

Abstract
Rationale
Pro-cognitive agents for chronic psychotic disorders (CPDs) might be detected via experimental medicine models, in which neural targets engaged by the drug predict sensitivity to the drug’s pro-cognitive effects.
Objective
This study aims to use an experimental medicine model to test the hypothesis that “target engagement” predicts pro-cognitive effects of the NMDA antagonist, memantine (MEM), in CPDs.
Methods
MATRICS Consensus Cognitive Battery (MCCB) performance was assessed in CPD (n = 41) and healthy subjects (HS; n = 41) in a double-blind, randomized cross-over design of acute (single dose) MEM (placebo vs. 10 or 20 mg p.o.). Measures of prepulse inhibition (PPI) and mismatch negativity previously reported from this cohort substantiated target engagement. Biomarkers predicting MEM neurocognitive sensitivity were assessed.
Results
Testing confirmed MCCB deficits associated with CPD diagnosis, age, and anticholinergic exposure. MEM (20 mg p.o.) reduced MCCB performance in HS. To control for significant test order effects, an “order-corrected MEM effect” (OCME) was calculated. In CPD subjects, greater age, positive MEM effects on PPI, and SNP rs1337697 (within the ionotropic NMDA receptor gene, GRIN3A) predicted greater positive OCME with 20 mg MEM.
Conclusions
An experimental medicine model to assess acute pro-cognitive drug effects in CPD subjects is feasible but not without challenges. A single MEM 20 mg dose had a negative impact on neurocognition among HS. In CPD patients, age, MEM effects on PPI, and rs1337697 predicted sensitivity to the neurocognitive effects of MEM. Any potential clinical utility of these predictive markers for pro-cognitive effects of MEM in subgroups of CPD patients cannot be inferred without a validating clinical trial.
Keywords: Biomarker, Experimental medicine, MATRICS, Memantine, Neurocognition, Schizophrenia, Prepulse inhibition
Introduction
Neurocognitive deficits contribute strongly to functional disability in chronic psychotic disorders (CPDs), including schizophrenia (SZ) (Green 1996; Velligan et al. 1997). While antipsychotics (APs) can blunt the most severe acute psychotic symptoms, any salubrious effects of APs on neurocognitive deficits are generally of small effect sizes (Keefe et al. 2007). Several studies investigating pro-cognitive interventions for SZ have targeted specific neurotransmitter systems including nicotine (AhnAllen et al. 2008; Barr et al. 2008) and glutamate (Heresco-Levy et al. 2004, 2005; Buchanan et al. 2007; Patil et al. 2007; Zink and Correll 2015), based on their hypothesized role in the pathophysiology of neurocognitive deficits, but to date no clear functional benefits from pro-cognitive agents have been demonstrated in SZ patients.
Clinical trials assessing potential pro-cognitive drug effects on neurocognition, symptoms, and function in SZ patients are lengthy and expensive, and ultimately may yield inconclusive results based on many factors. One of these factors is sample heterogeneity: inconsistent or negligible benefits identified in a large cohort of patients might reflect the admixture of subgroups, including some that are sensitive, and others that are insensitive, to pro-cognitive effects. Conceivably, it might be possible to use an experimental medicine approach (Insel 2015) to both accelerate identification of potentially useful pro-cognitive candidates, and to reduce sample heterogeneity by using biomarkers to identify individuals most likely to be sensitive to specific pro-cognitive drug effects. This proof of concept study explores a model for such an experimental medicine approach.
One drug that has yielded inconsistent results in clinical trials in CPD populations is memantine (MEM), a moderate affinity, non-competitive, voltage-dependent NMDA antagonist (Kishi et al. 2013). MEM is FDA-approved to treat dementia and preferentially blocks excessive NMDA receptor activity without disrupting normal activity (Johnson and Kotermanski 2005). Several lines of evidence suggest that MEM might have pro-cognitive effects in disorders other than dementias that are also characterized by cognitive deficits (Alizadeh et al. 2015; Abbasinazari et al. 2015). For example, MEM 20 mg acutely enhances cortical metabolic efficiency (Willenborg et al. 2011) and improves neurocognition after brain injury, and these effects are strongly associated with increased left frontal and parietal lobe glucose metabolism (Kim et al. 2010). To the degree that CPD is accompanied by dysregulation of cortical NMDA release (Javitt 2007; Deutsch et al. 2001), one might predict that pharmacological interventions that selectively block excessive synaptic glutamate transmission might have beneficial effects for CPD patients. Other hypotheses link CPDs to excitotoxins (Schwarcz and Hunter 2007; Braidy et al. 2009), whose effects might be opposed by MEM (Keilhoff and Wolf 1992); indeed, the combined use of MEM and galantamine as a pro-cognitive intervention in SZ has been proposed on this basis (Koola et al. 2014; Geerts et al. 2015).
However, among the small number of placebo-controlled studies of MEM’s pro-cognitive effects in patients with SZ (Lee et al. 2012; de Lucena et al. 2009; Lieberman et al. 2009; Krivoy et al. 2007; Zdanys and Tampi 2008), results are inconsistent. Importantly, these studies did not focus specifically on potential neurocognitive benefits of MEM or on the engagement of neural targets linked with neurocognitive processes. Conceivably, a biomarker predicting sensitivity to the central nervous system effects of MEM might allow investigators to stratify patient cohorts, and thereby increase the likelihood of identifying patients in whom MEM would have pro-cognitive effects.
One critical requirement for a successful experimental medicine approach is the engagement of symptom-relevant neural targets—e.g., some evidence that the drug is active within brain systems relevant to neurocognition. In healthy subjects (HS), NMDA antagonists such as MEM, amantadine, and ketamine enhance performance in measures of early information processing such as prepulse inhibition (PPI; Duncan et al. 2001; Abel et al. 2003; Swerdlow et al. 2002, 2009) and mismatch negativity (MMN; Korostenskaja et al. 2007). Immediately prior to the testing described in the present paper, we detected significant effects of acute administration of MEM (10 and 20 mg p.o.) on PPI and MMN in the present CPD subjects; this provides evidence in the present test subjects that MEM engaged forebrain neural circuitry regulating PPI and MMN (Swerdlow et al. 2016).
While MEM-enhanced PPI and MMN in these test subjects provide evidence of “target engagement” within brain systems that regulate early information processing (Swerdlow et al. 2016), it is not known whether these MEM-induced neurophysiological changes are “predictive biomarkers” of sensitivity to MEM’s putative pro-cognitive effects. Neurocognitive performance in this same cohort was assessed via the MATRICS Consensus Cognitive Battery (MCCB), considered a gold standard for cognitive assessment in pro-cognitive clinical trials of SZ (Green et al. 2004); we chose to report these findings with a detailed neurocognitive assessment separately from those with neurophysiological measures (PPI, MMN) to permit a full analysis of both sets of measures. Here, we report the effects of a single acute “challenge” dose of either 10 or 20 mg MEM on MCCB performance in these CPD subjects and HS, and our efforts to identify predictive factors that distinguish “MEM-sensitive” subgroups. The primary goal of this study was to test the hypothesis that “target engagement” predicts pro-cognitive effects of MEM in CPDs.
Methods
The study was approved by the UCSD Human Subject Institutional Review Board. Subjects were recruited via public advertisements and were paid for study participation. Written informed consent was obtained from all subjects. This laboratory-based experiment was not associated with any clinical trial. Healthy adult subjects and clinically stable CPD patients participated in this study. Ninety percent of CPD subjects were taking antipsychotic (AP) medications at the time of testing (Table S3). Non-experimental data (e.g., demographics, clinical assessments) from this study sample approximates those in our recent report of PPI and MMN measures (Table S3; Swerdlow et al. 2016), save for a small number of subjects whose PPI or MMN data were not usable (Table S2).
After passing a phone interview (assessing current and past medical and psychiatric history, medication and recreational drug use, and family history of psychosis), subjects were invited for an in-person screening session that included the modified Structured Clinical Interview for DSM-IV, Non-Patient Edition (SCID NP) for HS and Mini International Neuropsychiatric Interview (M.I.N.I plus 6) (Sheehan et al. 1998) for patients, the reading component of Wide Range Achievement Test (WRAT version 4; Wilkinson and Robertson 2006) to estimate premorbid level of cognitive ability, the positive and negative syndrome scale (PANSS) for SZ (Kay et al. 1987), a physical examination (medical exclusion criteria: pregnancy, current substance abuse, history of significant medical illness such as hepatitis C, HIV, seizures, open head injury or closed head injury with loss of consciousness >1 min), electrocardiogram, and hearing test (Saico Audiometer, Assens Denmark; excluded for threshold >40 dB(A) at 1000 Hz in either ear). Because subjects were also to be tested on measures of acoustic startle, a screening session was used to confirm reliable startle reflex magnitude (for startle-related methods, see Swerdlow et al. (2016)). In addition, subjects completed several questionnaires related to ethnicity, sexual preference, smoking, and caffeine consumption.
Subjects were told to refrain from consuming psychoactive substances from 1 week prior to testing through completion of the study, other than those prescribed for their illness. Urine toxicology was performed at screening and on the morning of each test day to rule out recent recreational drug use. CPD subjects were asked to maintain compliance with their prescribed medications and to inform the research team of any medication changes during the study.
Subjects meeting inclusion criteria (Table S1) returned for their first test day after 5–10 days, and for a second test day 1 week after the first test day. Eighty-nine subjects completed at least one test day; four patients and three HS were excluded from the final data analyses, as shown in Table S2. There were 82 completers (10 mg MEM—HS/CPD = 19:20; 20 mg MEM—HS/CPD = 22:21) (Table S3).
Across the two test days, all subjects received MEM and PBO in a randomized, double-blind, counterbalanced, within-subject design. On each test day, subjects received either MEM HCl (10 or 20 mg) or PBO. The timing of procedures is detailed in Table S4. Vital signs (VS) and symptom rating scale (SRS) scores were obtained at baseline and periodically after pill administration. MCCB testing began 240 min post pill ingestion, based on the reported pharmacokinetics of MEM in HS (Sonkusare et al. 2005). On test day 2, subjects also provided 5 ml of blood for genotyping.
The MCCB was developed to evaluate neurocognition in trials of pro-cognitive therapies for SZ and is accepted by the FDA as a primary endpoint (Kern et al. 2008; Nuechterlein et al. 2008). The MCCB measures seven key domains relevant to cognitive deficits in SZ and includes ten tests that assess the following: speed of processing (SP), attention/vigilance (AV), working memory (WM), verbal learning (VL), visual learning (VsL), reasoning and problem solving (RP), and social cognition (SC), and provides T-scores for each domain and a composite score of all domains. While alternate forms of some MCCB tests can be used to blunt practice effects, they also introduce variability in PBO vs. active drug conditions; thus, the a priori decision was made to maintain consistency in MCCB versions across tests.
Genotyping assays
We reviewed a list of 13 single nucleotide polymorphisms within 8 genes that were previously associated with measures of PPI and/or related neurophysiological or neurocognitive measures in mixed samples of SZ patients and HS (Greenwood et al. 2011, 2013); of these, 4 SNPs within 4 genes yielded sufficient minor allele frequencies to permit meaningful analyses in the present sample: rs158337 (GRID2), rs40184 (SLC6A3), rs1394785 (ERBB4), and rs1337697 (GRIN3A). Genotyping of SNPs was performed using iPLEX Gold chemistry on Agena Bioscience MassARRAY® System (Agena Bioscience, San Diego, CA) at the Roswell Park Cancer Institute Genomics Shared Resource facility. Two multiplex assays were designed using Assay Design Suite v2.0 (Agena) software and genotyping was performed as per manufacturer protocol. Briefly, a locus-specific PCR reaction was carried out, followed by an allele-specific primer extension reaction (iPLEX assay primers in Supplemental Methods). The genotypes for each SNP were obtained after visual inspection and corrections of the spectra using MassARRAY Typer Analyzer v4 (Agena).
PPI and MMN
The effects of MEM on PPI and MMN in this study cohort were reported in full in a recent publication (Swerdlow et al. 2016). Details of the PPI- and MMN-enhancing effects of MEM in CPD subjects and HS are found in Fig. 1.
Fig. 1.
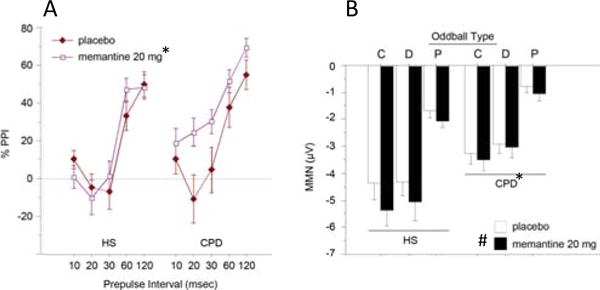
Effects of MEM (20 mg) on PPI (a) and MMN (b) in the present cohort of HS and CPD subjects, as reported recently (Swerdlow et al. 2016). (Number sign) significant main effect of drug; (asterisk) significant effect of diagnosis; oddball type: (P)—pitch, (D)—duration, and (C)—combined P and D. Importantly, these effects provide evidence that MEM engaged neural “targets” in the present cohort, i.e., forebrain circuitry regulating PPI and MMN, immediately prior to the MCCB testing reported in this paper
Statistical analyses
The main effect of MEM (10 and 20 mg) on MCCB performance was analyzed using repeated measures analysis of variance (RMANOVA) with MEM (PBO vs. active) as a within-subject factor, and diagnosis (HS vs. CPD) and dose as between-subject factors. Significant two-way (drug × diagnosis or drug × MCCB domain) or three-way interactions (drug × MCCB domain × diagnosis) were followed by appropriate post-hoc analyses. In addition, categorical (ANOVA after median split) and continuous (regression) analyses were used to assess the effects of age, test order, gender, placebo neurocognitive performance (henceforth, “baseline”), MEM-induced MMN changes (MEM minus PBO values), and MEM-induced PPI changes (MEM minus PBO values) on MCCB performance and MEM sensitivity.
Analyses progressed in a systematic manner: (1) We first compared our sample’s baseline performance with published MCCB patterns, examining sample characteristics, and the effects of age, sex, and diagnosis on MCCB performance. This was done to test the validity and sensitivity of this MCCB assessment; (2) We next assessed the effects of MEM on MCCB performance using RMANOVA, starting with the most global metric: the MCCB composite T-score, followed by individual MCCB domains. We included age, test order (active drug administered on day 1 vs. day 2), and baseline performance as covariates in these analyses; (3) To address significant effects of test order (due to increased MCCB performance from test day 1 to test day 2), we empirically calculated an “order-corrected MEM effect” (OCME) on each MCCB domain. To accomplish this, a simple mean “Day effect” (mean (day 2 minus day 1)) was calculated for each dose group and diagnosis. A “MEM effect” (active minus PBO) was also calculated for each subject, for each MCCB measure (composite and each domain). The MEM effect for each subject was then “order-corrected” by either (a) adding the mean Day effect to the MEM effect (if active drug was administered on day 1) or (b) subtracting the mean Day effect from MEM effect (if active drug was administered on day 2).
One-way ANOVAs were used to calculate the effects of age, SNP genotype, dose, and diagnosis on OCME values. Simple regression analyses were performed to correlate effects of MEM-induced PPI changes, MEM-induced MMN changes, age, duration of illness, and anticholinergic load (calculated as picomole of atropine equivalents per milliliter (pmol/ml) according to Chew et al. 2008) with OCME values. Alpha for planned comparisons and empirical findings were set at 0.05 and 0.01, respectively.
Results
Subjects
Table S3 shows demographic and clinical characteristics of the 10 and 20 mg groups. Two CPD subjects carried a diagnosis of schizoaffective disorder, depressed type; all others were diagnosed with SZ. Most (37/41) patients were taking atypical APs; only one was taking clozapine. No subjects were taking medications with known NMDA antagonist properties.
Subjective and physiological measures
Compared to pre-pill baseline levels, analyses of autonomic measures before and after MCCB testing (Fig. S1) revealed no significant main or interaction effects of diagnosis, MEM, or dose group on change in heart rate and systolic or diastolic blood pressure. Analyses of SRS scores before and after MCCB testing (Fig. S1) generally yielded no meaningful main or interaction effects. When inspection of the data revealed non-normal distributions, with many subjects providing extreme baseline SRS scores of 100 (e.g., “Happy”) or 0 (e.g., “Drowsy”), non-parametric comparisons confirmed the general lack of drug effects on subjective measures before and after MCCB testing.
MCCB performance
Analyses of MCCB performance after PBO (baseline MCCB) (Table 1) support the validity and sensitivity of this neurocognitive assessment. ANOVA of T-scores across the seven MCCB domains confirmed significant main effects of diagnosis (HS > CPD), domain and test order (active pill day 1 vs. 2). There were no significant differences in 10 vs. 20 mg dose groups, nor significant two-way interactions. Post-hoc contrasts supported the sensitivity of MCCB measures to known impairing effects of age (young > old: p < 0.015) and both anticholinergic activity (scores by CPD with <15 pmol/ml exceeds those with ≥15 pmol/ml; p = 0.005) and global functioning in CPD subjects (median split: high GAF > low GAF, p < 0.025).
Table 1.
PBO MCCB performance (post-placebo)
| PBO MCCB domain T-score | All subjects |
|---|---|
| Main analysis with diagnosis as between-subject factor | |
| Diagnosis (Dx) | F = 38.71, df (1,78), p < 0.0001 |
| Domain | F = 10.72, df (6, 468), p < 0.0001 |
| Domain × Dx | F = 4.13, df (6, 468), p = 0.0005 |
| Additional analyses with either sex/age/test order/dose as between-subject factor | |
| 1. Gender | F < 1 |
| 2. Age (median split) | F = 6.35, df (1,78), p = 0.01 |
| Age × domain | F = 2.83, df (6, 468), p = 0.01 |
| Age × Dx | F < 1 |
| Age × domain × Dx | F < 1 |
| 3. Test order | F = 4.94, df (1,78), p < 0.03 |
| Test order × domain | F = 2.46, df (6, 468), p < 0.025 |
| Test order × Dx | F < 1 |
| 4. Dose | F < 1 |
| Dose × Dx | F = 5.23, df (1,78), p < 0.025 |
| Dose × domain | NS |
| Dose × domain × Dx | NS |
| In CPD subjects only | |
| 5. Smoking status (SS) | F < 1 |
| Domain × SS | F = 3.82, df (6, 234), p < 0.002 |
| PBO composite T-score | CPD subjects |
| Anticholinergic activity (low vs. high, split >15/≤ 15) | F = 8.71, df (1,35), p = 0.005 |
| Correlation(s) vs. PBO composite T-score | |
| Global assessment of function score | R = 0.5, p = 0.001 |
| Duration of illness (years) | R = 0.04, NS |
Analysis of MCCB performance including both MEM and PBO (Table 2) revealed significant main effects of diagnosis (p < 0.0001), drug (PBO vs. active: p < 0.001), and MCCB domain (p < 0.0001). There were significant interactions of dose × drug (p < 0.005), dose × test order (p < 0.05), and dose × drug × test order (p < 0.05), in addition to several other significant two- and three-way interactions involving drug, diagnosis, test order, and domain.
Table 2.
MEM effect on MCCB performance
| MCCB cognitive domain T-score | All subjects |
|---|---|
| Main analysis with diagnosis, dose, and test order as between-subject factor | |
| Diagnosis (Dx) | F = 56.17, df (1,72), p < 0.0001 |
| Drug | F = 12.26, df (1,72), p = 0.0008 |
| Domain | F = 16.23, df (6, 432), p < 0.0001 |
| Dose | F < 1 |
| Test order | F < 1 |
| Dose × test order | F = 4.09, df (1,72), p < 0.05 |
| Drug × dose | F = 8.73, df (1,72), p = 0.004 |
| Drug × test order | F = 85.74, df (1,72), p < 0.0001 |
| Domain × Dx | F = 4.57, df (6, 432), p = 0.0002 |
| Drug × dose × test order | F = 5.45, df (1,72), p < 0.05 |
| Drug × domain × dose | F = 3.06, df (6, 432), p = 0.006 |
| Drug × domain × test order | F = 16.39, df (6, 432), p < 0.0001 |
| Drug × domain × test order × Dx | F = 2.09, df (6, 432), p = 0.05 |
| MCCB composite T-score | All subjects |
| Main analysis with diagnosis, dose, and test order as between-subject factors | |
| Dx | F = 55.71, df (1,72), p < 0.0001 |
| Drug | F < 1 |
| Drug × test order | F = 39.58, df (1,72), p < 0.0001 |
| Drug × dose | F = 4.15, df (1,72), p < 0.05 |
| OCME cognitive domain T-score | All subjects |
| Main analysis with diagnosis and dose as between-subject factor | |
| Diagnosis | F < 1 |
| Dose | F = 7.84, df (1,76), p = 0.006 |
| Domain × dose | F = 2.51, df (6, 456), p = 0.02 |
Based on these interactions, post-hoc analyses first examined drug effects for each MEM dose. For the 10-mg group, ANOVA of MCCB T-scores confirmed significant main effects of diagnosis and domain, and significant interactions of drug × test order (p<0.0001), drug × domain (p<0.05), drug × domain × test order (p< 0.0001), and drug × diagnosis × domain × test order (p<0.006) (Fig. 2a). Across several MCCB domains, there were significant interactions of drug × test order, and in some cases, drug × diagnosis × test order. Similarly, for the 20-mg group, ANOVA confirmed significant main effects of diagnosis and domain, as well as drug (p<0.0001), and significant three-way interactions of drug × diagnosis × domain (p<0.05) and drug × test order × domain (p<0.0001). The interaction of drug × diagnosis × domain reflected a MEM-induced suppression in verbal learning in HS but not in CPD subjects, best explained as a “floor effect” in CPD subjects resulting at least in part from their greater age (Fig. 2b).
Fig. 2.
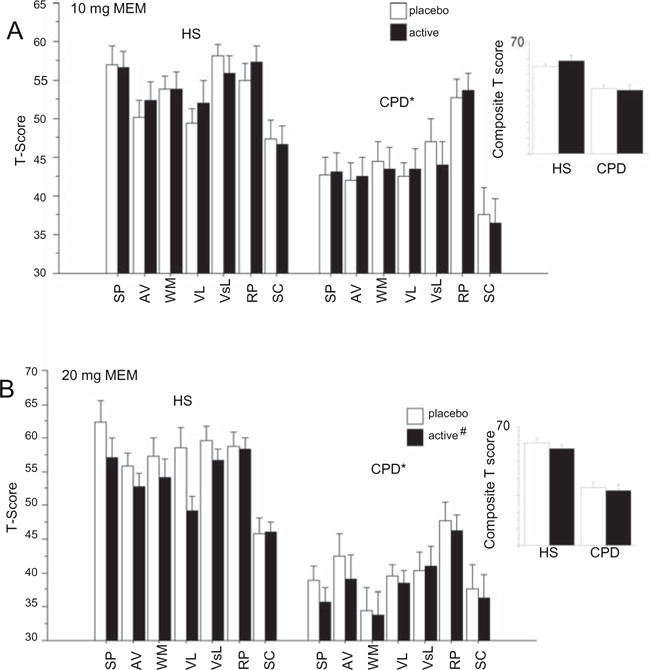
Effects of acute administration of MEM (PBO vs. 10 mg po (a) and PBO vs. 20 mg po (b)) on MCCB performance (cognitive domain T-score and composite score) in HS (n = 41) and CPD subjects (n = 41). MCCB domains: SP—speed of processing; AV—attention/vigilance; WM—working memory; VL—verbal learning; VsL—visual learning; RP—reasoning and problem solving; SC—social cognition. (Number sign) significant main effect of drug; (asterisk) significant effect of diagnosis. Alternative strategies were explored for understanding differential MEM effects across domains, including parsing MCCB domains into those incorporating a timing/speed demand (SP, AV, RP) vs. those without such a demand (WM, VL, VisL, SC) (Fig. S2)
These results suggest that acute MEM effects on MCCB performance were weak, at best, and that the degree to which MEM impacted performance at either dose was obscured by strong effects of test order (Table 2) and practice effects (Table S7). These effects of test order and practice were expected, based on the 7-day inter-test interval and a priori decision to not use alternative MCCB versions on different test days. To mitigate these effects of test order and practice, we empirically generated an OCME variable, by calculating for each subject’s T-score the “MEM effect” (i.e., MEM minus PBO), and adjusting for the group mean order effect (day 2 minus 1) (see “Methods” section and Fig. 3a). Group mean order effects varied across MCCB domains, reflecting the magnitude of “practice effects” for each domain; for the full cohort, these values ranged from 0.06 T-score units (social cognition) to 8.46 T-score units (verbal learning), with a Composite Score value of 5.26 T-score units. With these values extracted, the OCME values approximated “order-neutral” indices of MEM effects on MCCB performance; distributions of domain and composite scores tended to be bimodal for MEM effects (reflecting the impact of the two test orders) and unimodal and normally distributed for the OCME (Fig. 3a).
Fig. 3.
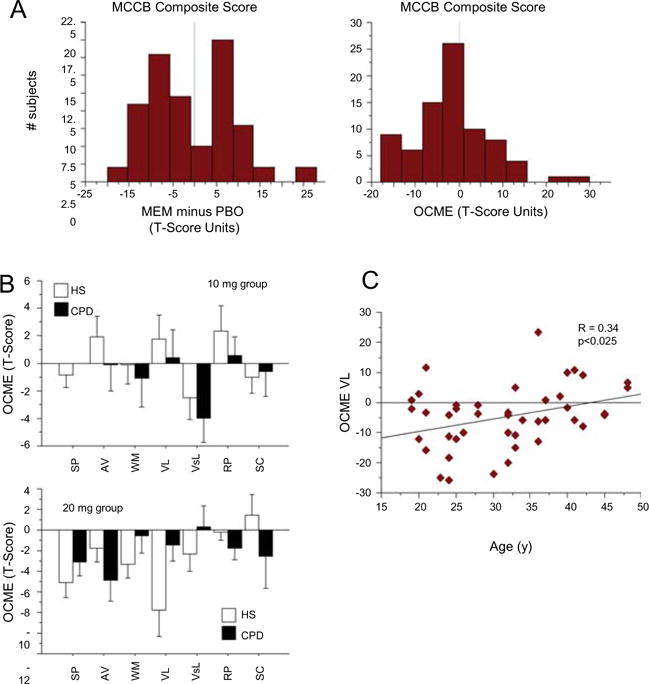
Correcting MEM effects to account for impact of test order via an “order-corrected MEM effect” (OCME) variable. OCME was calculated from each subject’s T-score “MEM effect” (i.e., MEM minus PBO), and adjusting for the group mean order effect (day 2 minus day 1) for that domain or composite value. a Left histogram shows bimodal distribution of MEM effects on MCCB scores (MEM minus PBO, composite T-scores in this example) reflecting populations distinguished by those who received MEM on test day 1 vs. 2; right histogram shows unimodal distribution of OCME scores. b OCME scores for 10 mg group (top) and 20 mg group (bottom), showing mixed neurocognitive effects of 10 mg MEM across MCCB domains, and largely neurocognitive-impairing effects of 20 mg MEM, particularly across domains of SP, AV, WM, and VL. The high dose of MEM impaired VL to a greater degree in HS than in CPD subjects, an effect that cannot be easily parsed from the age sensitivity of this MEM effect (c), due to the older age of CPD vs. HS subjects in this 20 mg dose group
For the combined dose groups, this OCME analysis detected a significant effect of MEM dose (p < 0.007) and dose × domain interaction (p = 0.02) (Table 2). For the 10-mg MEM dose group, ANOVA revealed no significant effect of diagnosis (F < 1), a near-significant main effect of MCCB domain (p = 0.06), but no other significant two- or three-way interactions. The near-significant domain effect for the 10-mg dose group reflected medium-to-large effect size MEM-induced increases in performance in some domains (attention/vigilance, verbal learning, reasoning/problem solving) and medium-to-large effect size impairments in others (working memory, visual learning, social cognition) (Fig. 3b). For the 20-mg MEM dose group, this OCME analysis revealed no significant effect of diagnosis (F < 1) or domain, but a significant interaction of diagnosis × domain (p < 0.02). As with the “order-uncorrected” analyses (above), this interaction reflected a greater MEM-suppressed verbal learning among HS vs. CPD subjects, which appeared attributable to a floor effect in CPD subjects, associated with their greater age vs. HS. Consistent with this interpretation, there was a significant positive correlation between age and the OCME for verbal learning, i.e., MEM-enhanced verbal learning in older subjects, but suppressed verbal learning in younger subjects (Fig. 3c).
Other clinical, demographic, and neurocognitive variables were explored to determine whether they moderated the magnitude of OCMEs on neurocognitive performance (Tables S5A and B). Among these (age, duration of illness, age of onset, GAF score, PANSS score, and PBO-level MCCB performance), there were significant correlations of all age-related variables (age, age of onset, and duration of illness) with performance in several different MCCB domains, as might be predicted based on ANOVA comparisons described above (MEM-enhanced performance associated with older age and longer illness duration). Neither global function nor symptom levels strongly predicted OCME, and PBO-level MCCB performance modestly predicted OCME for composite but not domain-specific scores (Table S5A and B).
As noted above (Fig. 1), MEM (20 mg) significantly enhanced PPI and MMN in this study cohort (Swerdlow et al. 2016), presumably reflecting the activity of this NMDA antagonist within brain systems that regulate early information processing. One key hypothesis being tested in this study is that these laboratory-based indices of forebrain circuitry “target engagement” by MEM could identify individuals most sensitive to MEM-induced changes in MCCB performance (Table 3). For the 20-mg MEM dose that significantly increased PPI, ANOVA of MCCB performance, using diagnosis and MEM-induced PPI change (increase vs. decrease) as categorical grouping factors, yielded the expected significant effects of diagnosis and drug, as well as a significant three-way interaction of diagnosis × drug × PPI group (F = 4.81, df 1,30, p < 0.04); a separate analysis among CPD subjects detected a significant interaction of drug × PPI group (F = 6.67, df 1,15, p = 0.02). Among CPD subjects whose PPI was reduced by MEM, MCCB performance was impaired by MEM, but this was not the case among subjects whose PPI was increased by MEM (Cohen’s d = −0.38 vs. −0.04) (Fig. 4a).
Table 3.
MEM effect on MCCB performance in subjects with low vs. high MEM sensitivity on measures of PPI and MMN
| MCCB cognitive domain T-score | All subjects | |
|---|---|---|
| Main analysis with diagnosis and MEM effect on PPI as between-subject factor | ||
| 10 mg | 20 mg | |
| Diagnosis (Dx) | F = 20.41, p < 0.0001 | F = 14.97, p = 0.0005 |
| Drug | F < 1 | F = 17.22, p < 0.0005 |
| Domain | F = 9.36, p < 0.0001 | F = 5.22, p < 0.0001 |
| Lo/hi MEM effect PPI (PPI) | F < 1 | F < 1 |
| Drug × PPI × Dx | NS | F = 4.81, p < 0.04 |
| Domain × diagnosis | NS | F = 2.82, p < 0.025 |
| Drug × Dx | NS | F = 2.16, p < 0.05 |
| Drug × domain × PPI × Dx | F = 2.26, p < 0.05 | NS |
| Main analysis with diagnosis and MEM effect on MMN as between-subject factor | ||
| 10 mg | 20 mg | |
| Diagnosis | F = 25.26, p < 0.0001 | F = 30.56, p < 0.0001 |
| Lo/hi MEM effect MMN (MMN) | F < 1 | F < 1 |
| Drug | F < 1 | F = 7.85, p < 0.01 |
| Domain | F = 8.11, p < 0.0001 | F = 4.13, p < 0.001 |
| Domain × Dx | F < 1 | F = 3.12, p < 0.01 |
| Domain × Dx × MMN | F = 2.17, p < 0.05 | F < 1 |
Fig. 4.
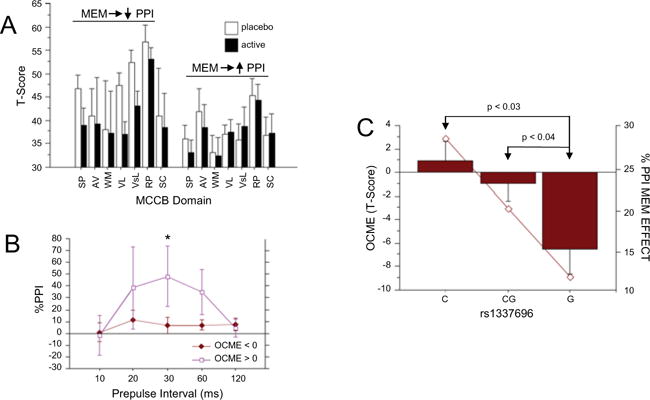
Exploring the relationship between MEM neurocognitive effects and biomarkers form the present study. a MCCB-impairing effects of acute MEM challenge were evident among CPD subjects who experienced PPI-reducing vs. PPI-enhancing effects of MEM; those subjects whose PPI was enhanced by MEM on average had lower basal (PBO) MCCB scores that were unaffected by MEM. b The inverse relationship from that shown in a: elevated PPI among subjects (here HS and CPD) whose MCCB composite score was enhanced vs. impaired by MEM (20 mg). Asterisk, significantly greater MEM effect on PPI for 30 ms prepulse trials (p < 0.035) and 60 ms trials (p < 0.04) among subjects whose MCCB was enhanced by MEM. c Bars show composite MCCB OCME score after 20 mg MEM among CPD subjects, based on the identity of their SNP rs1337697, within the glutamate receptor gene, ionotropic N-methyl-D-aspartate receptor 3A (GRIN3A). Subjects homozygous for the G allele were more sensitive to the MCCB-disruptive effects of MEM, compared to C-homozygotes or CG-heterozygotes. The solid line overlapping these bars represents values for MEM-enhanced PPI in these subjects, showing an arithmetically parallel relationship with large effect size differences between GG and CC individuals (see text)
We examined the “inverse” side of this relationship of MEM-enhanced PPI vs. MCCB performance by assessing the magnitude of MEM-enhanced PPI among subjects whose overall MCCB performance was either enhanced vs. impaired by MEM (i.e., those with positive vs. negative OCME values for the Composite MCCB score). ANOVA yielded no significant main effects of diagnosis or MCCB group, but there was a significant interaction of MCCB group × prepulse interval (F = 2.77, df 4120, p = 0.03), reflecting significantly greater MEM-induced increases in 30-ms (p < 0.035) and 60-ms (p < 0.04) PPI among subjects whose MCCB performance was enhanced vs. impaired by MEM (Fig. 4b).
A similar relationship was not detected between MEM-enhanced MMN and MCCB effects, likely due to the small MEM-induced reduction in MCCB performance among CPD patients whose MMN deficits were either exacerbated (d = −0.11) or normalized (d = −0.09) by MEM.
SNPs
Moderating effects of four SNPs (rs40184, rs1394785, rs1583337, rs1337697) on MEM sensitivity were explored in ANOVAs of composite OCMEs, using genotype, diagnosis, and dose as grouping factors, revealing no significant main effects of SNP (F < 1). For rs1337697 (a SNP within the glutamate receptor gene, ionotropic NMDA receptor 3A (GRIN3A)), there was a significant two-way diagnosis × dose interaction (p = 0.01) and three-way interaction of diagnosis × dose × SNP (F = 3.70, df 2,66, p = 0.02). Among CPD subjects in the 20-mg dose group, analyses revealed a significant SNP rs1337697 effect (F = 3.70, df 2,17, p < 0.05; G < CG < C) for the OCME composite score, reflecting greater sensitivity to the MCCB-disruptive effects of MEM in CPD subjects homozygous for the G allele compared to heterozygous CG (d = 1.10; p < 0.04) or CC homozygotes (d = 1.69; p < 0.03). A parallel but arithmetically smaller SNP pattern was evident in the magnitude of MEM effects on PPI (G< CG; d = 0.20; G < C; d = 0.59) (Fig. 4c).
Discussion
Populations defined by clinical symptoms are likely to include subgroups whose symptoms reflect distinct underlying mechanisms. Variable therapeutic drug responses based on this heterogeneity can contribute to negative outcomes in clinical trials; the cost of failed trials and the resulting dearth of novel therapeutics underscores the need for biomarkers that identify patient subgroups that share characteristics rendering them more vs. less sensitive to specific drug effects. Identifying these biomarkers and demonstrating their predictive value are two challenges to an experimental medicine paradigm. We asked whether a biomarker of drug sensitivity—enhanced sensorimotor gating after acute dosing with MEM—predicted sensitivity to the neurocognitive effects of this drug in CPD subjects. The findings highlight both the limitations of our study design and the challenges inherent to this experimental medicine paradigm.
We report that a single, acute dose of MEM (20 mg) that increased PPI in CPD subjects actually impaired MCCB performance, at a time point associated with robust blood levels (Sonkusare et al. 2005). In keeping with the experimental medicine model, MEM effectively engaged neural targets associated with neurocognition—PPI and MMN—but was ineffective in enhancing neurocognition. Of course, the effects of a single, relatively high dose on neurocognitive performance may not reflect a drug’s clinical impact after sustained dosing, using an escalating dose schedule, as MEM is typically used in patients with dementia. Thus, a limitation of the present design is that the clearest interpretation of the results relates to an acute drug-induced change in neurocognition that might have little relevance—mechanistic, predictive, or otherwise—to the ultimate clinical value of the drug. Many acute drug effects are useful biomarkers for therapeutics ranging from hormones (Biller 2007) to anti-Parkinsonian therapies (Hughes et al. 1990) to bronchodilators (Fruchter and Yigla 2009), but this is not always the case, and the most informative study design might have assessed the relationship of MEM-enhanced PPI to the clinical benefits of MEM exhibited after sustained daily dosing (e.g., for 12 weeks (Lieberman et al. 2009)) with appropriate dose titration. While such a design is feasible (Bhakta et al. 2015), its financial and logistical demands are substantial and would not have been justifiable prior to testing the effects of MEM on PPI and MMN in CPD patients, which was first accomplished with this present cohort.
Another challenge to our study design was evident in the substantial effect of test order (active drug day 1 vs. 2) on the measurement of MCCB changes, most likely due to known practice effects on MCCB performance (Nuechterlein et al. 2008; Wesnes and Pincock 2002). With 7 days separating MCCB tests, drug effects on neurocognitive performance were not easily separated from experiential effects; this potential confound can impact any within-subject study of acute pro-cognitive drug effects (e.g., Chou et al. 2013). While some MCCB measures have multiple versions that reduce practice effects (Nuechterlein et al. 2008), these multiple versions also are a source of variability across drug tests, and the considered approach of the present study was to accept larger practice effects while carefully measuring these effects and testing ways to minimize their impact on the dependent measures. The use of an OCME was an empirical, blunt attempt to extract from the dependent measure (MEM effect on an MCCB T-score) the degree to which any specific MCCB domain was impacted by practice effects in each subject group; this approach requires additional empirical confirmation. Nonetheless, some evidence suggested that this approach was valid (e.g., “normalized” distributional properties of MCCB T-scores) and sensitive (e.g., relationship of OCME scores to PPI MEM sensitivity). The use of longer inter-test intervals, alternative test forms and multiple doses with randomized orders, and more sophisticated statistical disentangling of potential practice or order effects may offer both advantages and disadvantages compared to the use of the proposed order-correction approach.
A third limitation of this study approach is that sample size is limited by the need for repeated testing, within-subject dose comparisons and the collection of numerous brain-based measures for evaluation as potential biomarkers. While our sample met power demands for individual dependent measures, it was inadequate for more complex statistical approaches to identify multivariate relationships and latent factors that might moderate drug sensitivity. Instead, the effects of several variables (e.g., demographic, clinical, and performance variables, specific SNPs, PPI, and MMN) on MEM sensitivity were considered individually, in some cases based on a priori predictions. Sample constraints clearly limit the ability to detect interactions among dependent and latent variables, though it is less obvious how such information could be incorporated into clinically useful predictive models. The small sample also limited the ability to utilize genetic information as potential biomarkers, and the identification of a single SNP that was associated with MEM sensitivity for MCCB (and to a lesser degree, PPI), and which may be mechanistically linked to MEM-like effects in models of neuroprotection (Nakanishi et al. 2016), must be viewed with caution, to be revisited in future studies.
Despite these limitations, some findings supported the potential utility of this experimental medicine model. First, even though the MCCB was embedded within this complex experimental model—that included repeated testing, double-blind pill administration, psychophysiological and neurophysiological measures, repeated symptom ratings, and autonomic measures in chronically psychotic patients—it was possible to confirm a number of predicted relationships between independent variables and PBO MCCB performance, including diagnosis, age, global functioning, anticholinergic burden, and practice (test order). Second, one pattern previously reported with MEM effects on PPI (Swerdlow et al. 2016)—greater enhancement among older vs. young CPD subjects—was reproduced to some degree in the present data set: in several of the current measures of MEM effects in CPD subjects, older age was associated with either less impairment or greater improvement. For example, the MEM effect on verbal learning was positive among older CPD subjects but negative among younger ones. As was the case with PPI (Swerdlow et al. 2016), some age effects on MCCB MEM sensitivity could not be neatly parsed from the associated variable of illness duration.
We predicted that any observed effects of MEM on neurocognitive performance might be associated with the magnitude to which MEM “engaged” measures of neural circuit function relevant to neurocognition. This prediction follows the simplest line of reasoning, e.g., that a drug is most likely to change neurocognition if it gets into the brain and changes circuitries that regulate neurocognition. Because brain mechanisms underlying both PPI and MMN are implicated in the regulation of neurocognition and its impairment in CPDs, we tested the hypothesis that MEM-induced changes in PPI or MMN were associated with acute MEM effects on MCCB performance. While such a relationship was not detected for MMN (perhaps due to small effect size increases in MMN observed with MEM (Swerdlow et al. 2016)), there was some evidence for a relationship between MEM-induced PPI changes and changes in MCCB performance. CPD subjects whose PPI was reduced after 20 mg MEM exhibited substantial sensitivity to MCCB-impairing effects of MEM, while those whose PPI was increased after 20 mg MEM exhibited no overall reductions in MCCB performance, and in some domains, exhibited small increases in MCCB performance. Perhaps this should not be surprising, since patients’ age was positively associated with greater enhancing (or less degrading) effects of MEM on both PPI and MCCB performance. The inverse pattern was also observed: CPD subjects exhibiting positive OCME scores for Composite MCCB performance also exhibited significantly greater sensitivity to the PPI-enhancing effects of MEM, compared to patients with negative OCME scores. Such categorical grouping strategies (PPI increased vs. decreased; OCME positive vs. negative) are arbitrary, and other strategies (e.g., quartile splits) might be more clinically useful and in some cases yielded greater group separation.
Nonetheless, there is practical value for such a dichotomous prediction within a clinical trial, and based on the present findings, it would be rational to predict that the subgroup of CPD subjects whose PPI was elevated by an acute dose of 20 mg MEM might be “enriched” with subjects most sensitive to positive effects of MEM on neurocognition. If such pro-cognitive effects could be marshaled in the service of cognitive remediation or other therapeutic interventions, this subgroup of patients might be expected to benefit clinically from the addition of MEM to their existing AP regimen. Conversely, based on the present findings, it would seem rational to predict that the CPD subjects whose PPI was reduced after 20 mg MEM might not be optimal candidates for experiencing such clinical benefits from adjunctive MEM treatment. Of course, an agnostic but equally defensible hypothesis is that any robust brain-based “signal” after acute MEM administration—whether it is increased or decreased MCCB performance, or increased or decreased PPI—reflects MEM’s CNS penetration and neural engagement that might ultimately predict clinical sensitivity after long-term administration.
Lastly, it is worth noting that the critical “biomarker” in this experimental medicine approach (PPI) is not diagnostically specific: MEM increases PPI not just in CPD subjects but also in HS (Swerdlow et al. 2009), and the rationale for selecting this “biomarker” is primarily that it provides evidence that an individual or group are sensitive to the ability of MEM to access forebrain circuitries that regulate higher cognition. Thus, PPI is used as a “blunt” instrument, simply to demonstrate forebrain “target engagement” by MEM, agnostic to any clinical, neurocognitive, or psychophysiological implications of this measure in CPD patients. The only other predictor of positive MEM effects in the present study or in our recent report (Swerdlow et al. 2016) is greater age. Thus, there is no reason to believe that this PPI biomarker would predict drug sensitivity that is limited to one diagnostic group over another, or one neurocognitive domain over another, and it is conceivable that PPI MEM sensitivity might be useful for predicting positive therapeutic responses in other clinical populations, including those in whom MEM is known to exhibit some benefits, such as patients with dementia.
Supplementary Material
Acknowledgments
The authors thank Ms. Sarah Lamb for her technical assistance in data collection, Ms. Michelle Breier for assistance in data collation, and Ms. Maria Bongiovanni in manuscript preparation.
Supported by MH059803, MH094320, and MH093453 (NRS, PI). SB and GAL supported by the VISN 22 MIRECC, GAL by the Sidney R. Baer Jr. Foundation and Brain & Behavior Research Foundation, SB and HHC by the Brain & Behavior Research Foundation, SB by the APF Kempf Award, and HHC by the APF/Merck Early Academic Career Award and T32-MH018399. SNP genotyping was performed by the Genomics Shared Resource supported by Roswell Park Cancer Institute and National Cancer Institute (NCI) grant P30CA016056. In the past 3 years, NRS has had support from Neurocrine, Inc. and Genco, Inc., and GAL has received support from Astellas and Forum Pharmaceuticals. Neither NRS nor GAL have ever received support from companies that develop or market memantine.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00213-016-4291-0) contains supplementary material, which is available to authorized users.
Conflict of interest The remaining authors declare no conflict of interest.
References
- Abbasinazari M, Adib-Eshgh L, Rostami A, Beyraghi N, Dabir S, Jafari R. Memantine in the prevention or alleviation of electroconvulsive therapy induces cognitive disorders: a placebo controlled trial. Asian J Psychiatry. 2015;15:5–9. doi: 10.1016/j.ajp.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Abel KM, Allin MP, Hemsley DR, Geyer MA. Low doses of ketamine increases prepulse inhibition in healthy men. Neuropharmacology. 2003;44:729–737. doi: 10.1016/s0028-3908(03)00073-x. [DOI] [PubMed] [Google Scholar]
- AhnAllen CG, Nestor PG, Shenton ME, McCarley RW, Niznikiewicz MA. Early nicotine withdrawal and transdermal nicotine effects on neurocognitive performance in schizophrenia. Schizophr Res. 2008;100:261–269. doi: 10.1016/j.schres.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh NS, Maroufi A, Jamshidi M, Hassanzadeh K, Gharibi F, Ghaderi E. Effect of memantine on cognitive performance in patients under electroconvulsive therapy: a double-blind randomized clinical trial. Clin Neuropharmacol. 2015;38:236–40. doi: 10.1097/WNF.0000000000000109. [DOI] [PubMed] [Google Scholar]
- Barr RS, Culhane MA, Jubelt LE, Mufti RS, Dyer MA, Weiss AP, Deckersbach T, Kelly JF, Freudenreich O, Goff DC, Evins AG. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008;33:480–490. doi: 10.1038/sj.npp.1301423. [DOI] [PubMed] [Google Scholar]
- Bhakta S, Twamley E, Light G, Talledo J, Chou HH, Balvaneda B, Gaddis L, Swerdlow N. Memantine’s acute effects on neurocognition in schizophrenia (SZ) as a predictor of neurocognitive benefits from Compensatory Cognitive Training (CCT) Neuropsychopharmacology. 2015;40(Supp):S368–369. [Google Scholar]
- Biller BM. Concepts in the diagnosis of adult growth hormone deficiency. Horm Res. 2007;68(Suppl 5):59–65. doi: 10.1159/000110478. [DOI] [PubMed] [Google Scholar]
- Bond AJ, James DC, Lader MH. Physiological and psychological measures in anxious patients. Psychol Med. 1974;4:364–373. doi: 10.1017/s0033291700045803. [DOI] [PubMed] [Google Scholar]
- Braidy N, Grant R, Adams S, Brew BJ, Guillemin GJ. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox Res. 2009;16(1):77–86. doi: 10.1007/s12640-009-9051-z. [DOI] [PubMed] [Google Scholar]
- Buchanan RW, Javitt DC, Marder SR, Schooler NR, Gold JM, McMahon RP, Heresco-Levy U, Carpenter WT. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry. 2007;164:1593–1602. doi: 10.1176/appi.ajp.2007.06081358. [DOI] [PubMed] [Google Scholar]
- Bunney WE, Jr, Hetrick WP, Bunney BG, Patterson JV, Jin Y, Potkin SG, Sandman CA. Structured Interview for Assessing Perceptual Anomalies (SIAPA) Schizophr Bull. 1999;25:577–592. doi: 10.1093/oxfordjournals.schbul.a033402. [DOI] [PubMed] [Google Scholar]
- Chew ML, Mulsant BH, Pollock BG, Lehman ME, Greenspan A, Mahmoud RA, Kirshner MA, Sorisio DA, Bies RR, Gharabawi G. Anticholinergic activity of 107 medications commonly used by older adults. J Am Geriatr Soc. 2008;56:1333–1341. doi: 10.1111/j.1532-5415.2008.01737.x. [DOI] [PubMed] [Google Scholar]
- Chou HH, Talledo JA, Lamb SN, Thompson WK, Swerdlow NR. Amphetamine effects on MATRICS Consensus Cognitive Battery performance in healthy adults. Psychopharmacology (Berlin) 2013;227:165–176. doi: 10.1007/s00213-012-2948-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lucena D, Fernandes BS, Berk M, Dodd S, Medeiros DW, Pedrini M, Kunz M, Gomes FA, Giglio LF, Lobato MI, Belonte-de-Abreu PS, Gama CS. Improvement of negative and positive symptoms in treatment refractory schizophrenia: a double-blind, randomized, placebo-controlled trial with memantine as add-on therapy to clozapine. J Clin Psychiatry. 2009;70:1416–1423. doi: 10.4088/JCP.08m04935gry. [DOI] [PubMed] [Google Scholar]
- Deutsch SI, Rosse RB, Schwartz BL, Mastropaolo J. A revised excitotoxic hypothesis of schizophrenia: therapeutic implications. Clin Neuropharmacol. 2001;24:43–49. doi: 10.1097/00002826-200101000-00008. [DOI] [PubMed] [Google Scholar]
- Duncan EJ, Madonick SH, Parwani A, Angrist B, Rajan R, Chakravorty S, Efferen TR, Szilagyi S, Stephanides M, Chappell PB, Gonzenbach S, Ko GN, Rotrosen JP. Clinical and sensorimotor gating effects of ketamine in normals. Neuropsychopharmacology. 2001;25:72–83. doi: 10.1016/S0893-133X(00)00240-2. [DOI] [PubMed] [Google Scholar]
- Fruchter O, Yigla M. Bronchodilator response after negative methacholine challenge test predicts future diagnosis of asthma. J Asthma. 2009;46:722–725. doi: 10.1080/02770900903067903. [DOI] [PubMed] [Google Scholar]
- Geerts H, Roberts P2, Spiros A3. Assessing the synergy between cholinomimetics and memantine as augmentation therapy in cognitive impairment in schizophrenia. Avirtual human patient trial using quantitative systems pharmacology. Front Pharmacol. 2015;6:198. doi: 10.3389/fphar.2015.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153:321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- Green MF, Kern RS, Heaton RK. Longitudinal studies of cognition and functional outcome in schizophrenia: implications for MATRICS. Schizophr Res. 2004;72:41–51. doi: 10.1016/j.schres.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Greenwood TA, Lazzeroni LC, Murray SS, Cadenhead KS, Calkins ME, Dobie DJ, Green MF, Gur RE, Gur RC, Hardiman G, Kelsoe JR, Leonard S, Light GA, Nuechterlein KH, Olincy A, Radant AD, Schork NJ, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Swerdlow NR, Tsuang DW, Tsuang MT, Turetsky BI, Freedman R, Braff DL. Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the Consortium on the Genetics of Schizophrenia. Am J Psychiatry. 2011;168:930–946. doi: 10.1176/appi.ajp.2011.10050723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood TA, Swerdlow NR, Gur RE, Cadenhead KS, Calkins ME, Dobie DJ, Freedman R, Green MF, Gur RC, Lazzeroni LC, Nuechterlein KH, Olincy A, Radant AD, Ray A, Schork NJ, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Sugar CA, Tsuang DW, Tsuang MT, Turetsky BI, Light GA, Braff DL. Genome-wide linkage analyses of 12 endophenotypes for schizophrenia from the Consortium on the Genetics of Schizophrenia. Am J Psychiatry. 2013;170:521–532. doi: 10.1176/appi.ajp.2012.12020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heresco-Levy U, Ermilov M, Lichtenberg P, Bar G, Javitt DC. High dose glycine added to olanzapine and risperidone for the treatment of schizophrenia. Biol Psychiatry. 2004;55:165–171. doi: 10.1016/s0006-3223(03)00707-8. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, Bar G, Catinari S, Ermilov M. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiatry. 2005;57:577–585. doi: 10.1016/j.biopsych.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Hughes AJ, Lees AJ, Stern GM. Apomorphine test to predict dopaminergic responsiveness in parkinsonian syndromes. Lancet. 1990;336:32–34. doi: 10.1016/0140-6736(90)91531-e. [DOI] [PubMed] [Google Scholar]
- Insel TR. The NIMH experimental medicine initiative. World Psychiatry. 2015;14:151–153. doi: 10.1002/wps.20227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69–108. doi: 10.1016/S0074-7742(06)78003-5. [DOI] [PubMed] [Google Scholar]
- Johnson JW, Kotermanski SE. Mechanism of action of memantine. Curr Opin Pharmacol. 2005;6:61–67. doi: 10.1016/j.coph.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261–276. doi: 10.1093/schbul/13.2.261. [DOI] [PubMed] [Google Scholar]
- Keefe RS, Bilder RM, Davis SM, Harvey PD, Palmer BW, Gold JM, Meltzer HY, Green MF, Capuano G, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Davis CE, Hsiao JK, Lieberman JA, CATIE Investigators; Neurocognitive Working Group Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007;64:633–647. doi: 10.1001/archpsyc.64.6.633. [DOI] [PubMed] [Google Scholar]
- Keilhoff G, Wolf G. Memantine prevents quinolinic acid-induced hippocampal damage. Eur J Pharmacol. 1992;219:451–454. doi: 10.1016/0014-2999(92)90487-o. [DOI] [PubMed] [Google Scholar]
- Kern RS, Nuechterlein KH, Green MF, Baade LE, Fenton WS, Gold JM, Keefe RS, Mesholam-Gately R, Mintz J, Seidman LJ, Stover E, Marder SR. The MATRICS Consensus Cognitive Battery, part 2: co-norming and standardization. Am J Psychiatry. 2008;165:214–220. doi: 10.1176/appi.ajp.2007.07010043. [DOI] [PubMed] [Google Scholar]
- Kim YW, Shin JC, An YS. Changes in cerebral glucose metabolism in patients with osttraumatic cognitive impairment after memantine therapy: a preliminary study. Ann Nucl Med. 2010;24:363–369. doi: 10.1007/s12149-010-0360-3. [DOI] [PubMed] [Google Scholar]
- Kishi T, Iwata N. NMDA receptor antagonists interventions in schizophrenia: meta-analysis of randomized, placebo-controlled trials. J Psychiatr Res. 2013;47:1143–1149. doi: 10.1016/j.jpsychires.2013.04.013. [DOI] [PubMed] [Google Scholar]
- Koola MM, Buchanan RW, Pillai A, Aitchison KJ, Weinberger DR, Aaronson ST, Dickerson FB. Potential role of the combination of galantamine and memantine to improve cognition in schizophrenia. Schizophr Res. 2014;157:84–89. doi: 10.1016/j.schres.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korostenskaja M, Nikulin VV, Kicić D, Nikulina AV, Kähkönen S. Effects of NMDA receptor antagonist memantine on mismatch negativity. Brain Res Bull. 2007;72:275–283. doi: 10.1016/j.brainresbull.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Krivoy A, Weizman A, Laor L, Hellinger N, Zemishlany Z, Fischel T. Addition of memantine to antipsychotic treatment in schizophrenia inpatients with residual symptoms: a preliminary study. Eur Neuropsychopharmacol. 2007;18:117–121. doi: 10.1016/j.euroneuro.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Lee JG, Lee SW, Lee BJ, Park SW, Kim GM, Kim YH. Adjunctive memantine therapy for cognitive impairment in chronic schizophrenia: a placebo-controlled pilot study. Psychiatry Investig. 2012;9:166–173. doi: 10.4306/pi.2012.9.2.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman JA, Papadakis K, Csernansky J, Litman R, Volavka J, Jia XD, Gage A, MEM-MD-29 Study Group A randomized, placebo-controlled study of memantine as adjunctive treatment in patients with schizophrenia. Neuropsychopharmacology. 2009;34:1322–1329. doi: 10.1038/npp.2008.200. [DOI] [PubMed] [Google Scholar]
- Nakanishi N, Kang YJ, Tu S, McKercher SR, Masliah E, Lipton SA. Differential effects of pharmacologic and genetic modulation of NMDA receptor activity on HIV/gp120-induced neuronal damage in an in vivo mouse model. J Mol Neurosci. 2016;58:59–65. doi: 10.1007/s12031-015-0651-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris H. The action of sedation on brain-stem oculomotor systems in man. Neuropharmacology. 1971;10:181–191. doi: 10.1016/0028-3908(71)90039-6. [DOI] [PubMed] [Google Scholar]
- Nuechterlein KH, Green MF, Kern RS, Baade LE, Barch DM, Cohen JD, Essock S, Fenton WS, Frese FJ, 3rd, Gold JM, Goldberg T, Heaton RK, Keefe RS, Kraemer H, Mesholam-Gately R, Seidman LJ, Stover E, Weinberger DR, Young AS, Zalcman S, Marder SR. The MATRICS Consensus Cognitive Battery, part 1: test selection, reliability, and validity. Am J Psychiatry. 2008;165:203–213. doi: 10.1176/appi.ajp.2007.07010042. [DOI] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova M, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Hunter CA. Toxoplasma gondii and schizophrenia: linkage through astrocyte-derived kynurenic acid? Schizophr Bull. 2007;33:652–653. doi: 10.1093/schbul/sbm030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini- International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(Suppl 20):22–33. quiz 34–57. [PubMed] [Google Scholar]
- Sonkusare SK, Kaul CL, Ramarao P. Dementia of Alzheimer’s disease and other neurodegenerative disorders—memantine, a new hope. Pharmacol Res. 2005;51:1–17. doi: 10.1016/j.phrs.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Eastvold A, Karban B, Ploum Y, Stephany N, Geyer MA, Cadenhead K, Auerbach PP. Dopamine agonist effects on startle and sensorimotor gating in normal male subjects: time course studies. Psychopharmacology. 2002;161:189–201. doi: 10.1007/s00213-002-1040-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, van Bergeijk DP, Bergsma F, Weber E, Talledo J. The effects of memantine on prepulse inhibition. Neuropsychopharmacology. 2009;34:1854–1864. doi: 10.1038/npp.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Bhakta S, Chou HH, Talledo JA, Balvaneda B, Light GA. Memantine effects on sensorimotor gating and mismatch negativity in patients with chronic psychosis. Neuropsychopharmacology. 2016;41:419–30. doi: 10.1038/npp.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velligan DI, Mahurin RK, Diamond PL, Hazleton BC, Eckert SL, Miller AL. The functional significance of symptomatology and cognitive function in schizophrenia. Schizophr Res. 1997;25:21–31. doi: 10.1016/S0920-9964(97)00010-8. [DOI] [PubMed] [Google Scholar]
- Wesnes K, Pincock C. Practice effects on cognitive tasks: a major problem? Lancet Neurol. 2002;1:473. doi: 10.1016/s1474-4422(02)00236-3. [DOI] [PubMed] [Google Scholar]
- Wilkinson GS, Robertson GJ. WRAT4: wide range achievement test professional manual. 4th. Psychological Assessment Resources; Lutz: 2006. [Google Scholar]
- Willenborg B, Schmoller A, Caspary J, Melchert UH, Scholand-Engler HG, Jauch-Chara K. Memantine prevents hypoglycemia-induced decrements of the cerebral energy status in healthy subjects. J Clin Endocrinol Metab. 2011;96:E384–E388. doi: 10.1210/jc.2010-1348. [DOI] [PubMed] [Google Scholar]
- Zdanys K, Tampi RR. A systematic review of off-label uses of memantine for psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1362–1374. doi: 10.1016/j.pnpbp.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Zink M, Correll CU. Glutamatergic agents for schizophrenia: current evidence and perspectives. Expert Rev Clin Pharmacol. 2015;8:335–52. doi: 10.1586/17512433.2015.1040393. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.