observed microscopic reversibility between methanol and methyl acetate (Section SIII in the Supporting Information) provides justification in support of the formation of methyl acetate, DMM, surface formate, and acetate directly from methanol during the induction period of the MTO reaction.
Similar “Koch‐type” carbonylation of the zeolite surface could also lead to the formation of surface formate (Scheme S3),19 as observed by ssNMR spectroscopy (Figure 3 b).46 The highly electrophilic surface formate species then becomes available for a nucleophilic attack by methanol to form an acetal of formaldehyde, DMM, on the solid acid (Scheme S2 and Figure 2 a,d).24 Therefore, in addition to the existence of methyl acetate, DMM, surface formate and acetate, the spectroscopic identification of surface intermediate A/A′ (Schemes 1 and Figure 2 c) provides evidence in favor of the direct C−C bond formation route from methanol during the early MTO stages. The first few olefins from the “direct mechanism” (paths a and b in Scheme 1) then participate in the alkene part of the HCP mechanism (Scheme S1 and Table S1) and hence, initiate the auto‐catalytic part of the MTO process.
In conclusion, our data provide direct spectroscopic evidence for the formation of surface‐bound acetate (i.e., a direct C−C bond‐containing reaction intermediate), methyl acetate (i.e., the first C−C bond containing molecule) as well as dimethoxymethane during the MTO process over H‐SAPO‐34. We propose the surface species‐assisted “direct mechanism”, as outlined in Scheme 1 as the most likely candidate for the formation of the first C−C bond during the initial stages of the MTO process.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research work was funded by a European Research Council (ERC) Advanced grant (number 321140 to BMW) and supported by NWO (Middelgroot program, grant number 700.58.102 to MB) as well as a grant from the Deanship of Scientific Research (DSR) of King Abdulaziz University (grant number T‐002‐431 to BMW). We thank Dr. Pieter C. A. Bruijnincx for fruitful discussions.
A. D. Chowdhury, K. Houben, G. T. Whiting, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, M. Baldus, B. M. Weckhuysen, Angew. Chem. Int. Ed. 2016, 55, 15840.
Contributor Information
Prof. Dr. Marc Baldus, Email: M.Baldus@uu.nl
Prof. Dr. Bert M. Weckhuysen, Email: b.m.weckhuysen@uu.nl
References
- 1. Tian P., Wei Y., Ye M., Liu Z., ACS Catal. 2015, 5, 1922–1938. [Google Scholar]
- 2. Olsbye U., Svelle S., Lillerud K. P., Wei Z. H., Chen Y. Y., Li J. F., Wang J. G., Fan W. B., Chem. Soc. Rev. 2015, 44, 7155–7176. [DOI] [PubMed] [Google Scholar]
- 3. Ilias S., Bhan A., ACS Catal. 2013, 3, 18–31. [Google Scholar]
- 4. Vogt E. T. C., Whiting G. T., Dutta Chowdhury A., Weckhuysen B. M., Adv. Catal. 2015, 58, 143–314. [Google Scholar]
- 5. Olsbye U., Svelle S., Bjrgen M., Beato P., Janssens T. V. W., Joensen F., Bordiga S., Lillerud K. P., Angew. Chem. Int. Ed. 2012, 51, 5810–5831; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5910–5933. [Google Scholar]
- 6. Stöcker M., Microporous Mesoporous Mater. 1999, 29, 3–48. [Google Scholar]
- 7. Dahl I. M., Kolboe S., Catal. Lett. 1993, 20, 329–336. [Google Scholar]
- 8. Lercher J. A., ACS Cent. Sci. 2015, 1, 350–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Song W., Marcus D. M., Fu H., Ehresmann J. O., Haw J. F., J. Am. Chem. Soc. 2002, 124, 3844–3845. [DOI] [PubMed] [Google Scholar]
- 10. Jiang Y., Wang W., Reddymarthala V., Huang J., Sulikowski B., Hunger M., J. Catal. 2006, 238, 21–27. [Google Scholar]
- 11. Dai W., Wang C., Dyballa M., Wu G., Guan N., Li L., Xie Z., Hunger M., ACS Catal. 2015, 5, 317–326. [Google Scholar]
- 12. Dai W., Wu G., Li L., Guan N., Hunger M., ACS Catal. 2013, 3, 588–596. [Google Scholar]
- 13. Dai W., Dyballa M., Wu G., Li L., Guan N., Hunger M., J. Phys. Chem. C 2015, 119, 2637–2645. [Google Scholar]
- 14. Wang W., Hunger M., Acc. Chem. Res. 2008, 41, 895–904. [DOI] [PubMed] [Google Scholar]
- 15. Wang W., Buchholz A., Seiler M., Hunger M., J. Am. Chem. Soc. 2003, 125, 15260–15267. [DOI] [PubMed] [Google Scholar]
- 16. Yamazaki H., Shima H., Imai H., Yokoi T., Tatsumi T., Kondo J. N., J. Phys. Chem. C 2012, 116, 24091–24097. [Google Scholar]
- 17. Li J., Wei Z., Chen Y., Jing B., He Y., Dong M., Jiao H., Li X., Qin Z., Wang J., Fan W., J. Catal. 2014, 317, 277–283. [Google Scholar]
- 18. Wei Z., Chen Y.-Y., Li J., Wang P., Jing B., He Y., Dong M., Jiao H., Qin Z., Wang J., Fan W., Catal. Sci. Technol. 2016, 6, 5526–5533. [Google Scholar]
- 19. Comas-Vives A., Valla M., Copéret C., Sautet P., ACS Cent. Sci. 2015, 1, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Y., Müller S., Berger D., Jelic J., Reuter K., Tonigold M., Sanchez-Sanchez M., Lercher J. A., Angew. Chem. Int. Ed. 2016, 55, 5723–5726; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5817–5820. [Google Scholar]
- 21. Lesthaeghe D., Van Speybroeck V., Marin G. B., Waroquier M., Ind. Eng. Chem. Res. 2007, 46, 8832–8838. [DOI] [PubMed] [Google Scholar]
- 22. Wang W., Seiler M., Hunger M., J. Phys. Chem. B 2001, 105, 12553–12558. [Google Scholar]
- 23. Hutchings G. J., Gottschalk F., Hall M. V. M., Hunter R., J. Chem. Soc. Faraday Trans. 1 1987, 83, 571–583. [Google Scholar]
- 24. Novakova J., Kubelkova L., Dolejsek Z., J. Catal. 1987, 108, 208–213. [Google Scholar]
- 25. Tajima N., Tsuneda T., Toyama F., Hirao K., J. Am. Chem. Soc. 1998, 120, 8222–8229. [Google Scholar]
- 26. Chang C., Silvestri A. J., J. Catal. 1977, 47, 249–259. [Google Scholar]
- 27. Andrew E. R., Bradbury A., Eades R. G., Nature 1958, 182, 1659–1659. [Google Scholar]
- 28. Qian Q., Vogt C., Mokhtar M., Asiri A. M., Al-Thabaiti S. A., Basahel S. N., Ruiz-Martínez J., Weckhuysen B. M., ChemCatChem 2014, 6, 3396–3408. [Google Scholar]
- 29. Borodina E., Meirer F., Lezcano-Gonzalez I., Mokhtar M., Asiri A. M., Al-Thabaiti S. A., Basahel S. N., Ruiz-Martinez J., Weckhuysen B. M., ACS Catal. 2015, 5, 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wulfers M. J., Jentoft F. C., ACS Catal. 2014, 4, 3521–3532. [Google Scholar]
- 31. Bjørgen M., Bonino F., Kolboe S., Lillerud K. P., Zecchina A., Bordiga S., J. Am. Chem. Soc. 2003, 125, 15863–15868. [DOI] [PubMed] [Google Scholar]
- 32. Dai W., Scheibe M., Guan N., Li L., Hunger M., ChemCatChem 2011, 3, 1130–1133. [Google Scholar]
- 33. Li J., Wei Y., Chen J., Tian P., Su X., Xu S., Qi Y., Wang Q., Zhou Y., He Y., Liu Z., J. Am. Chem. Soc. 2012, 134, 836–839. [DOI] [PubMed] [Google Scholar]
- 34. Xu S., Zheng A., Wei Y., Chen J., Li J., Chu Y., Zhang M., Wang Q., Zhou Y., Wang J., Deng F., Liu Z., Angew. Chem. Int. Ed. 2013, 52, 11564–11568; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11778–11782. [Google Scholar]
- 35. Wang C., Wang Q., Xu J., Qi G., Gao P., Wang W., Zou Y., Feng N., Liu X., Deng F., Angew. Chem. Int. Ed. 2016, 55, 2507–2511; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2553–2557. [Google Scholar]
- 36. Bloembergen N., Physica 1949, 15, 386–426. [Google Scholar]
- 37. Weingarth M., van der Cruijsen E. A. W., Ostmeyer J., Lievestro S., Roux B., Baldus M., J. Am. Chem. Soc. 2014, 136, 2000–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salehirad F., Anderson M. W., J. Catal. 1996, 164, 301–314. [Google Scholar]
- 39. Kazansky V., Senchenya I. N., J. Catal. 1989, 119, 108–120. [Google Scholar]
- 40. Yamazaki H., Shima H., Imai H., Yokoi T., Tatsumi T., Kondo J. N., Angew. Chem. Int. Ed. 2011, 50, 1853–1856; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1893–1896. [Google Scholar]
- 41. Paik Y., Kim S.-S., Han O. H., Angew. Chem. Int. Ed. 2008, 47, 94–96; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 100–102. [Google Scholar]
- 42. Anderson J. E., Heki K., Hirota M., Jørgensen F. S., J. Chem. Soc. Chem. Commun. 1987, 554–555. [Google Scholar]
- 43. Migda W., Rys B., Magn. Reson. Chem. 2004, 42, 459–466. [DOI] [PubMed] [Google Scholar]
- 44. Juaristi E., Cuevas G., Tetrahedron 1992, 48, 5019–5087. [Google Scholar]
- 45. Blaszkowski S. R., van Santen R. A., J. Am. Chem. Soc. 1997, 119, 5020–5027. [Google Scholar]
- 46. Jiang Y., Hunger M., Wang W., J. Am. Chem. Soc. 2006, 128, 11679–11692. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary