

Summary
Cancer‐germline genes in both humans and mice have been shown to encode antigens susceptible to targeting by cytotoxic CD8 T effector cells (CTL). We analysed the ability of CTL to kill different tumour cell lines expressing the same cancer‐germline gene P1A (Trap1a). We previously demonstrated that CTL expressing a T‐cell receptor specific for the P1A35–43 peptide associated with H‐2Ld, although able to induce regression of P1A‐expressing P815 mastocytoma cells, were much less effective against P1A‐expressing melanoma cells. Here, we analysed parameters of the in vitro interaction between P1A‐specific CTL and mastocytoma or melanoma cells expressing similar levels of the P1A gene and of surface H‐2Ld. The mastocytoma cells were more sensitive to cytolysis than the melanoma cells in vitro. Analysis by video‐microscopy of early events required for target cell killing showed that similar patterns of increase in cytoplasmic Ca2+ concentration ([Ca2+]i) were induced by both types of P1A‐expressing tumour cells. However, the use of CTL expressing a fluorescent granzyme B (GZMB‐Tom) showed a delay in the migration of cytotoxic granules to the tumour interaction site, as well as a partially deficient GZMB‐Tom exocytosis in response to the melanoma cells. Among surface molecules possibly affecting tumour–CTL interactions, the mastocytoma cells were found to express intercellular adhesion molecule‐1, the ligand for LFA‐1, which was not detected on the melanoma cells.
Keywords: cytolytic T lymphocyte, fluorescent granzyme B, melanoma, tumour
Abbreviations
- CTL
cytolytic T lymphocyte
- CTV
Cell Tracker Violet
- GZMB
granzyme B
- MFI
mean fluorescence intensity
- Tom
tdTomato
Introduction
CD8 T cells specific for melanoma tumour‐associated antigens expressed as MHC class I‐bound peptides have been detected in most melanoma patients. These cells, however, were generally unable to control tumour development (reviewed in ref. 1). The inefficiency of tumour antigen‐specific T cells developing in cancer patients has been attributed to: (i) their inefficient differentiation into effector cells; (ii) immune control mechanisms intrinsic to the T cells or dependent on regulatory T cells, the latter being relieved by antibody reagents targeting proteins involved in ‘immune checkpoint controls’; and (iii) the tumour environment being conducive to ‘exhaustion’ of the T cells and producing immunomodulatory/immunosuppressive cytokines. Treatments with monoclonal antibodies (mAb) blocking the engagement of T‐cell‐expressed inhibitory receptors such as cytotoxic T‐lymphocyte antigen 4 (CTLA‐4) and programmed death 1 (PD‐1) were recently reported to provide long‐term clinical benefit for some cancer patients, emphasizing the potential role of the immune system in tumour eradication.2, 3, 4 Furthermore, the increase in the frequency and diversity of tumour‐specific CD8 T cells in anti‐CTLA‐4 mAb‐treated patients5 and the observed proliferation of intra‐tumoral CD8 T cells in patients that responded to anti‐PD‐1 ligand mAb treatment6 underline the importance of CD8 T cells as anti‐tumour effector cells in humans. Regardless of the type of therapy administered, a favourable prognosis was also found associated with the presence of cytotoxic T cells and expression of transcripts associated with cytolytic function (i.e. perforin, granzymes) in patients' primary tumours.7
The effector functions of differentiated CD8 T cells include their capacity to kill target cells through the release of cytolytic granules at the immune synapse (reviewed in ref. 8). Indeed, when optimally stimulated, CD8 T cells acquire the expression of exocytic granules belonging to the lysosome family, which contain the cytolytic enzymes perforin and granzymes. The different steps involved in the process of tumour cell killing have been clearly identified. Hence, on recognition of target cells via the T‐cell receptor (TCR), cytotoxic granules are anchored to microtubules and migrate towards the immune synapse, together with the microtubule organizing centre. Polarized cytotoxic granules then fuse with the cell membrane, which leads to the release of the granule content (including perforin and granzymes) in the synaptic cleft.9 The released perforin forms calcium‐dependent pores in the target cell membrane. This allows for granzymes to access the target cell cytoplasm and induce cell death either directly (reviewed in ref. 8) or after their endocytosis following a membrane repair response in the target cell.10 Although the mechanistic details of CTL activation leading to exocytosis of lytic granules have been extensively described,11, 12 the molecular basis for the resistance of some tumour cells to CTL‐mediated killing is less clear.
Calcium ion signals crucially control functional T‐lymphocyte differentiation and activation.13 They are initiated by engagement of the TCR, which results in activation of phospholipase Cγ, production of inositol 1,4,5‐trisphosphate and diacylglycerol. In turn the inositol 1,4,5‐trisphosphate causes the release of Ca2+ from the endoplasmic reticulum stores and leads thereby to the activation of Ca2+ release‐activated Ca2+ (CRAC) channels and extracellular Ca2+ entry into the cytoplasm (reviewed in ref. 14). Store‐operated Ca2+ entry (SOCE) is required for cytolysis by CD8 T cells and, in particular, for TCR‐induced lytic granule exocytosis.15 Additionally, L‐type calcium channels have also been implicated in the development of cytolytic effector cells.16
We analysed the efficiency of CD8 T effector cells to kill different tumour cell lines expressing the same cancer‐germline gene P1A (Trap1a). We previously demonstrated that CD8 T effector cells expressing a TCR‐specific for the P1A35–43 peptide (TCRP1A) associated with H‐2Ld that are able to induce regression of P1A‐expressing mastocytoma P815 cells,17 were much less efficient against P1A‐expressing melanoma cells.18, 19 Here, we analysed parameters of the in vitro interaction between P1A‐specific TCRP1A CD8 T effector cells and mastocytoma or melanoma cells expressing similar levels of the P1A gene and of surface H‐2Ld. We previously generated knock in (KI) mice expressing Granzyme B (GZMB) as a fusion protein with red fluorescent tdTomato (GZMB‐Tom).20 Using these mice, we here derived P1A‐specific TCRP1A CD8 T effector cells (CTL) expressing GZMB‐Tom and monitored the early events of CTL activation by video‐microscopy. These events were measured by changes in [Ca2+]i followed by the re‐localization of granules containing the fluorescent GZMB‐Tom upon CTL interaction with distinct P1A‐expressing tumour target cells.
Material and methods
Mice
Mice (Gzmb‐Tom) genetically modified by homologous recombination to express GZMB‐Tomato instead of GZMB have been described20 and are registered as (EM:05732) at EMMA http://strains.emmanet.org/mutant_types.php#keyword=5732.
For this study, Gzmb‐Tom mice were crossed with TCRP1A mice that express as a transgene the H‐2Ld/P1A35‐43‐specific TCR on the Rag‐1−/− B10.D2 background.21 The derived TCRP1A Rag‐1−/− B10.D2 mice expressing GZMB‐Tom are designated as TCRP1A‐GZMB‐Tom. All mice were kept at the CIML animal facility in specific pathogen‐free conditions. Mouse genotyping was performed by PCR as described previously.20
Mice and ethics statement
All procedures were approved by the Regional ‘Provence‐Alpes‐Cote d'Azur’ Committee on Ethics for Animal Experimentation (authorization: no 13.521, date: 08/10/2012) and were in accordance with French and European directives. All efforts were made to minimize animal suffering.
Cell lines
Melanoma cell lines were derived from either TiRP (Tyr‐iRas‐P1A‐transgenic Ink4a/Arf flox/flox) mice22 kept on the B10.D2 background18 (line T‐1236) or TiRP‐RFP mice (line T‐RFP‐69)23 obtained after crossing TiRP B10.D2 mice with a Cre‐reporter mouse expressing a tandem‐dimer red fluorescent protein (RFP).24 Melanoma cells cultured in RPMI‐1640 complete medium (10% fetal calf serum, antibiotics, glutamine 2 mm, 2‐mercaptoethanol 50 µm) were collected before confluence after one wash with PBS followed by treatment with PBS, EDTA Trypsin (Gibco) for 5 min at 37° and one wash in RPMI‐1640 culture medium. P1A‐positive (P815 subline) mastocytoma P511 and its P1A‐negative variant P1.204 obtained from Dr B. Van den Eynde (Ludwig Institute for Cancer Research, Brussels, Belgium)25 were cultured in RPMI‐1640 complete medium.
Quantitative RT‐PCR
Quantitative RT‐PCR for expression of endogenous P1A (Trap1a) and for the TiRP transgene‐encoded HRasG12V and P1A (Trap1a) transcripts in melanoma cell lines was performed as previously described (see Supplementary methods in refs 18, 23).
T‐cell activation and differentiation
CD8 T cells from lymph nodes and spleen of TCRP1A‐Gzmb‐Tom mice were purified by negative selection. They were activated in 24‐well culture plates (105 cells/well) by splenocytes from congeneic Rag−/− mice (2 × 105 cells) loaded for 1 hr with peptide P1A35‐43 (P1Ap) 10−7 m. For some experiments lymph node cells were directly activated by addition of P1Ap (10−7 m final). After 3 days in culture during which TCRP1A CD8 T cells divide and differentiate into CTL, T cells were further expanded in the presence of interleukin‐2 (IL‐2; murine IL‐2 at 104 units/ml or human IL‐2 at 105 units/ml) and were used within 1 week.
T‐cell analysis
FACS analysis on live or fixed and permeabilized cells, labelling with Cell Tracer Violet (CTV; Molecular Probes, Eugene, OR) to follow cell division, measure of degranulation using anti‐Lamp1‐APC mAb, Fluo‐4 acetoxymethylester (Molecular Probes) labelling to measure Ca2+ flux, and video microscopy analyses were as previously described.20 When indicated, tumour target cells were pre‐loaded with calcein violet acetoxymethylester (1 µm final; Molecular Probes) or CTV (2·5 µm final). For video‐microscopy using adherent melanoma cells as target cells, eight‐well plates (Lab‐Tek chamber slides, NUNC) were seeded with the tumour cells to be used before full confluence.
Video analyses
Time‐lapse analysis was performed using a Zeiss780 confocal microscope (40×/0·7 NA oil immersion objective) as described previously.20 Images were analysed with the FIJI software. The complete videos were analysed by the recently described methods for automated and accurate analysis of cell signals (MAAACS) software.26 Briefly, the software allowed the automated tracking and analysis of the calcium response of each individual cell. The fluorescence intensity was normalized to the median of the values of signal intensity of each CTL. Thresholding was determined with the aim of maximizing the detection of truly activated cells while minimizing the probability of false alarm, as described elsewhere.26 The fluorescence amplitude (FA) threshold was set to a value of 2·5 in combination with a response fraction threshold of 0·1 (RF; time fraction where fluorescence signal is greater than the FA threshold) to classify calcium response types (sustained: FA > 2·5 and RF > 0·6; oscillating: FA > 2·5 and 0·2 < RF < 0·6, unique: FA > 2·5, RF < 0·2 and a single burst).
Results
Characteristics of tumour cell lines expressing the P1A tumour antigen
Mastocytoma cell line P511 and melanoma lines established from TiRP mice express transcripts encoded by the endogenous P1A gene (P511) or by the transgene construct in the melanoma lines (T‐1236 and T‐RFP‐69) (see Supplementary material, Fig. S1a). In agreement with our previous findings18, 23 TiRP‐derived melanoma lines generally express similar (here line T‐1236) or higher (here line T‐RFP‐69) P1A transcript levels than P511 mastocytoma cells. As expected,27 no P1A transcripts were detected in the mastocytoma antigen‐loss variant P1.204 (see Supplementary material, Fig. S1a). In Fig. 1 we determine the level of surface expression of some of the markers of the tumour lines that may be important for immunogenicity and T‐cell activation. H‐2Ld is highly expressed on the mastocytoma cells P511 and P1.204, as well as on the T‐1236 melanoma line (Fig. 1 and results not shown), whereas it is not detectable on the surface of melanoma line T‐RFP‐69 (Fig. 1).23 The CD28 ligand B7‐1 (CD80) was not expressed on the mastocytoma cells, whereas it was highly expressed on T‐1236 and on part of the T‐RFP‐69 melanoma cells. The LFA‐1 adhesion ligand intercellular cell adhesion molecule 1 (ICAM‐1), on the other hand, was highly expressed on the mastocytoma cells but very poorly expressed on the melanoma lines.
Figure 1.
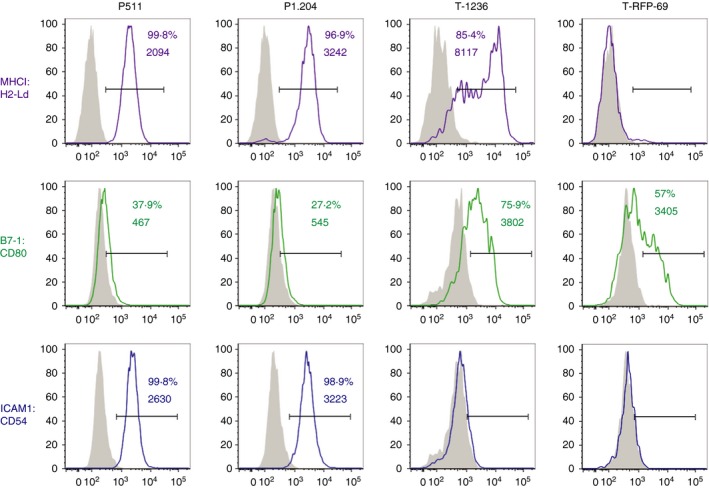
Characteristics of different P1A‐expressing tumour cells. FACS analysis for expression of H‐2Ld, B7.1 (CD80) and ICAM‐1 (CD54) was performed as described in the Material and methods. Numbers correspond to % positive cells and mean fluorescence intensity (MFI) of the positive cells.
As previously shown,17, 28 TCRP1A CD8 T effector cells were efficient at killing specifically the P1A‐expressing P511 target cells, but not the P1A‐deficient P1.204 mastocytoma cells (see Supplementary material, Fig. S1b). Lysis by the same CTL effector cells of P1A‐positive melanoma lines expressing similar levels of the P1A gene and surface H‐2Ld as P511 cells was much less efficient (see Supplementary material, Fig. S1b). To address which steps in tumour target cell killing by the CTL were responsible for the difference in sensitivity of the P1A‐expressing melanoma versus mastocytoma cells, we developed TCRP1A T cells expressing a fluorescent granzyme B (GZMB) (see Material and methods section) to allow monitoring of the process of lytic granule polarization towards the CTL/tumour contact zone, a process required for target cell killing.
In vitro differentiation of CD8 T cells from TCRP1A‐Gzmb‐Tom mice
Data in Fig. 2 show the kinetics of activation of purified CD8 T cells from TCRP1A‐Gzmb‐Tom mice when stimulated in culture by P1A‐peptide‐loaded splenocytes from congenic Rag−/− mice. A fraction of the T cells secreted IL‐2 and interferon‐γ at 24 hr. Dilution of CTV (see Material and methods) indicates that the T cells have undergone two cycles of division at 48 hr with acquisition of GZMB‐Tom expression as well as surface expression of CD25 (the IL‐2Rα chain) and CD44. Upon further divisions of the T cells after 72 hr, the levels of expression of CD25 as well as of GZMB‐Tom decreased and no IL‐2 production was detected. This pattern is consistent with that observed for stimulation of naive CD8 T cells with a weak agonist, a condition in which IL‐2 production is limiting.29 Indeed, signalling through the IL‐2 receptor is required for sustained expression of CD25 as well as GZMB.29 Accordingly, addition of IL‐2 at day 3 to the peptide‐stimulated TCRP1A‐Gzmb‐Tom CD8 T cells allowed for a sustained expression of GZMB‐Tom. This mode of stimulation was used to generate TCRP1A‐Gzmb‐Tom CTL to be used in the following sections (see Material and methods).
Figure 2.
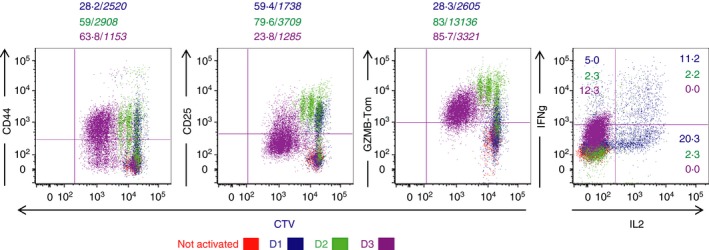
In vitro differentiation of TCRP1A‐GZMB‐Tom CD8 T cells. CD8 T cells from P1A‐GZMB‐Tom mice labelled with Cell Tracer Violet (CTV) were cultured with 10−7 m P1Ap‐preloaded splenocytes from congenic rag−/− mice (see Materials and methods) for 1 (blue), 2 (green) or 3 (bordeaux) days. Non‐activated CD8 T cells (red) were cultured for 1 day in the absence of P1Ap. CTV and GZMB‐Tom fluorescence as well as staining for CD25 and CD44 expression were measured by FACS (see Materials and methods) on the gated CD8 T cells. Interleukin‐2 (IL‐2) and interferon‐γ (IFN‐γ) production were measured by FACS after reactivation of the CD8 T cells for 4 hr with ionomycin and PMA in the presence of monensin followed by fixation and permeabilization. Numbers indicate % positive cells /(in italic) MFI of the positive cells. Results are representative of three and two experiments, respectively, for surface markers and for cytokines.
CTL degranulation induced by different tumour cells
We next measured the capacities of the different tumour cells to induce degranulation of the TCRP1A‐GZMB‐Tom CTL. CTL degranulation can be monitored by the resulting exposure of Lamp‐1 that is measured by anti‐Lamp‐1 mAb binding.30 Cytolytic granule exocytosis can also be measured here by the decrease of GZMB‐Tom red fluorescence from CTL.20, 31 Resting CTL (Fig. 3a in red) are not labelled by anti‐Lamp1 mAb added to the incubation medium (< 5% Lamp1 + cells, Fig. 3a,b), and present a high red fluorescence of the GZMB‐Tom protein (Fig. 3a,b). P511 mastocytoma cells induce the degranulation of TCRP1A‐GZMB‐Tom CTL as indicated by the Lamp1 staining of 55% of the cells and by the decrease in GZMB‐Tom fluorescence of the CTL (to an MFI around 3800 for the total cells and to a loss of about 45% of GZMB‐Tom fluorescence within the Lamp1 + CTL, Fig. 3a,b). As expected, the P1A‐negative variant P1.204 did not induce degranulation. T‐1236 melanoma cells induced some CTL degranulation as measured by Lamp1 staining (about 25% of the CTL, Fig. 3a,b), but this was not associated with a significant loss of GZMB‐Tom fluorescence (to an MFI of around 5700 for the total cells, Fig. 3b), even if the GZMB‐Tom fluorescence of the Lamp1 + CTL was analysed (loss of < 12% of GZMB‐Tom fluorescence within the Lamp1 + CTL, Fig. 3b, top right). The H‐2d‐low T‐RFP‐69 melanoma cells did not induce degranulation as measured by either Lamp1 staining or decrease in GZMB‐Tom fluorescence (Fig. 3a,b). Pre‐incubation of tumour cells with P1Ap slightly increased CTL degranulation whether measured by increase in Lamp1 detection or decrease in GZMB‐Tom fluorescence for the P511 tumour cells, whereas for P1.204 tumour cells, which did not induce degranulation in the absence of added peptide, a clear increase in anti‐Lamp1 and decrease in GZMB‐Tom fluorescence was observed. For T‐1236 cells, P1Ap pre‐incubation led to an increase in anti‐Lamp1 fluorescence and to detection of a decrease in GZMB‐Tom fluorescence. For T‐RFP‐69 cells peptide pre‐incubation also led to previously undetected increase in anti‐Lamp1 fluorescence but no significant decrease in GZMB‐Tom was observed (Fig. 3a,b).
Figure 3.
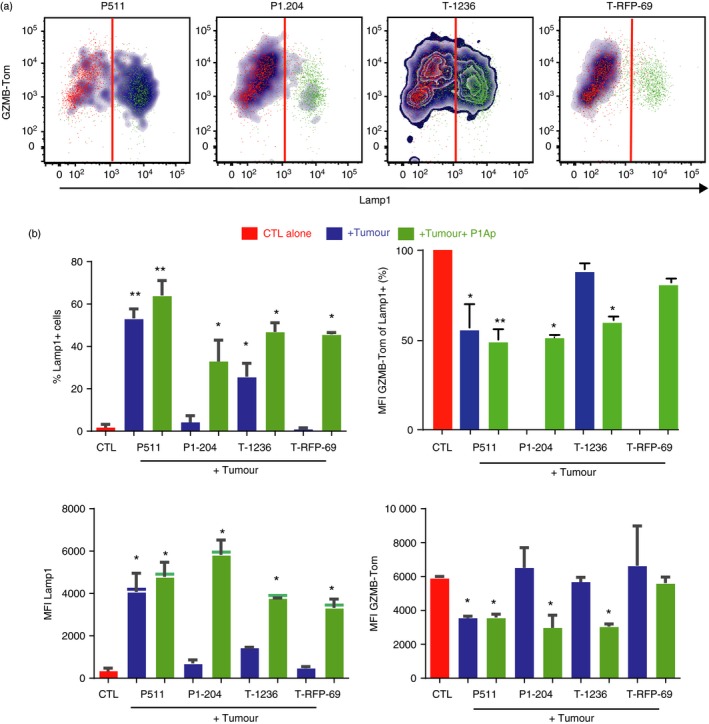
Differential extent of degranulation of TCRP1A‐GZMB‐Tom CTL induced by different P1A‐expressing tumour cells. CTL were incubated alone (red) or mixed with the tumour cells (untreated: blue, or pre‐loaded with peptide P1Ap: green) at a 1 : 1 ratio in 100 μl culture medium containing the anti‐Lamp1‐APC monoclonal antibody (mAb), before mild centrifugation and 30 min incubation at 37°. FACS analysis of anti‐Lamp1 mAb and GZMB‐Tom fluorescence was performed. (a) Density plots representing the binding of Lamp1 mAb and the expression of GZMB‐Tom on the CTL. (b) CTL degranulation measured by increase in Lamp1 staining on the CTL, expressed as % Lamp1‐positive CTL (upper left graph) and MFI of Lamp1 on all CTL (lower left graph) or by decrease in GZMB‐Tom fluorescence, expressed as % of MFI of GZMB‐Tom on Lamp‐1‐positive CTL (upper right graph, where MFI for all CTL alone was set at 100%) and MFI of GZMB‐Tom on all CTL (lower right graph). Results are the mean of three (without peptide) or two (with peptide) experiments. Statistical analysis was performed using t‐test: *P < 0·05; **P < 0.01.
These results suggest that the availability of the relevant peptide–MHC complexes on melanoma cells may be limiting the CTL response. Melanoma cells expressing high levels of surface H‐2Ld could induce some CTL exocytosis as measured by Lamp1 staining, albeit sub‐optimally and without detectable loss in GZMB‐Tom expression in the absence of exogenous peptide addition. For melanoma cells expressing low levels of surface H‐2Ld, exogenous peptide addition was mandatory.
Time‐lapse live microscopy of early activation events induced by different P1A‐expressing tumours in TCRP1A‐GZMB‐Tom CTL.
To monitor early changes in [Ca2+]i induced by tumour cells in TCRP1A‐GZMB‐Tom CTL by video microscopy, the CTL were loaded with the [Ca2+]i sensitive fluorescent dye Fluo‐4, the intensity of which is represented in false colours in Fig. 4(a,b) (upper lanes), whereas GZMB‐Tom containing granules visualized in red (Fig. 4a,b, upper and lower lanes) can be clearly observed when Fluo‐4 fluorescence is deleted (Fig. 4a,b, lower lanes). The P511 and P1.204 mastocytoma target cells were pre‐loaded with Calcein Violet‐AM to detect dye leakage upon damage to the target plasma membrane (Fig. 4a).
Figure 4.
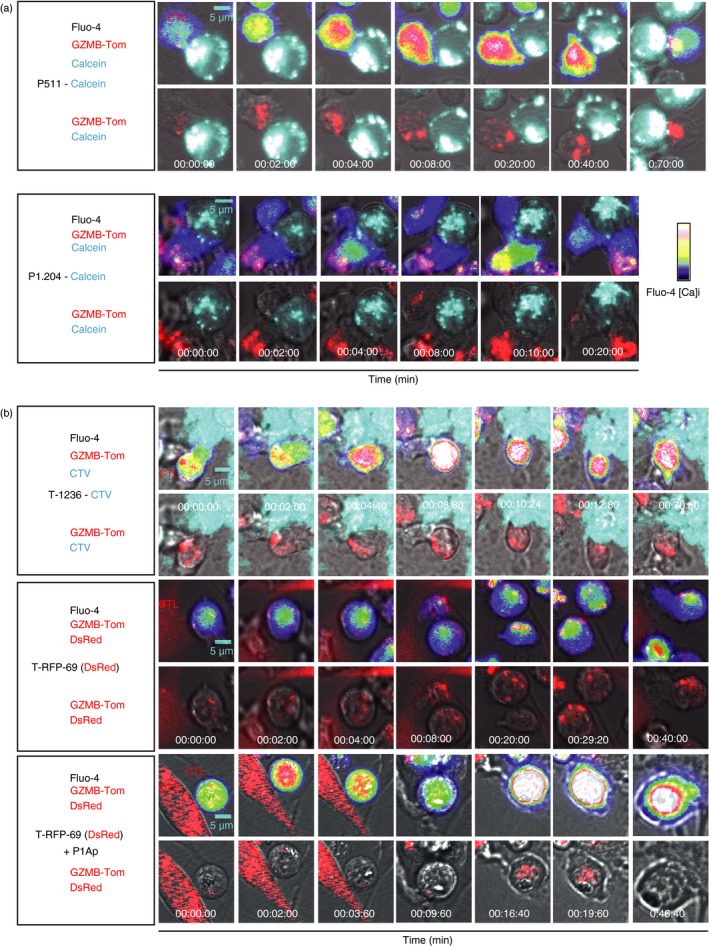
Visualization of the kinetics of CTL activation and granule re‐localization upon CTL/tumour cell contact by video microscopy. Images from video microscopy show Ca2+ fluxes and fluorescence of GZMB‐Tom containing granules in Fluo‐4‐labelled TCRP1A‐GZMB‐Tom CTL added to various tumour targets. The first image corresponds to that before the first increase in Ca2+ (except for the P1.204 P1A‐negative tumour cells). (a) P511 and P1.204 non‐adherent tumour cells were labelled with Violet Calcein and deposited onto polylysine‐activated Labtek wells, before addition of the CTL. Upper images show Fluo‐4 and GZMB‐Tom fluorescence in the CTL and Calcein Violet fluorescence in the tumour cells. Lower images are devoid of Fluo‐4 fluorescence to better visualize GZMB‐Tom. (b) Adherent tumour cells T‐1236 and T‐RFP‐69 were grown on Labtek wells 2 days before the experiment. T‐1236 cells were labelled with CTV whereas T‐RFP‐69 cells endogenously express DsRed. When indicated (two last lanes), T‐RFP69 cells were loaded with peptide P1Ap. For T‐1236 tumour cells, upper images show Fluo‐4, GZMB‐Tom and Calcein Violet fluorescence and lower images are devoid of Fluo‐4 fluorescence. For T‐RFP‐69 cells, upper images show Fluo‐4, GZMB‐Tom and DsRed fluorescence, lower images are devoid of Fluo‐4 fluorescence. Video microscopy recordings (90 min at 37°) were performed and analysed as described in Materials and methods.
One representative sequence of events (Fig. 4a; and see Supplementary material, Video S1) shows that the contact (time 0) of TCRP1A‐GZMB‐Tom CTL with P1A‐expressing mastocytoma P511 induces an important [Ca2+]i (sustained from 2 to 40 min after cell contact) associated with a change in the CTL shape and formation of an immune synapse, leading to massive re‐localization of red GZMB‐Tom granules to the contact zone (starting 6 min after the [Ca2+]i burst – Fig. 4a). The average time for granule re‐localization for a number of observed CTL/P511 cell conjugates (see Supplementary material, Fig. S2) was around 8 min (range 0·8–20 min) after the [Ca2+]i burst, which is consistent with previous observations.20, 32, 33 P511 killing as measured by calcein release was not observed for all conjugates (see Supplementary material, Fig. S2). When observed, it occurred with a mean time required for half calcein release of 20 min (see Supplementary material, Fig. S2). These kinetics are in the range of those previously reported for efficient CTL activity (see Supplementary material, Table S1).20, 32 For P1A‐negative P1.204 mastocytoma cells, no granule re‐localization occurred in the CTL and no calcein leakage from the tumour was observed (Fig. 4a). A transient [Ca2+]i was occasionally observed (see later section – Fig. 5). Melanoma cells T‐1236 were labelled with CTV because these tumour cells failed to take up Calcein Violet. The representative sequence of events (Fig. 4b, and see Supplementary material, Video S2) following contact between TCRP1A‐GZMB‐Tom CTL and melanoma T‐1236 shows the induction of a strong [Ca2+]i with a maximal response at 8 min and with granule re‐localization starting at 10 min and complete at 12 min. The average time for granule re‐localization for a number of observed CTL/T‐1236 cell conjugates (8/11, see Supplementary material, Fig. S2) was around 20 min, indicating that this process was slower in response to the melanoma than in response to the mastocytoma cells. Additionally, leakage of CTV from T‐1236 melanoma cells was not observed during the time of video recording. Of note, we previously observed that CTV leakage could be observed when other CTL/target cell pairs (OT1/EL4‐OVA) were monitored (Mouchacca, Boyer, results not shown).
Figure 5.
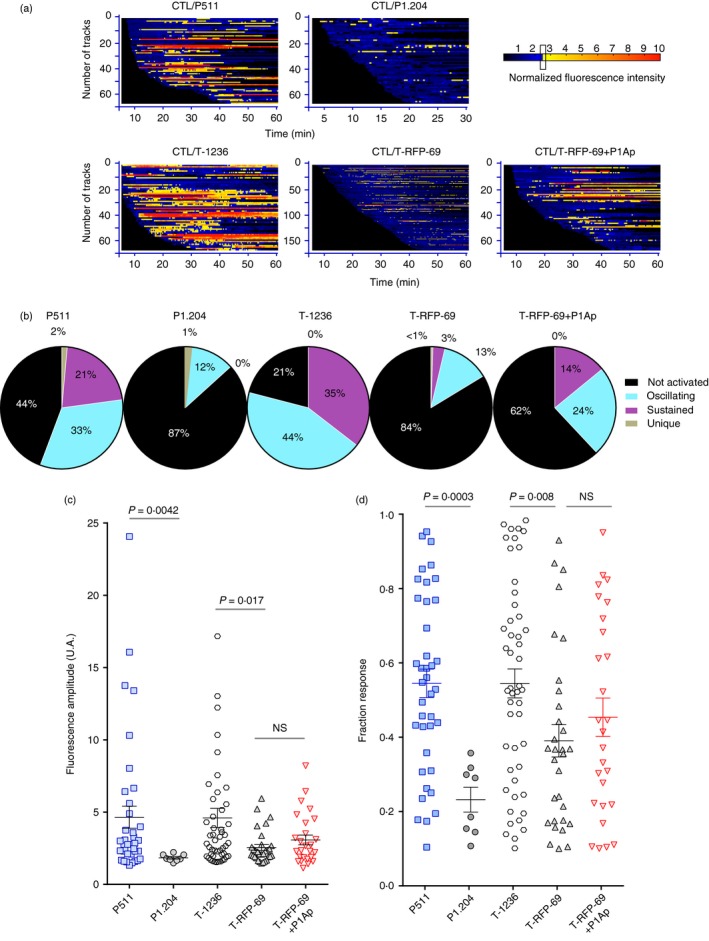
Barcoding the TCRP1A‐GZMB‐Tom CTL Ca2+ fluxes in response to the different tumour cells. The videos corresponding to the CTL responses to tumour cells shown in Fig. 4 were analysed for Fluo‐4‐positive cell tracking with the MAAACS software described by Salles et al.26. The raw fluorescence recordings were normalized to the median of Fluo‐4 fluorescence values for each individual CTL. (a) Normalized Fluo‐4 intensity is displayed for individual cells as a barcoded response over time (colour‐code is function of the fluorescence amplitude (FA) threshold set to FA > 2·5) of the CTL in response to the indicated tumour cells. (b) The pattern of the Ca2+ response of the CTL towards each of the indicated tumour cells is represented in pie charts (non‐activated: black, oscillating: blue, sustained: purple, unique: grey) (see Materials and methods for classification of criteria). (c, d) Statistical representation of the fluorescence signal amplitude (c) and of the proportion of Ca2+ signal above the threshold for each cell per unit time (response fraction) (d). Mean ± SEM is shown. Statistical analysis was carried out with the Mann–Whitney non‐parametric test.
Melanoma cells T‐RFP‐69 express RFP and very low surface H‐2Ld. Their interaction with TCRP1A‐GZMB‐Tom CTL did not induce high [Ca2+]i in the CTL, nor granule re‐localization (Fig. 4b). When pre‐pulsed with P1Ap, however, the T‐RFP‐69 melanoma cells induced CTL activation with an increase in [Ca2+]i, a change in cell shape and re‐localization of a few granules at the CTL–target contact zone (at time 9 min 50 seconds – Fig. 4b, and see Supplementary material, Video S3).
As diverse patterns of [Ca2+]i appeared to be induced in the CTL by the different tumour cells, we used a software, MAAACS, established in the laboratory26 to sort these patterns in detail for a number of CTL–tumour cell conjugates (Fig. 5). The software allows the automated analysis of individual CTL presenting a [Ca2+]i change greater than an activation threshold set to optimize the detection of activated cells. Based upon the ‘fluorescence amplitude’ of the response (measure of the mean of the intensity of the [Ca2+]i over time) and the ‘fraction response’ (ratio of time during which the [Ca2+]i was above the FA threshold) MAAACS classifies the calcium responses as sustained, oscillating (or transient) and unique.
As the plating efficiency of the melanoma cells (plastic‐adherent cells) was generally higher than that of the non‐adherent mastocytoma cells, the chances for a CTL to interact with a target cell was higher for the melanoma compared with mastocytoma cells. Therefore the comparison of the fraction of responding CTL is probably underestimated for the mastocytoma compared with the melanoma cells. We think therefore that the frequency of CTL activated with a given type of [Ca2+]i response (sustained versus oscillating) is a more important parameter than the frequency of responding CTL in the comparison between melanoma and mastocytoma cells.
The analysis shows that mastocytoma cells P511 induce [Ca2+]i changes in 56% of the CTL, of which 38% presented a sustained response, 59% an oscillating response and 3% a unique transient response (Fig. 5a,b). The amplitude of the response could reach a 10‐ to 25‐fold increase, but was around fivefold on average (Fig. 5c) with a fraction response around 0·55 on average (Fig. 5d). By comparison, the P1A‐negative P1.204 mastocytoma cells induced a weak amplitude response (Fig. 5c) in 13% of the CTL mostly of the oscillating type with no sustained response (Fig. 5a,b) and a very low fraction response (Fig. 5d). In these conditions no granule re‐localization was observed.
Melanoma cells T‐1236 induced a [Ca2+]i increase in 79% of the CTL, 44% of which presented a sustained response, whereas 56% presented an oscillating response (Fig. 5a,b). The mean amplitude of the response and the fraction response were in the same range as those induced by the P511 cells (Fig. 5c,d). It should be noted that no saturation of the Fluo‐4 signal was observed for the highest amplitude responses to either P511 or T‐1236 (see Supplementary material, Fig. S3). The H‐2Ld low T‐RFP‐69 melanoma cells induced responses of low amplitude and fraction time in 16% of the CTL with 81% being oscillating and 19% sustained (Fig. 5a–d). Although of similar low amplitude as the response to the P1A‐negative mastocytoma cells, the H‐2low melanoma cells elicited some ‘sustained’ [Ca2+]i responses, as also indicated by their slightly higher fraction response (Fig. 5d). When T‐RFP‐69 melanoma cells were pre‐incubated with P1Ap, 38% of the CTL responded, 37% of them with a ‘sustained’ profile and 63% with an oscillating profile (Fig. 5a,b). The mean amplitude and fraction responses were slightly, but not statistically significantly, increased compared with those induced in the absence of P1Ap (Fig. 5c,d). This result suggests that even with low MHC expression on the melanoma cells, addition of exogenous cognate peptide may lead to sufficient relevant peptide–MHC complexes to allow activation with a sustained [Ca2+]i response of a fraction of the CTL.
In conclusion, we described different behaviours of the same CTL specific for a tumour antigen expressed in different tumour cell lines. P1A‐expressing mastocytoma cells were able to efficiently activate the TCRP1A CTL, leading to granule exocytosis and target cell lysis. TCRP1A CTL activation by the P1A‐expressing melanoma cells was less efficient, even for the H‐2Ld high T‐1236 tumour cells, which induced activation up to re‐localization of the granules to the target contact zone, but in which no lethal hit was observed. The lower efficiency for induction of cytolysis by the melanoma as compared to the mastocytoma cells occurred in spite of their similar induction of sustained [Ca2+]i responses in about 40% of the activated CTL. The frequency of the [Ca2+]i bursts was lower, however, in the response to the P1A‐expressing mastocytoma compared with melanoma cells (see later section, and see Supplementary material, Fig. S6), a point to be discussed later. T‐RFP‐69 H‐2Ld low P1A‐expressing melanoma cells were inefficient at activating the CTL, but addition of exogenous P1Ap allowed the activation of some of the CTL up to the step of granule re‐localization, presumably because a required threshold of P1Ap/H‐2Ld expression was reached. The P1Ap‐induced effect was correlated with the ability of the melanoma cells to induce a sustained versus an oscillating [Ca2+]i response in a higher proportion of the CTL.
Comparison of the patterns of early activation events of P1A‐tumour antigen reactive TCRP1A CTL with those of OT1 CTL optimally stimulated by peptide OVAp
To evaluate how the early activation events of CTL directed at the P1A‐encoded cancer‐germline antigen compare with the optimal simulation described for the OT1 CD8 T cells/OVAp model, MAAACS analysis was applied to our previously reported time‐lapse live microscopy experiments of OT1 GZMB‐Tom CTL interacting with either OVAp‐pulsed RMA‐S thymoma cells or with EL4 thymoma expressing the OVA gene (EG7) whereas RMA‐S cells pulsed with an irrelevant H‐2Kb binding peptide (pKB1) were used as a control.20 Data (see Supplementary material, Fig. S4 and Table S1A) report that the OT1 response to OVAp/RMA‐S or to EG7 cells was of similar amplitude as the TCRP1A response to P511 or to T‐1236. However, a higher proportion of the OT1 cells presented a sustained [Ca2+]i rise (64% and 73%, respectively against OVAp/RMA‐S and EG7 target cells). Altogether, the frequency of [Ca2+]i bursts was much lower in the OT1 compared with the TCRP1A response (see Supplementary material, Fig. S6). When comparing data for degranulation (see Supplementary material, Table S1B), it was found that 90% of the OT1 cells expressed Lamp‐1 and a 66% decrease in GzmB‐Tom MFI was observed in response to RMA‐S/OVAp. Hence a sustained [Ca2+]i response for a higher proportion of the CTL and a more extensive degranulation as measured by either Lamp‐1 exposure or loss of GzmB appear to characterize the OT1 response to OVAp. This was associated with granule re‐localization to the target cell after about 5 min (similar to the response of TCRP1A CTL to P511) but led to a measurable target hit (calcein release) within < 15 min for all CTL observed (see Supplementary material, Fig. S5, Table S1A). In comparison only about half of the P511 target cells leaked calcein (see Supplementary material, Fig. S2, Table S1A).
The high affinity OT1 TCR/OVAp led to a sustained [Ca2+]i rise correlated with GzmB exocytosis in a majority of the CTL, resulting in calcein release from all hit target cells.
Discussion
To optimize immunotherapies against neoplasms, it is important to understand to which effector mechanisms a particular tumour is sensitive or resistant. These characteristics may differ depending on the expressed tumour antigens (qualitative/quantitative) and degree of MHC expression, as well as on the level of expression of ligands for adhesion receptors (LFA‐1) or co‐stimulatory or inhibitory receptors (such as PD‐1). Parameters that affect the efficiency of adoptive CD8 T‐cell therapy in pre‐clinical models include the capacity of the transferred T cells to infiltrate the tumour34 and to survive in the tumour microenvironment35, 36 as well as extended proliferative capacity of the T cells.37, 38 Although a correlation has been observed between the affinity of TCR/tumour antigen–MHC interaction and efficiency in adoptive T‐cell therapy,36 it is not clear which parameters affect the differential capacity of CD8 T effector cells to induce regression of tumour cells through the same TCR/tumour antigen–MHC interaction.
Here, we analysed how tumour cells of distinct origin expressing the same cancer‐germline antigen are differentially sensitive to tumour antigen‐specific CTL obtained by in vitro stimulation with peptide antigen. The P1A‐encoded tumour antigen to which partial CD8 T‐cell tolerance exists39 represents a model natural tumour antigen with moderate stimulatory capacity for TCRP1A CD8 T cells when compared with the often used OTI‐TCR/OVA‐peptide model.20
By monitoring in vitro the interaction of CTL specific for the P1A encoded tumour antigen on mastocytoma or melanoma cells, we observed differences in the response elicited in the same CTL by the different tumour cells. A surge in cytosolic Ca2+ concentration by entry of extracellular Ca2+ is a hallmark of CTL activation.15, 40 Both tumour types expressing similar levels of P1A‐encoded transcripts and surface H‐2Ld induced a sustained [Ca2+]i rise in a similar proportion of the CTL (40%), with most responses (60%) being of the oscillating type. Although engagement of the adhesion receptor LFA‐1 has previously been implicated in the sustained Ca2+ entry in CTL,41, 42, 43, 44 the absence of expression on the melanoma cells of the ICAM‐1 ligand of LFA‐1 did not appear to affect the pattern of [Ca2+]i response to the melanoma compared with the mastocytoma cells. Previous reports pointed to the unique role for the LFA‐1/ICAM‐1 interaction as providing co‐activation to CD3/TCR‐mediated lysis by CTL through both an enhanced CTL‐target cell binding45 and the delivery of post‐conjugate co‐stimulatory signals.46, 47 It should be noted that the response to both P1A‐expressing mastocytoma and melanoma cells contrasted with the ‘optimal’ response of OT1 CTL to OVAp on EL4 tumour cells that was mostly of the sustained type. This difference is possibly to be attributed to the lower strength of TCR signalling in the P1A compared with the OVAp models. In the latter case it was shown that the strength of engagement of the OT1 TCR (by altered peptide G4 instead of OVA257–264) controlled the recruitment of the cytotoxic granules to the immune synapse, although centrosome polarization was not affected.33 In this example, however, the interaction of the TCR with the altered G4 peptide was of such low avidity that it failed to induce any [Ca2+]i response.48
In our model, both P1A‐expressing mastocytoma and melanoma cells induced similar patterns of [Ca2+]i in the CTL. The question as to whether the frequency of [Ca2+]i bursts in the CTL response is a relevant parameter for CTL activation remains to be addressed, as it appeared to be inversely correlated to the extent of degranulation induced by different tumour cells (see Supplementary material, Fig. S6, Table S1). Tumour‐induced re‐localization of cytolytic granules toward the CTL–target cell contact zone occurred in response to both P1A‐expressing mastocytoma and melanoma cells. The kinetics of this process were much delayed, however, for melanoma compared with mastocytoma cells. The next steps involving the fusion of cytolytic granules with the plasma membrane and the exocytosis leading to the release of cytolytic enzymes appeared to take place in response to both mastocytoma and melanoma cells, although less efficiently in the latter case. In particular, while degranulation induced by the mastocytoma cells could be measured by either the exposure of Lamp1 or by the decrease in GZMB‐Tom content in the CTL, the latter event was not detected upon interaction of the CTL with the melanoma cells. Finally, although the lethal hit in the mastocytoma cells could be visualized, this was not the case for the melanoma cells, an observation in line with the poor 51Cr‐release observed in classical 4‐hr CTL assays.
At this point we are unable to formally discriminate whether the CTL granule content was not delivered efficiently to the melanoma cells or whether the melanoma cells presented an intrinsic resistance to the cytolytic effectors. Relevant to the first possibility, it has been suggested that LFA‐1 may deliver a distinct signal essential for directing released cytolytic granules to the surface of target cells to mediate lysis.49 The nature of this potential signal has not been defined, however, and it is not clear whether lack of the ICAM‐1 ligand on the melanoma target cell may contribute to such a defective delivery. Other data lend support to the second, not exclusive possibility. Notably, a study on human melanoma cells50 showed that ICAM‐1‐negative melanoma cells could induce cytolytic granule polarization as well as ICAM‐1‐expressing melanomas, but failed to be killed by the same CTL. The authors further observed that the level of ICAM‐1 expression by melanoma cells correlated positively with decreased phosphatase and tensin homolog (PTEN) activity and decreased activity of the PI3K/AKT pathway, suggesting that low expression of ICAM‐1 was intrinsically associated with the activation of a survival pathway.50 In another study comparing the efficiency of the same human CTL effector cells on tumour target cells from different tissue origins, it was concluded that melanoma cells were intrinsically more resistant than ‘classically’ used Epstein–Barr virus‐transformed B‐cell lines to killing via cytolytic granule delivery.51 In that study, the kinetics of CTL activation, polarization of cytolytic granules and lethal hit delivery appeared similar for the two types of tumours. However, repetitive hits from the CTL appeared to be required to achieve melanoma cell destruction. The status of ICAM‐1 expression by the tumour cells was not discussed in that study and fine differences in the pattern of CTL activation (as observed in our study) may have been bypassed due to use of high concentrations of exogenous antigenic peptide.51 More recently, this group identified a defence mechanism of human melanoma cells based on their exacerbated lysosome secretion and cathepsin B‐mediated perforin degradation at the lytic synapse.52 Over‐expression of serpin‐9 (gene PI‐9), an inhibitor of GzmB activity, has also been described in metastatic melanoma. However, its depletion did not enhance the melanoma susceptibility to CTL killing.50
Whether LFA‐1/ICAM‐1 interactions may affect the exocytosis step is still to be determined. Indeed, a recent report suggests that cytolytic secretory events occur in an intermediate domain that overlaps with the region of strongest force exertion.53 In the ‘Immune Synapse’ (IS), this zone is occupied by the clusters of integrins (LFA‐1)54 corresponding to the adhesive and cytoskeletal machinery required to transmit force. The authors favour a model in which degranulation occurs at the border between the IS central F‐actin‐depleted area and the peripheral F‐actin ring (LFA‐1). This would balance the physical requirements of exocytosis (actin hypodense region of the plasma membrane suitable for vesicle fusion and granule secretion12) and synaptic mechanopotentiation provided by the integrin‐associated cytoskeletal machinery.53
Consistent with the low number of peptide–MHC complexes on target cells required to activate CTL,55, 56 we observed that while melanoma cells from line T‐RFP‐69 with undetectable surface MHC were unable to activate the P1A‐reactive CTL, addition of P1Ap to the T‐RFP‐69 cells allowed a threshold of peptide–MHC complexes to be reached sufficient to induce a [Ca2+]i response of low amplitude with a fraction of the CTL (about 40%) with a sustained [Ca2+]i profile. It appears that different sensors Stomal Interaction Molecule (STIM1 or STIM2) may regulate SOCE depending on the degree of receptor stimulation and endow the STIM/CRAC‐channel complex to be differentially regulated by feedback mechanisms, thereby affecting the pattern of the [Ca2+]i response.14, 57 Furthermore, additional Ca2+ channels may influence the variations in [Ca2+]i.58, 59 It will therefore be important to further establish which parameters (strength of TCR engagement/co‐stimulatory receptors) affect the regulation of Ca2+‐channel functioning in CTL and how this impacts on the various steps of granule dynamics and exocytosis.
It is not clear at this point whether the observed difference in killing efficiency by TCRP1A effector cells of mastocytoma versus melanoma tumour cells is responsible for the difference in tumour regression observed in vivo upon transfer of TCRP1A effector cells in tumour‐bearing mice.19 Additional mechanisms independent of GzmB/perforin, such as those mediated by TnfSF molecules like Fas‐ligand may be involved in tumour cell killing in vivo. No significant Fas‐mediated killing was detected in in vitro stimulated TCRP1A effector cells, however.60 Tumour regression may also derive from indirect actions by CTL‐secreted cytokines such as interferon‐γ that exert angiostatic effects,61 and can promote stroma destruction.62 CTL‐initiated cytokine‐mediated activation of innate‐type effectors within the tumour microenvironment may also contribute to tumour regression.17
Together with available data from the literature,50, 63 our study suggests that lack of ICAM‐1 expression by tumour cells may represent an unfavourable factor for potential use of adoptive immunotherapy using killer cells unless the T cells are engineered to present heightened effector functions19 or to express an affinity‐enhanced TCR against the tumour antigen.36
Use of the GZMB‐Tom CTL would further be of interest – to establish whether LFA‐1 engagement may contribute to improve the killing process of melanoma cells by CTL (comparing melanoma cells expressing or not ICAM‐1) and decipher the step at which this may occur and whether a correlative change in the dynamics of granule re‐localization and exocytosis of GzmB would be observed – to observe the respective effects of enhanced CTL effector function or TCR affinity on the killing process – and finally to monitor the killing events within the tumour microenvironment in vivo.
Contribution to authorship
CBo and AMSV designed the experiments. MFr and CBo performed most experiments and NAA provided some data and discussion. PM developed the GzmB‐Tom mice. GV and MB established the melanoma lines, MFa helped with the video microscopy and YH and CBi helped with Ca2+ signalling data analyses. CBo and AMSV wrote the paper.
Animal care ethics
All procedures were approved by the Regional ‘Provence‐Alpes‐Cote d'Azur’ Committee on Ethics for Animal Experimentation (authorization: no 13.521, date: 08/10/2012) and were in accordance with French and European directives. All efforts were made to minimize animal suffering.
Disclosures
The authors have no potential conflicts of interest.
Supporting information
Figure S1. Characteristics of different P1A‐expressing tumour cells.
Figure S2. Kinetics of GZMB‐Tom containing granule re‐localization to the CTL‐tumour contact zone during the video experiments.
Figure S3. Individual recordings of TCRP1A‐GZMB‐Tom CTL Ca2+ fluxes in response to the different tumour cells.
Figure S4. Barcoding the TCR‐OT1‐GZMB‐Tom CTL Ca2+ fluxes in response to OVAp/H‐2Kb.
Figure S5. Time required for GZMB‐Tom granule redistribution in the CTL and for calcein release from the target cells during the videos using the TCR‐OT1‐GZMB‐Tom CTL.
Figure S6. Frequency of bursts in Ca2+ fluxes in the response of CTL to different tumour cells.
Table S1. Comparison of type of Ca2+ fluxes and of degranulation for the TCRP1A‐GZMB‐Tom and TCR‐OT1‐GZMB‐Tom CTL.
Video S1. Kinetics of activation of TCRP1A‐GZMB‐Tom CTL in response to P511 mastocytoma cells (as described in Fig. 4a).
Video S2. Kinetics of activation of TCRP1A‐GZMB‐Tom CTL in response to T‐1236 melanoma cells (as described in Fig. 4b).
Video S3. Kinetics of activation of TCRP1A‐GZMB‐Tom CTL in response to T‐RFP‐69 melanoma cells pre‐pulsed with P1Ap (as described in Fig. 4b).
Acknowledgements
This work was supported by institutional funding from INSERM and CNRS, and by grants from ‘Association pour la Recherche sur le Cancer’ (ARC‐SL220110603479) (to AMSV). It was performed using France‐BioImaging infrastructure supported by the Agence Nationale de la Recherche (ANR‐10‐INSB‐04‐01, call ‘Investissements d'Avenir’). MFr was the recipient of a Fulbright fellowship, PM obtained a doctoral fellowship from ARC and GV a grant from ANR ‘retour post‐doc’. We thank Amandine Mas for screening of mice and the CIML imaging and animal facilities personnel for assistance.
Contributor Information
Anne‐Marie Schmitt‐Verhulst, Email: verhulst@ciml.univ-mrs.fr.
Claude Boyer, Email: boyer@ciml.univ-mrs.fr.
References
- 1. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014; 14:135–46. [DOI] [PubMed] [Google Scholar]
- 2. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P et al Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB et al Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF et al Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012; 366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph‐Pietras D et al Anti‐CTLA‐4 therapy broadens the melanoma‐reactive CD8+ T cell response. Sci Transl Med 2014; 6:254ra128. [DOI] [PubMed] [Google Scholar]
- 6. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L et al PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 2013; 39:11–26. [DOI] [PubMed] [Google Scholar]
- 8. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 2015; 15:388–400. [DOI] [PubMed] [Google Scholar]
- 9. Stinchcombe JC, Griffiths GM. Secretory mechanisms in cell‐mediated cytotoxicity. Annu Rev Cell Dev Biol 2007; 23:495–517. [DOI] [PubMed] [Google Scholar]
- 10. Thiery J, Keefe D, Boulant S, Boucrot E, Walch M, Martinvalet D et al Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat Immunol 2011; 12:770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Saint Basile G, Menasche G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol 2010; 10:568–79. [DOI] [PubMed] [Google Scholar]
- 12. Ritter AT, Asano Y, Stinchcombe JC, Dieckmann NM, Chen BC, Gawden‐Bone C et al Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity 2015; 42:864–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oh‐hora M, Rao A. Calcium signaling in lymphocytes. Curr Opin Immunol 2008; 20:250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shaw PJ, Feske S. Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity. J Physiol 2012; 590(Pt 17):4157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maul‐Pavicic A, Chiang SC, Rensing‐Ehl A, Jessen B, Fauriat C, Wood SM et al ORAI1‐mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci U S A 2011; 108:3324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matza D, Badou A, Jha MK, Willinger T, Antov A, Sanjabi S et al Requirement for AHNAK1‐mediated calcium signaling during T lymphocyte cytolysis. Proc Natl Acad Sci U S A 2009; 106:9785–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shanker A, Verdeil G, Buferne M, Inderberg‐Suso EM, Puthier D, Joly F et al CD8 T cell help for innate antitumor immunity. J Immunol 2007; 179:6651–62. [DOI] [PubMed] [Google Scholar]
- 18. Soudja SM, Wehbe M, Mas A, Chasson L, de Tenbossche CP, Huijbers I et al Tumor‐initiated inflammation overrides protective adaptive immunity in an induced melanoma model in mice. Cancer Res 2010; 70:3515–25. [DOI] [PubMed] [Google Scholar]
- 19. Grange M, Buferne M, Verdeil G, Leserman L, Schmitt‐Verhulst AM, Auphan‐Anezin N. Activated STAT5 promotes long‐lived cytotoxic CD8+ T cells that induce regression of autochthonous melanoma. Cancer Res 2012; 72:76–87. [DOI] [PubMed] [Google Scholar]
- 20. Mouchacca P, Schmitt‐Verhulst AM, Boyer C. Visualization of cytolytic T cell differentiation and granule exocytosis with T cells from mice expressing active fluorescent granzyme B. PLoS ONE 2013; 8:e67239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shanker A, Auphan‐Anezin N, Chomez P, Giraudo L, Eynde BVD, Schmitt‐Verhulst A‐M. Thymocyte‐intrinsic genetic factors influence CD8 T cell lineage commitment and affect selection of a tumor‐reactive TCR. J Immunol 2004; 172:5069–77. [DOI] [PubMed] [Google Scholar]
- 22. Huijbers IJ, Krimpenfort P, Chomez P, van der Valk MA, Song JY, Inderberg‐Suso EM et al An inducible mouse model of melanoma expressing a defined tumor antigen. Cancer Res 2006; 66:3278–86. [DOI] [PubMed] [Google Scholar]
- 23. Buferne M, Chasson L, Grange M, Mas A, Arnoux F, Bertuzzi M et al IFNγ producing CD8 T cells modified to resist major immune checkpoints induce regression of MHC class I‐deficient melanomas. Oncoimmunology 2015; 4:e974959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ. Faithful activation of an extra‐bright red fluorescent protein in “knock‐in” Cre‐reporter mice ideally suited for lineage tracing studies. Eur J Immunol 2007; 37:43–53. [DOI] [PubMed] [Google Scholar]
- 25. Brandle D, Bilsborough J, Rulicke T, Uyttenhove C, Boon T, Van den Eynde BJ. The shared tumor‐specific antigen encoded by mouse gene P1A is a target not only for cytolytic T lymphocytes but also for tumor rejection. Eur J Immunol 1998; 28:4010–9. [DOI] [PubMed] [Google Scholar]
- 26. Salles A, Billaudeau C, Serge A, Bernard AM, Phelipot MC, Bertaux N et al Barcoding T cell calcium response diversity with methods for automated and accurate analysis of cell signals (MAAACS). PLoS Comput Biol 2013; 9:e1003245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uyttenhove C, Maryanski J, Boon T. Escape of mouse mastocytoma P815 after nearly complete rejection is due to antigen‐loss variants rather than immunosuppression. J Exp Med 1983; 157:1040–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shanker A, Buferne M, Schmitt‐Verhulst AM. Cooperative action of CD8 T lymphocytes and natural killer cells controls tumour growth under conditions of restricted T‐cell receptor diversity. Immunology 2010; 129:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Verdeil G, Puthier D, Nguyen C, Schmitt‐Verhulst AM, Auphan‐Anezin N. STAT5‐mediated signals sustain a TCR‐initiated gene expression program toward differentiation of CD8 T cell effectors. J Immunol. 2006; 176:4834–42. [DOI] [PubMed] [Google Scholar]
- 30. Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M et al Sensitive and viable identification of antigen‐specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 2003; 281:65–78. [DOI] [PubMed] [Google Scholar]
- 31. Kannan K, Stewart RM, Bounds W, Carlsson SR, Fukuda M, Betzing KW et al Lysosome‐associated membrane proteins h‐LAMP1 (CD107a) and h‐LAMP2 (CD107b) are activation‐dependent cell surface glycoproteins in human peripheral blood mononuclear cells which mediate cell adhesion to vascular endothelium. Cell Immunol 1996; 171:10–9. [DOI] [PubMed] [Google Scholar]
- 32. Faroudi M, Utzny C, Salio M, Cerundolo V, Guiraud M, Muller S et al Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc Natl Acad Sci U S A 2003; 100:14145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jenkins MR, Tsun A, Stinchcombe JC, Griffiths GM. The strength of T cell receptor signal controls the polarization of cytotoxic machinery to the immunological synapse. Immunity 2009; 31:621–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH et al Vascular normalization in Rgs5‐deficient tumours promotes immune destruction. Nature 2008; 453:410–4. [DOI] [PubMed] [Google Scholar]
- 35. Auphan‐Anezin N, Verdeil G, Grange M, Soudja SM, Wehbe M, Buferne M et al Immunosuppression in inflammatory melanoma: can it be resisted by adoptively transferred T cells? Pigment Cell Melanoma Res 2013; 26:167–75. [DOI] [PubMed] [Google Scholar]
- 36. Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen HN, Cuevas C et al T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell 2015; 28:638–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ et al Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298:850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z et al Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest 2005; 115:1616–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huijbers IJ, Soudja SM, Uyttenhove C, Buferne M, Inderberg‐Suso EM, Colau D et al Minimal tolerance to a tumor antigen encoded by a cancer‐germline gene. J Immunol 2012; 188:111–21. [DOI] [PubMed] [Google Scholar]
- 40. Poenie M, Tsien RY, Schmitt‐Verhulst A‐M. Sequential activation and lethal hit measured by [Ca2+]i in individual cytolytic T cells and targets. EMBO J 1987; 6:2223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim K, Wang L, Hwang I. LFA‐1‐dependent Ca2+ entry following suboptimal T cell receptor triggering proceeds without mobilization of intracellular Ca2+ . J Biol Chem 2009; 284:22149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schwarz EC, Qu B, Hoth M. Calcium, cancer and killing: the role of calcium in killing cancer cells by cytotoxic T lymphocytes and natural killer cells. Biochim Biophys Acta 2013; 1833:1603–11. [DOI] [PubMed] [Google Scholar]
- 43. Bachmann MF, McKall‐Faieenza K, Schmits R, Bouchard D, Beach J, Speiser DE et al Distinct roles for LFA‐1 and CD28 during activation of naive T cells: adhesion versus stimulation. Immunity 1997; 7:549–57. [DOI] [PubMed] [Google Scholar]
- 44. Wulfing C, Sjaastad MD, Davis MM. Visualizing the dynamics of T cell activation: intracellular adhesion molecule 1 migrates rapidly to the T cell/B cell interface and acts to sustain calcium levels. Proc Natl Acad Sci U S A 1998; 95:6302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Waal Malefyt R, Verma S, Bejarano MT, Ranes‐Goldberg M, Hill M, Spits H. CD2/LFA‐3 or LFA‐1/ICAM‐1 but not CD28/B7 interactions can augment cytotoxicity by virus‐specific CD8+ cytotoxic T lymphocytes. Eur J Immunol 1993; 23:418–24. [DOI] [PubMed] [Google Scholar]
- 46. Braakman E, Goedegebuure PS, Vreugdenhil RJ, Segal DM, Shaw S, Bolhuis RL. ICAM‐ melanoma cells are relatively resistant to CD3‐mediated T‐cell lysis. Int J Cancer 1990; 46:475–80. [DOI] [PubMed] [Google Scholar]
- 47. Deeths MJ, Mescher MF. ICAM‐1 and B7‐1 provide similar but distinct costimulation for CD8+ T cells, while CD4+ T cells are poorly costimulated by ICAM‐1. Eur J Immunol 1999; 29:45–53. [DOI] [PubMed] [Google Scholar]
- 48. Rosette C, Werlen G, Daniels MA, Holman PO, Alam SM, Travers PJ et al The impact of duration versus extent of TCR occupancy on T cell activation: a revision of the kinetic proofreading model. Immunity 2001; 15:59–70. [DOI] [PubMed] [Google Scholar]
- 49. Anikeeva N, Somersalo K, Sims TN, Thomas VK, Dustin ML, Sykulev Y. Distinct role of lymphocyte function‐associated antigen‐1 in mediating effective cytolytic activity by cytotoxic T lymphocytes. Proc Natl Acad Sci U S A 2005; 102:6437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hamai A, Meslin F, Benlalam H, Jalil A, Mehrpour M, Faure F et al ICAM‐1 has a critical role in the regulation of metastatic melanoma tumor susceptibility to CTL lysis by interfering with PI3K/AKT pathway. Cancer Res 2008; 68:9854–64. [DOI] [PubMed] [Google Scholar]
- 51. Caramalho I, Faroudi M, Padovan E, Muller S, Valitutti S. Visualizing CTL/melanoma cell interactions: multiple hits must be delivered for tumour cell annihilation. J Cell Mol Med 2009; 13:3834–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khazen R, Muller S, Gaudenzio N, Espinosa E, Puissegur MP, Valitutti S. Melanoma cell lysosome secretory burst neutralizes the CTL‐mediated cytotoxicity at the lytic synapse. Nat Commun 2016; 7:10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Basu R, Whitlock BM, Husson J, Le Floc'h A, Jin W, Oyler‐Yaniv A et al Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 2016; 165:100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dustin ML, Chakraborty AK, Shaw AS. Understanding the structure and function of the immunological synapse. Cold Spring Harb Perspect Biol 2010; 2:a002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Valitutti S, Müller S, Calla M, Padovana E, Lanzavecchia A. Serial triggering of many T‐cell receptors by a few peptide‐MHC complexes. Nature 1995; 375:148–51. [DOI] [PubMed] [Google Scholar]
- 56. Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN. Evidence that a single peptide‐MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 1996; 4:565–71. [DOI] [PubMed] [Google Scholar]
- 57. Thiel M, Lis A, Penner R. STIM2 drives Ca2+ oscillations through store‐operated Ca2+ entry caused by mild store depletion. J Physiol 2013; 591(Pt 6):1433–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Badou A, Jha MK, Matza D, Flavell RA. Emerging roles of L‐type voltage‐gated and other calcium channels in T lymphocytes. Front Immunol 2013; 4:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nohara LL, Stanwood SR, Omilusik KD, Jefferies WA. Tweeters, woofers and horns: the complex orchestration of calcium currents in T lymphocytes. Front Immunol 2015; 6:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Grange M, Verdeil G, Arnoux F, Griffon A, Spicuglia S, Maurizio J et al Active STAT5 regulates T‐bet and eomesodermin expression in CD8 T cells and imprints a T‐bet‐dependent Tc1 program with repressed IL‐6/TGF‐β1 signaling. J Immunol 2013; 191:3712–24. [DOI] [PubMed] [Google Scholar]
- 61. Qin Z, Schwartzkopff J, Pradera F, Kammertoens T, Seliger B, Pircher H et al A critical requirement of interferon‐γ‐mediated angiostasis for tumor rejection by CD8+ T cells. Cancer Res 2003; 63:4095–100. [PubMed] [Google Scholar]
- 62. Zhang B, Karrison T, Rowley DA, Schreiber H. IFN‐γ‐ and TNF‐dependent bystander eradication of antigen‐loss variants in established mouse cancers. J Clin Invest 2008; 118:1398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jenkinson SR, Williams NA, Morgan DJ. The role of intercellular adhesion molecule‐1/LFA‐1 interactions in the generation of tumor‐specific CD8+ T cell responses. J Immunol 2005; 174:3401–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Characteristics of different P1A‐expressing tumour cells.
Figure S2. Kinetics of GZMB‐Tom containing granule re‐localization to the CTL‐tumour contact zone during the video experiments.
Figure S3. Individual recordings of TCRP1A‐GZMB‐Tom CTL Ca2+ fluxes in response to the different tumour cells.
Figure S4. Barcoding the TCR‐OT1‐GZMB‐Tom CTL Ca2+ fluxes in response to OVAp/H‐2Kb.
Figure S5. Time required for GZMB‐Tom granule redistribution in the CTL and for calcein release from the target cells during the videos using the TCR‐OT1‐GZMB‐Tom CTL.
Figure S6. Frequency of bursts in Ca2+ fluxes in the response of CTL to different tumour cells.
Table S1. Comparison of type of Ca2+ fluxes and of degranulation for the TCRP1A‐GZMB‐Tom and TCR‐OT1‐GZMB‐Tom CTL.
Video S1. Kinetics of activation of TCRP1A‐GZMB‐Tom CTL in response to P511 mastocytoma cells (as described in Fig. 4a).
Video S2. Kinetics of activation of TCRP1A‐GZMB‐Tom CTL in response to T‐1236 melanoma cells (as described in Fig. 4b).
Video S3. Kinetics of activation of TCRP1A‐GZMB‐Tom CTL in response to T‐RFP‐69 melanoma cells pre‐pulsed with P1Ap (as described in Fig. 4b).