

SUMMARY
Anion exchanger 1 (AE1) is a critical transporter and the primary structural scaffold for large macromolecular complexes responsible for erythrocyte membrane flexibility and integrity. We used zero-length crosslinking and mass spectrometry to probe AE1 structures and interactions in intact erythrocyte membranes. An experimentally verified full-length model of AE1 dimers was developed by combining crosslink-defined distance constraints with homology modeling. Previously unresolved cytoplasmic loops in the AE1 C-terminal domain are packed at the domain-domain interface on the cytoplasmic face of the membrane where they anchor the N-terminal domain’s location and prevent it from occluding the ion channel. Crosslinks between AE1 dimers and ankyrin-1 indicate the likely topology for AE1 tetramers and suggest that ankyrin-1 wraps around AE1 tetramers, which may stabilize this oligomer state. This interaction and interactions of AE1 with other major erythrocyte membrane proteins show that protein-protein contacts are often substantially more extensive than previously reported.
In Brief
Rivera-Santiago et al. use zero-length chemical crosslinking of erythrocyte membranes, homology modeling, and known domain structures to deduce a structure for full-length anion exchanger 1. This model shows extensive interactions between domains and protein-protein interaction sites that are much larger than previous biochemical studies suggested.
INTRODUCTION
The major function of erythrocytes is delivery of oxygen to, and removal of carbon dioxide from cells throughout the body. Mammalian erythrocytes have evolved to an enucleated state and developed a biconcave cell shape to maximize surface area and enable efficient gas exchange. They have also developed a flexible membrane that can withstand the high amounts of shear stress involved in squeezing through capillaries (Romero et al., 2004). The most abundant protein in the erythrocyte membrane is anion exchanger 1 (AE1) with approximately one million copies per cell (Burton and Bruce, 2011), also known as band 3, which is encoded by the SLC4A1 gene (Choi, 2012). An isoform of AE1 is expressed in the kidney and the related homologs, AE2 and AE3, serve similar functions in most other tissues (Alper, 2009; Bonar and Casey, 2008). AE1 plays two central roles in the erythrocyte membrane by participating in the gas-exchange process and by serving as the primary protein scaffold for other transmembrane proteins as well as the spectrin-actin membrane skeleton that is responsible for the biconcave cell shape and membrane elasticity. Pathogenic mutations to AE1 cause a number of different hereditary hemolytic anemias in erythrocytes and acidosis in the kidney (Chu et al., 2010; Escobar et al., 2013; Jarolim et al., 1991).
Erythrocyte AE1 is a large, polytopic membrane protein consisting of 911 amino acids that comprise two major domains: a soluble N-terminal 400 amino acid domain, also known as the band 3 cytoplasmic domain or cdb3 (hereafter cytoplasmic domain), and a C-terminal anion exchanger domain that is largely embedded in the lipid bilayer (hereafter ion channel domain) (Figure 1). The cytoplasmic domain has been shown to interact with a large number of other proteins on the cytoplasmic face of the membrane, including the ankyrin-1-spectrin and actin junctional complex (van den Akker et al., 2010), which are the major components of the erythrocyte membrane skeleton. The specific partners anchored by AE1 are apparently regulated in part by the oligomeric state of AE1 in the membrane, where it exists as a mixture of dimers and tetramers (Jennings and Nicknish, 1985; Steck, 1972). The ion channel domain plays an important role in carbon dioxide transport. In distal tissues, carbon dioxide diffuses across the erythrocyte membrane, where the abundant cytoplasmic protein carbonic anhydrase converts it plus a water molecule to a proton and a bicarbonate ion (HCO3−). AE1 actively transports the bicarbonate out of the cell with neutral exchange of chloride (Cl−) ions into the cell (Choi, 2012). Several crystal structures of the cytoplasmic domain (Shnitsar et al., 2013; Zhang et al., 2000), as well as a structure of the ion channel domain (Arakawa et al., 2015), have been reported. However, these partial structures have not resolved several key structural questions with functional implications, including how these two domains fit together in membrane-bound dimers, and how the cytoplasmic domain interacts with the ion channel including whether it is involved in gating ion transport. In addition, the structures and potential functional roles of several small segments were not reported in the crystal structures, due to a lack of crystallographic data density (see Figure 1).
Figure 1. Schematic of AE1 Domains and Known Structures.

From left to right: the white region corresponds to the first 55 residues of AE1, which have not been determined in any structural study; the wheat regions indicate the portion of the N-terminal cytoplasmic domain resolved in two crystallographic studies indicated by the referenced PDB files; the brown region indicates a portion of the N-terminal cytoplasmic domain that was not structurally characterized in the crystallographic studies but is characterized in our model; the gray regions indicate transmembrane and extracellular regions of the anion channel domain that were structurally characterized (PDB: 4YZF); the orange and red regions indicate cytoplasmic loops of the C-terminal ion channel domain that were resolved in the structure (PDB: 4YZF); the green and blue regions indicate a cytoplasmic loop and the C-terminal cytoplasmic tail of the ion channel domain, respectively, that were not resolved (PDB: 4YZF) but were determined in our model.
One strategy for extending structural analysis beyond available crystal structures is the use of chemical crosslinking coupled with mass spectrometry (CX-MS) to identify crosslinked peptides after proteolysis of the modified protein or protein complex (Back et al., 2003; Paramelle et al., 2013; Rappsilber, 2011; Sinz, 2006). This technique has been combined with recent advances in MS instrument technology and complementary structural techniques, such as cryoelectron microscopy, to interrogate the structures of highly complex protein structures (Gaubitz et al., 2015; Greber et al., 2014; Lasker et al., 2012; Navare et al., 2015; Olson et al., 2014; Raveh et al., 2016). A seminal in silico study demonstrated that homology modeling could determine highly accurate structures when the modeling was combined with a sufficient density of distance constraints derived from chemical crosslinking (Leitner et al., 2010). Not surprisingly, that study showed that the most useful data were derived from “zero-length” crosslinks, such as those catalyzed by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) in conjunction with N-hydroxysuccinimide (NHS). Using this chemistry, crosslinks occur between a carboxyl residue and an amine residue that are within salt-bridging distances of each other. However, until recently the use of zero-length crosslinking strategies was limited primarily to small proteins or simple targeted questions due to the substantial challenges in identification of zero-length crosslinked peptides in complex peptide mixtures (Bumpus and Hollenberg, 2010; Kalkhof et al., 2005; Li et al., 2008, 2010; Marekov, 2007; Nagao et al., 2010; Schmidt et al., 2005). To expand the utility of zero-length crosslinking experiments, we recently developed a label-free differential liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis and a software package, ZXMiner that was specifically designed and optimized for analyzing zero-length crosslinked samples. We demonstrated the utility of this approach for determining structures that were too large or too flexible for crystallization (Harper et al., 2013; Rivera-Santiago et al., 2015a) or involved conformational changes in solution that could not be readily probed by nuclear magnetic resonance (Sriswasdi et al., 2014a). Furthermore, our recent adaptation of the method to newer, higher duty cycle mass spectrometers has enabled the examination of very large biological systems (Rivera-Santiago et al., 2015b).
In this study, our optimized zero-length CX-MS analysis strategy was used to probe the native structure of AE1 within intact erythrocyte membranes. The use of intact cell membranes ensures that proteins remain in a native physiological state with normal oligomeric and protein-protein interactions. Distance constraints from these experiments were combined with available crystal structures of the cytoplasmic (Shnitsar et al., 2013; Zhang et al., 2000) and ion channel domains (Arakawa et al., 2015) in order to elucidate an experimentally validated structure of full-length AE1 homodimers. The resulting structural model not only shows how these two large domains interact, but also provides the first structural and functional insights into the roles of multiple previously unresolved segments. This integrated structure will serve as a valuable, comprehensive scaffold for mapping the interaction interfaces between AE1 and other proteins. Initial analysis of major binding partners of AE1 demonstrates that protein-protein contact sites are much more extensive than determined previously using protein fragments and binary binding assays. This is the first experimental demonstration that zero-length crosslinking can be used for in-depth analysis of protein structures and interactions in intact cell membranes.
RESULTS AND DISCUSSION
Erythrocyte Membrane CX-MS
Two different preparations of freshly isolated erythrocyte membranes were crosslinked with EDC/sulfo-NHS using reaction time courses where aliquots of the reaction were collected after 15, 30, 60, and 120 min. Representative results of SDS gel patterns are shown in Figure S1. Gel patterns of control membranes primarily detect the most abundant membrane proteins including α- and β-spectrin, AE1, proteins 4.1 and 4.2, and actin, among others. AE1 migrates as a broad, diffuse band due to extensive and variable glycosylation. As expected, with increasing extent of crosslinking, these bands decrease in intensity and ultimately disappear with appearance of crosslinked complexes above 300 kDa (α-spectrin molecular weight = 281 kDa) and at the top of the gel. For most LC-MS/MS analyses, control samples and crosslink time course samples were cleaned up by running the sample into an SDS gel for 0.5 cm. The entire stained area of the gel was digested with trypsin including the top of the gel where most of the crosslinked proteins were located, particularly for later time points. Similar to our previous workflows on simpler protein complexes (Harper et al., 2013; Rivera-Santiago et al., 2015b; Sriswasdi et al., 2014a; Sriswasdi et al., 2014b), we used label-free LC-MS/MS comparisons of crosslinked and uncrosslinked controls samples to identify putative crosslinked peptides. However, in this study the increased capability of the Thermo Scientific Q Exactive Plus instrument to collect high-resolution data at high speed eliminated the need for subsequent targeted LC-MS/MS runs (Sriswasdi et al., 2014b).
A total of five sets of LC-MS/MS runs, where each set consisted of a control sample plus a crosslink time course, were analyzed separately in five ZXMiner experiments to identify crosslinked peptides and specific crosslink sites. Typically, multiple MS/MS spectra were acquired for a given precursor and most crosslinked peptides were detected at multiple charge states, e.g., +3, +4, and +5 and at multiple reaction time points. The ZXMiner analysis used a database of the 57 most abundant erythrocyte membrane proteins as operationally defined using a single conventional LC-MS/MS analysis to identify easily detectable proteins (see Experimental Procedures for details). This database of readily detected proteins ensured that if a cross-linked peptide resulted in an MS/MS spectrum, the sequence should be in the database. Crosslinked peptides are typically more difficult to detect than linear peptides, and therefore proteins where linear peptides are near the detection limit are unlikely to produce detectable crosslinked peptides. This is because crosslinked peptides are substoichiometric and usually produce weaker MS signals than linear peptides. At the same time, this restricted sequence database reduced the likelihood that an isobaric sequence from a low-abundance, undetectable protein could be assigned to a given crosslinked peptide by random chance, thus keeping the false-discovery rate relatively low. Within each experiment, the ZXMiner software reports the highest-scoring match for each detected charge state and methionine oxidation state. The putative crosslink assignments were verified as described in Experimental Procedures, and the verified results from the different experiments were combined into a master list. All verified crosslinked peptides assignments were detected multiple times and many crosslinks were detected in most or all experiments.
For this study, we focused on crosslinks within AE1 and cross-links between AE1 and other major membrane proteins. The 17 validated crosslinks between two AE1 peptides are summarized in Table 1. Some peptides contained multiple acidic residues and the MS/MS spectra did not always unambiguously support a single linkage site. In most cases the acidic residues were adjacent to each other and it is likely that both residues were within crosslinking distance with the partner lysine. Interestingly, some of the AE1 intra-protein crosslinks linked the protein’s well-characterized globular cytoplasmic N-terminal domain (Shnitsar et al., 2013; Zhang et al., 2000) to small cytoplasmic loops of the ion channel domain and some of these loops had not been resolved in the crystal structure (Arakawa et al., 2015). This provided valuable proximity data for confidently orienting these two domains and for locating and modeling the previously undefined loops (Figure 1).
Table 1.
AE1 Crosslinks Identified by Analysis of White Ghosts and AE1 Immunoprecipitation Reactions
| Crosslink Group |
za | Peptide Ab | Peptide A Sequence |
Peptide Bb | Peptide B Sequence |
Crosslinked Residue A |
Crosslinked Residue B |
Structure Distance (Å)c |
Model Distance (Å)c |
|---|---|---|---|---|---|---|---|---|---|
| Crosslinks Only Involving the AE1 N-Terminal Domain | |||||||||
| 1 | 3,4,5 | VYVELQELVMD[E]K | 57–69 | HSHAGELELEALGGV[K]PAVLTR | 161–180 | 68 | 174 | 5.9 | 8.4 |
| 2 | 5 | WVQLEENLG[E]NGAWGR | 81–96 | HSHAGELELEALGGV[K]PAVLTR | 161–180 | 90 | 174 | 9.5 | 9.7 |
| 3 | 4,5 | HSHAGELELEALGGV[K]PAVLTR | 161–180 | LQEAAEL[E]AVELPVPIR | 247–263 | 174 | 254 | 22.1 | 14.6 |
| 4 | 4 | FLFVLLGP(E)APHI(D)YTQLGR | 264–283 | YQSSPA[K]PDSSFYK | 347–360 | 272/277 | 353 | –d | –d |
| 5 | 5 | I[D]AYMAQSR | 296–304 | YQSSPA[K]PDSSFYKGLDLNGGPDDPLQQTGQLFGGLVR | 347–384 | 297 | 353 | 13.6 | 12.6 |
| Crosslinks Involving the AE1 N-Terminal Domain and Cytoplasmic Segments of the AE1 Anion Exchanger Domain | |||||||||
| 6 | 4 | VFT[K]GTVLLDLQETSLAGVANQLLDR | 113–138 | ATFD[E]EEGR | 893–901 | 116 | 897 | NA | 10.3 |
| 7 | 4,5 | FIFE[D]QIR | 139–146 | SVTHANALTVMG[K]ASTPGAAAQIQEVK | 731–757 | 143 | 743 | NA | 5.7 |
| 8 | 3,4,5 | FIFE[D]QIR | 139–146 | YHPDVPYV[K]R | 818–827 | 143 | 826 | NA | 13.0 |
| 9 | 4,5 | ADFL[E]QPVLGFVR | 234–246 | SVTHANALTVMG[K]ASTPGAAAQIQEVK | 731–757 | 238 | 743 | NA | 11.3 |
| 10 | 3,4,5 | ADFL[E]QPVLGFVR | 234–246 | YHPDVPYV[K]R | 818–827 | 238 | 826 | NA | 7.3 |
| 11 | 4 | ADFL[E]QPVLGFVR | 234–246 | NVELQCLDADDA[K]ATFDEEEGR | 880–901 | 238 | 892 | NA | 8.5 |
| 12 | 4 | YQSSPA[K]PDSSFYK | 347–360 | YHP[D]VPYVK | 818–826 | 353 | 821 | NA | 13.4 |
| 13 | 3,4 | YQSSPA[K]PDSSFYK | 347–360 | ATFDEE[E]GR | 893–901 | 353 | 899 | NA | 11.8 |
| 14 | 3 | YQSSPA[K]PDSSFYK | 347–360 | DEY[D]EVAMPV | 902–911 | 353 | 905 | NA | 10.9 |
| Crosslinks between Cytoplasmic Segments of the AE1 Anion Exchanger Domain | |||||||||
| 15 | 4 | F[K]NSSYFPGK | 591–600 | ATFDEE[E]GR | 893–901 | 592 | 899 | NA | 11.7 |
| 16 | 3,4,5 | YHPDVPYV[K]R | 818–827 | ATFD[E]EEGR | 893–901 | 826 | 897 | NA | 11.3 |
| 17 | 3 | YHPDVPYV[K]R | 818–827 | DEYD[E]VAMPV | 902–911 | 826 | 906 | NA | 9.3 |
Observed charge states of the crosslinked peptide.
[], crosslinked residue; (), potential crosslinked residue (ambiguous location).
All Cα-Cα distances calculated for residues within the same AE1 monomer using the AE1 N-terminal crystal structure by Zhang et al. (2000) (PDB: 1HYN) or the final model, respectively. See also Figures S3 and S4.
This is an inter-chain crosslink that connects two AE1 dimers (see Figure 5).
Immunoprecipitation and Evaluation of AE1 Oligomer States
As noted above, AE1 exists in erythrocyte membranes as a mixture of dimers and tetramers. Therefore, prior to using cross-link distance constraints for modeling, it was necessary to determine which crosslinks occurred within single polypeptide chains and which crosslinks might bridge between subunits in either dimers or tetramers. First, AE1 was enriched by immunoprecipitation from the uncrosslinked control and a crosslink time course. Aliquots of the bound fractions were initially separated on full-length SDS gels followed by western blotting with an anti-AE1 antibody (Figure S2A) to identify AE1-containing bands. In the presence of SDS, the untreated sample showed primarily AE1 monomers with a trace of dimers due to the intrinsic crosslinking that occurs in intact erythrocytes in vivo. The 30 and 60 min reaction time points showed substantial amounts of crosslinked AE1 dimers and tetramers as well as other complexes containing AE1 based on LC-MS/MS analysis of the entire sample excised from a 0.5 cm gel (Figure S2B, left panel). For the 120 min time point, the efficiency of the pull-down and elution from the beads were greatly reduced, apparently due to modifications to, and/or shielding of, antibody epitopes in the crosslinked complex. To reduce gel volumes for trypsin digestion of the different AE1 oligomer states, aliquots of each bound fraction were separated for 2 cm on SDS gels (Figure S2B) and the regions corresponding to AE1 monomers, dimers, and tetramers in the 2 cm gels were excised. These bands were digested and analyzed by LC-MS/MS and crosslinked peptides were identified using the contents of the 0.5 cm gels to determine what proteins and protein complexes were expected to be present, based on the results of a MaxQuant search (Cox and Mann, 2008). Most crosslinks identified in Table 1 were identified in all three bands corresponding to putative AE1 monomers, dimers, and tetramers, which indicated that these crosslinks occurred within a single polypeptide chain. The only exception was crosslink group 4, which was only detected in tetramers.
Development and Modeling of AE1 Full-Length Structure
A structure of the full-length AE1 protein was developed using the program MODELLER, the available crystal structures for the N-terminal cytoplasmic (Shnitsar et al., 2013; Zhang et al., 2000) and the C-terminal ion channel (Arakawa et al., 2015) domains of AE1 and the distance constraints defined by the 16 intra-chain crosslinks are shown in Table 1. The resulting models were evaluated for steric conflicts between residues, and the model with the lowest energy minimization scores was chosen. The final model was compared with the crystal structures of the cytoplasmic and membrane domains to evaluate whether any of the crosslinks and the modeling introduced perturbations to the known domain structures. As shown in Figure 2, overall folding of the AE1 N- and C-terminal domains resolved in the crystal structures were not significantly perturbed in the final dimeric full-length AE1 model as the structures superimposed with root-mean-square deviation of ~0.5 Å. However, one loop in the crystal structure of the N-terminal domain appears to have a substantially different orientation in the membrane as reflected by crosslink group 3 which had a Cα spacing of 22 Å in the crystal structure (Table 1). This crosslink involved two surface loops so it was not surprising that it was larger than the typical maximum spacing of up to 12 Å for well-ordered regions, but it was also somewhat beyond the typical maximum distance of up to ~16 Å observed for some flexible loop regions and subunit interfaces as reported previously (Harper et al., 2013; Rivera-Santiago et al., 2015a, 2015b; Sriswasdi et al., 2014b). However, we have occasionally observed other cases where zero-length crosslinks identified differences in orientation of surface loops relative to a crystal structure (Rivera-Santiago et al., 2015a). It is unlikely that this crosslink is a false positive as it was identified in all experiments with high confidence. It was also identified in the monomer, dimer, and tetramer bands from the immunoprecipitated AE1. Its identification in the monomer band rules out the possibility that is could be an inter-chain crosslink. We therefore conclude that it is another example of a local difference between a crystallographic structure and an in-solution, or in this case, in-membrane structure. Close-up images of the crosslinks in the N-terminal domain are shown in Figure S3 using both the N-terminal crystal structure and the final model. Similarly, close-up images of all other AE1 crosslinks in the final model are shown in Figure S4. The final model of the entire AE1 dimer is shown in Figure 3.
Figure 2. Evaluation of the AE1 Full-Length Model Relative to Its Template Structures.

(A) Comparison for the AE1 C-terminal anion exchanger domain between our final full-length model shown in gold and the template crystal structure (PDB: 4YZF; Arakawa et al., 2015) shown in cyan (root-mean-square deviation [RMSD] = 0.5 Å).
(B) Comparison for the AE1 N-terminal domain between our full-length model shown in gold and the template structure by Zhang et al. (2000) (PDB: 1HYN) in blue (RMSD = 0.5 Å) and the structure by Shnitsar et al. (2013) (PDB: 4KY9) in red (RMSD = 0.8 Å). AE1 model structure shown in gold. Structures are oriented with the cytoplasm at the bottom.
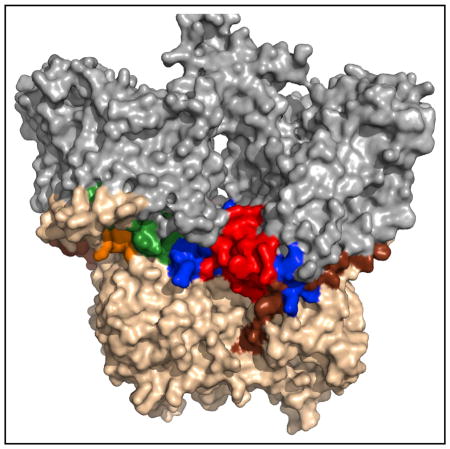
Figure 3. The Full-Length AE1 Dimer Structure.

The relationship between the N- and C-terminal domains and locations of cytoplasmic loops from the C-terminal ion channel domain are illustrated.
(A) Cartoon view of the full-length AE1 model. Color coding is the same as Figure 1: N-terminal cytoplasmic domain (wheat), previously uncharacterized linker domain (brown), and C-terminal ion channel (gray). Also, the following segments are shown using “sticks”: the cytoplasmic region between transmembrane spans 6 and 7, also referred to as C1 (orange), the cytoplasmic region between transmembrane spans 10 and 11, also referred to as C2 (green), the cytoplasmic region between transmembrane spans 12 and 13, also referred to as C3 (red), and the C-terminal tail following transmembrane span 14 also referred to as C-tail (blue). Approximate depth and width of the cytoplasmic and membrane structures are shown.
(B) Surface view of the full-length AE1 model. Color coding of domains and segments as described in (A). See also Movie S1.
Locating Cytoplasmic Loops from the AE1 Ion Channel Domain
As noted above, two related questions that the domain crystal structures did not address are how the cytoplasmic domain interacts with the ion channel domain, and the location/role of small cytoplasmic loops from the ion channel domain (see Figure 1). Consistent color coding is used in Figures 1, 3, and 4 and Table 1 to highlight the locations of these loops. The structure of the full-length AE1 dimer (Figure 3) shows how the cytoplasmic loops from the ion channel domain are packed at the interface between the cytoplasmic and ion channel domains. These loop regions form extensive contacts with both domains, which may explain the weak crystallographic density of these segments when only one of the two domains was present. These interactions also are most likely responsible for maintaining a fixed orientation between the ion channel domain and the cytoplasmic domain. Specifically, the cytoplasmic segments between TM6 and TM7 (hereafter C1 and color-coded orange), TM10 and TM11 (hereafter C2 and color-coded green), TM12 and TM13 (hereafter C3 and color-coded red), and the region following TM14 (hereafter C-tail and color-coded blue) probably stabilize the domain-domain interactions. In particular, the C3 and C-tail segments seem to be most responsible for the positioning of the cytoplasmic domain relative to the membrane-embedded ion channel domain, due to the larger density of crosslinks involving these two segments. C1 and C2 seem to be providing supporting interactions. The other cytoplasmic connectors between transmembrane helices are short turns comprising only a few residues and do not significantly contribute to this interface. Securing and stabilizing the orientation of the large N-terminal domain that acts as a scaffold for many cytoskeletal proteins (van den Akker et al., 2010) is probably a functionally important reason for the existence and location of these cytoplasmic loops.
Figure 4. The N-Terminal Domain of the Full-Length AE1 Model Does Not Occlude the C-Terminal Domain Ion Channel.

(A) Cartoon view of the N-terminal/ion channel domain interface. Areas shown in colors are previously characterized N-terminal domain (gold), previously un-characterized N-terminal domain (brown), C1 (orange), C2 (green), C3 (red), C-tail (blue), ion channel core helices (cyan), and ion channel exit residues (purple).
(B) Surface view of the AE1 N-terminal/ion channel domain interface. Colors are as described in (A).
The crystallographic structures of both the ion channel domain and the cytoplasmic domains showed that the dimers were much wider than deep when viewed head on. The pairing of these two domains in the current model shows that both narrow dimensions are oriented along the same axis producing a molecule of roughly equal depth (32–36 Å). In contrast, the cytoplasmic domain of the dimer is about 65 Å wide while the ion channel domain is about 99 Å wide.
The AE1 Ion Channel Exit Site Is Not Occluded by the N-Terminal Domain
Another previously unresolved question is how the very large cytoplasmic domain interacts with the ion channel. The current structure shows that the cytoplasmic face of the pore exits on the side of the cytoplasmic domain where it is exposed to the aqueous environment (Figure 4). It is apparent that the AE1 cytoplasmic domain does not have a substrate access tunnel as is sometimes found in other ion channel proteins containing an N-terminal domain that does not directly take part in ion transport (Shnitsar et al., 2013). The exit also is not near the dimer interface (Figures 3 and 4) and is probably not near the tetramer interface, if the tetramer is a dimer of dimers, which suggests that changes in oligomeric state will not affect transport function. However, the pore is surrounded by the cytoplasmic loops that form the interface between domains and it seems likely that conformational changes affecting these loops and/or binding of other proteins to the cytoplasmic face of AE1 could affect transport.
Orientation of AE1 Dimers in Tetramer Complexes
As indicated above, we only identified a single tetramer-specific crosslink (group 4 in Table 1), which was D272/E277 to K353, with D272-K353 being the more likely of the two possible linkages as these two residues are similar distances from the lipid bilayer (Figure 5A). Assuming that AE1 tetramers are a dimer of dimers, the observed crosslink is only consistent with one of the two possible orientations of dimers as shown in the left panel of Figure 5B. This schematic suggests further polymerization to higher oligomers would be possible; however, only dimers and tetramers are observed in erythrocyte membranes. Further association of additional dimers is probably prevented by stabilization of the tetramer by association of ankyrin (see below). The proposed orientation of AE1 tetramers is consistent with the asymmetric shape of AE1 dimers as the transmembrane domain is substantially larger than the cytoplasmic domain in the “head-on view” but of a similar size in the perpendicular (side view) dimension (Figure 3). The similar size of these two domains in the side view dimension suggests that assembly of two dimers by front-to-back association is likely to involve inter-dimer interactions of the membrane domains as well as the cytoplasmic domains. This also suggests that the cytoplasmic region of the tetramer will be approximately 65 × 65 Å as shown in Figure 5B.
Figure 5. Schematic Representations of AE1 Tetramer and Ankryin-AE1 Interactions.

(A) Cartoon depicting the AE1 tetramerization site interface. Cytoplasmic domains and loops shown in wheat, and membrane-bound regions shown in gray. E272 and D277 are shown in blue spheres, and K353 is shown in red spheres. Dashed lines indicate the approximate location of the lipid bilayer. The solid black arrow indicates the most probable site of tetramer-specific crosslinking, E272 to K353, whereas the dashed black arrow indicates the alternative site for the tetramer-specific crosslink, D277 to K353.
(B) Schematic showing the two most likely alternative pairing of dimers to form AE1 tetramers. Cyan ovals represent AE1 N-terminal domain dimers. The approximate locations of the residues that form the tetramer-specific crosslink are highlighted with a dark red triangle (K353) and a complementary dark blue icon (E272). The dimer-dimer association on the left is consistent with this crosslink, while the schematic on the right indicates incompatible binding pairs.
(C) Schematic representation of ankyrin-1 and its major domains. Ankyrin repeats are shown in purple, ZU5 domains are shown in green, and the “death domain” is shown in gold. Blue arrows indicate previously characterized interactions with AE1, as described by Grey et al. (2012), which are on residues N601 and K611 on ankyrin repeat 18, and Q634/E645 on ankyrin repeat 19. Red and black arrows indicate interactions with AE1 defined by crosslinks involving ankyrin-1 residues 68 and 1,494, respectively (Table 2).
(D) Structure of full-length AE1 dimer highlighting ankyrin-1 binding sites as follows: previously defined by recombinant peptide and mutagenesis by Grey et al. (2012) (blue spheres), sites that crosslink to ankyrin residue 68 (red spheres), and sites that crosslink to ankyrin 1,494 (black spheres).
(E) Extended model of the ankyrin repeat domain of ankyrin-1 (adapted from Michaely et al., 2002). Arrows showing interactions with AE1 are colored as in (A). The black arrow indicates the estimated dimension of the opening within the structure for regions of the ankyrin repeat domain.
(F) Schematic describing a possible arrangement for the AE1-ankyrin-1 interaction. Cyan ovals represent AE1 N-terminal domain dimers. The approximate locations of the residues that form the tetramer-specific crosslink are highlighted with a dark red triangle (K353) and a complementary dark blue icon (E272/D277). Ankyrin repeat domain shown in purple, ZU5 domain shown in green, death domain shown in gold, and uncharacterized regions shown as gray lines. Blue lines indicate previously characterized interactions with AE1, as described by Grey et al. (2012). The red line indicates an AE1-ankyrin-1 interaction (AE1 K353–ankyrin-1 E68) in the ankyrin repeat domain. The black line indicates AE1-ankyrin-1 interactions (AE1 E142/D143–ankyrin-1 K1494) in the C-terminal domain.
Full-Length AE1 Is a Critical Foundation for Building Structures of Erythrocyte Membrane Complexes
As noted above, AE1 is the scaffold for many other proteins including the two major membrane skeleton complexes, which are the ankyrin-spectrin and actin junctional complexes. A number of proteins in these two complexes have been mapped by several groups onto AE1 using conventional biochemical approaches such as protein truncation or expression of recombinant domains followed by protein-protein or membrane-protein binding assays (van den Akker et al., 2010). The full-length AE1 dimer structure and AE1 tetramer schematic provide templates upon which we can begin to build structures of larger protein complexes. In this context, the crosslinking experiments in this study provided a number of high-confidence crosslinks between AE1 and several membrane skeleton proteins (Table 2). These data provide a number of important insights into protein-protein contacts involving AE1, although their density is currently too low to reliably model structures for these multi-protein complexes.
Table 2.
Validated Crosslinks between AE1 and Other Erythrocyte Proteins Identified by Analysis of White Ghosts and AE1 Immunoprecipitation Reactions
| Crosslink Group |
Protein Crosslinked to AE1 |
AE1 Peptidea | AE1 Peptide Sequence |
Peptide Ba,b | Peptide B Sequence |
Crosslinked Residue A |
Crosslinked Residue B |
zc |
|---|---|---|---|---|---|---|---|---|
| 1 | ankyrin | FIF(ED)QIR | 139–146 | NL[K]PDRR | 1,492–1,498 | 142/143 | 1,494 | 4 |
| 1 | ankyrin | YQSSPA[K]PDSSFYK | 347–360 | [E]IILETTTK | 68–76 | 353 | 68 | 3,4 |
| 2 | band 4.2 | ALLL[K]HSHAGELEALGGVK | 156–174 | SVTVVAPELS[A] | 681–691 | 160 | 691 | 4 |
| 2 | band 4.2 | HSHAGELEALGGV[K]PAVLTR | 161–180 | SVTVVAPELS[A] | 681–691 | 174 | 691 | 4 |
| 2 | band 4.2 | NV(E)LQCLDA(DD)AK | 880–892 | PHLAI[K]MPEK | 588–597 | 882/889/890 | 593 | 3 |
| 3 | glycophorin A | YQSSPA[K]PDSSFYK | 347–360 | SPSDVKPLPSPDTDVPLSSV(E)I(E)NPETSD[Q] | 121–150 | 353 | 141/143 | 4 |
| 4 | protein 4.1 | PER[K]MVK | 692–698 | KEDEPP[E]QAEPEPTEAWK | 598–615 | 695 | 604 | 4 |
[], crosslinked residue; (), potential crosslinked residue (ambiguous location).
Peptide corresponding to the protein crosslinked to AE1.
Observed charge states of the crosslinked peptide.
Ankyrin-1 Interactions with AE1
Ankyrin-1 forms a high-affinity interaction with AE1 tetramers but does not associate with AE1 dimers. The domain composition of the 206 kDa ankyrin-1 protein is shown in Figure 5C. A single region of ankyrin was previously reported to interact with AE1 based on biochemical approaches (Grey et al., 2012). Interestingly, our data indicate that the AE1-ankyrin-1 interaction is much more extensive than these earlier biochemical binding assays suggested. Specifically, AE1 crosslinks near both the N terminus (residue 68) and the C terminus of ankyrin-1 (residue 1,494) were detected in our experiments, suggesting extensive interactions. These include multiple contacts that are quite distal in the ankyrin-1 sequence to the previously known interaction site (Table 2 and Figure 5C). Similarly, these crosslink sites correspond to different locations on AE1 (Figure 5D). The locations of the previously reported site and the ankyrin residue 68 crosslink are consistent with a previously reported simulation model of the AEI-ankyrin-1 interaction where the 24 ankyrin-type repeats were proposed to form a spiral (Figure 5E) that wraps around AE1 (Michaely et al., 2002). The sizes of the ankyrin-1 repeat domain model and the AE1 tetramer (Figures 5C–5E), strongly suggest that the ankyrin domain opens up slightly relative to the Michaely model in order to wrap around the AE1 tetramer, and the two mapped ankyrin repeat domain interactions with AE1 probably bridge across an AE1 tetramer as shown in Figure 5F. The ankyrin-1 residue 1,494 crosslink maps to a site in the AE1 cytoplasmic domain that is very close to the ankyrin-1 residue 68 crosslink site (Figure 5B), but there is insufficient structural and topological data for the C-terminal region of ankyrin-1 to indicate its location relative to the ankyrin repeat domain and AE1 tetramers. One possibility is that the C-terminal region of ankyrin-1 wraps back upon the ankyrin repeats domain. Alternatively, as shown in Figure 5F, the ankyrin-1 molecule may continue to wrap further around the AE1 tetramer, completely encircling the AE1 tetramer with the ankyrin residue 68 and 1,494 sites associating with the same AE1 subunit of the tetramer. Finally, as there is no structural or topological information for the C-terminal tail beyond the death domain, this region is arbitrarily shown as an extended coil continuing to wrap around this protein complex.
Stabilization of AE1 Tetramers by Ankyrin-1
The schematic in Figure 5F indicates that ankyrin-1 interacts with subunits of both AE1 dimers within a tetramer complex, which supports the observation that ankyrin-1 binds AE1 tetramers but not dimers. These results also address the issue of the tetramerization interface of AE1 that involves two dimers in the same orientation. Such an orientation could theoretically lead to higher-order oligomeric states, but the interaction of ankyrin-1 with the tetramer would sterically hinder polymerization of AE1 to higher oligomers. An issue that remains to be resolved is whether ankyrin-1 drives AE1 tetramer formation or if it simply binds preformed tetramers with higher affinity than dimers due to an extended interaction surface. Another issue that was noted in the course of these analyses is the several roles of K353. This residue was identified in crosslinks associated with AE1 tetramerization as well as in crosslinks detailing the interactions of AE1 with ankyrin-1 and glycophorin A (Table 2). Despite the apparent multitude of roles this residue seems to play, these can be addressed by considering that there are four AE1 K353 sites for every tetramer. Thus, two K353 sites can be involved in tetramerization, while a third K353 binds ankyrin-1 as shown in Figure 5F. The fourth K353 could potentially bind a glycophorin A dimer if the wrapping of ankyrin-1 around the tetramer does not obscure the fourth site on the tetramer. More accurate estimates of copy numbers of each membrane component as well as isolation and analysis of specific crosslinked complexes would help sort out these possibilities.
While all the above interactions of K353 might simultaneously occur on a single AE1 tetramer, K353 is also observed to cross-link to several loops of the anion exchanger domain (Table 1, Figure S4). Interestingly, such promiscuous reactive residues are observed with significant frequency when using zero-length crosslinkers. Another example of a promiscuous reactive residue on AE1 is E238, which was observed to crosslink to K743 (crosslink group 7), K826 (crosslink group 8), and K892 (crosslink group 9). The multiple crosslinks between a single reactive residue and partners on different sequences provides useful information about proximity in three-dimensional space as shown in Figures S3 and S4. These results illustrate one of the major advantages of using zero-length crosslinkers rather than the more commonly employed longer crosslinkers. That is, crosslinkable residues must be approximately within salt-bridging distance and therefore identification of crosslink sites indicates direct protein-protein contact and not simply close proximity.
It is actually surprising that crosslinks involving promiscuous reactive residues such as K353 and E238 are readily detected. The crosslinking of a peptide containing a promiscuous reactive residue to multiple partners would be expected to further reduce an already substoichiometric yield, making such peptides much harder to detect than a peptide that crosslinks to a single partner. One possible explanation for the observed facile detection of promiscuous reactive residues is that the carboxyl group(s) may be more reactive with carbodiimides than carboxyls that form salt bridges with a single partner. Single-partner pairs may spend more time in the salt bridge state where the carboxyl may be less nucleophilic due to shared electron density with the amine. In contrast, promiscuous reactive residues may be more solvent exposed and carboxyls that can salt bridge to multiple amines may be toggling between multiple salt bridges with higher reactivity when transitioning between partners. Promiscuous amines such as K353 may be particularly reactive because each of the partner carboxyls are likely to be highly solvent exposed and will only be in the salt bridge state for a limited percentage of time.
Contacts between Interacting Proteins Are Often Much More Extensive than Reported Previously
Similar to the extensive interactions between AE1 and ankyrin-1, other proteins that interact with AE1 have larger protein-protein contact sites than suggested by biochemical assays. Three additional proteins with well-characterized interactions with AE1 are glycophorin A (Che and Cherry, 1995; Groves and Tanner, 1994; Knowles et al., 1994; Nigg et al., 1980; Telen and Chasis, 1990; Young et al., 2000; Young and Tanner, 2003), protein 4.1 (Jons and Drenckhahn, 1992), and protein 4.2 (Bustos and Reithmeier, 2011). Similar to the AE1-ankyrin-1 interaction, our crosslinking data (Table 2) indicate that known (Figure 6A) and newly observed interactions (Figure 6B) are spread over large distances on both interacting proteins indicating that the contact surfaces are much larger than those previously defined using protein fragments. The locations of the known and new interaction sites for glycophorin A is consistent with the known properties of this protein. Specifically, it is a type 1 membrane protein with a helical transmembrane domain (residues 92–114) and a 36-residue cytoplasmic domain the structure of which is unknown but is most likely in an extended conformation. The previously defined interactions are between the ends of the glycophorin A transmembrane helix and topologically corresponding locations on the transmembrane region of AE1. The crosslink between the cytoplasmic domain of glycophorin A and the cytoplasmic domain of AE1 is consistent with an extended coil structure for the glycophorin A C-terminal domain that wraps partially around an AE1 monomer. Because only partial structures are available for proteins 4.1 and 4.2, it is not feasible to further interpret these structures at present. However, the sites of known and newly defined crosslinks indicate extensive protein-protein contact surfaces, similar to the extensive footprints suggested for interaction of AE1 with ankyrin-1 and glycophorin A.
Figure 6. CX-MS of Intact Membranes Suggests Protein-Protein Interfaces Are More Extensive than Defined Previously.

(A) Previously described interaction sites on an AE1 dimer for glycophorin A (residues 658 and 701, orange spheres), protein 4.1 (residues 343–347 and 386–390, blue spheres), and protein 4.2 (residues 40, 130, and 327, green spheres) are highlighted.
(B) New interaction sites on AE1 for glycophorin, proteins 4.1 and 4.2, are shown using the same color coding as in (A).
In summary, this study shows that zero-length CX-MS can be productively applied to intact cell membranes to assist modeling of a large transmembrane protein, to identify new interfaces between interacting proteins, and identify completely novel protein-protein interactions (data not shown). The unique sequences that needed to be considered for producing theoretical crosslinked peptide possibilities in these studies represented more than 3 MDa, which was a challenging but tractable problem. One advantage of using zero-length crosslinking is the advantage that tighter distance constraints confer for molecular modeling. Another important advantage is that because residues must be within salt bridge distances in order to crosslink, we can be confident that cross-linked domains or proteins are in direct contact and are not simply in close proximity. Based on the results described herein, it should be feasible to build high-confidence structures for large membrane-bound macromolecular complexes using zero-length CX-MS by increasing the density of cross-links to assist model building. This process will be facilitated as additional domain structures for the target proteins or their homologs become available.
EXPERIMENTAL PROCEDURES
Preparation of Erythrocyte Membrane White Ghost Samples
Fresh blood samples were collected from healthy volunteers with informed written consent, using protocols approved by the Wistar Institute institutional ethical review board. Erythrocyte membranes, commonly called “white ghosts” were prepared as described by Speicher et al. (1992) with minor modifications. In brief, blood was stored at 4°C for 2 days prior to processing to allow reticulocytes to partially mature, as described previously by Pasini et al. (2006). After centrifugation (2,000 × g, 20 min, 4°C) and removal of the serum and buffy coat, the packed red cells were diluted to 50% hematocrit in 10 mM phosphate and 130 mM sodium chloride (pH 7.4) and passed through a Plasmodipur Filter (Accurate Chemical & Scientific) to remove any contaminating leukocytes. The filtered sample was centrifuged at 2,000 × g for 20 min at room temperature. The supernatant was aspirated and the red cells diluted to 50% hematocrit in 10 mM phosphate and 130 mM sodium chloride (pH 7.4). The red cell suspension was layered onto Lympholyte-H Cell Separation Media (Accurate Chemical & Scientific) and centrifuged for 20 min and 740 × g at room temperature to remove any contaminating lymphocytes. The supernatant was aspirated and the red cells were diluted in 10 mM phosphate and 130 mM sodium chloride (pH 7.4). The remaining washing and lysis steps were carried out as described previously until membrane pellets were white, which indicated essentially complete removal of hemoglobin and other cytoplasmic proteins.
Crosslinking of Erythrocyte Membranes
Purified membranes were crosslinked using EDC/sulfo-NHS at 0°C with varying reagent concentrations and reaction times. All reactions were quenched at the indicated time points by adding 20 mM DTT followed by incubation on ice overnight to facilitate reversal of activated carboxyls that did not form cross-links. A moderate drop in pH during the crosslinking reaction occurred and the final pH was typically between 6.6 and 7.2 depending upon crosslinker concentration. Visible protein patterns were compared using full-length SDS gels stained with colloidal Coomassie blue (see Figure S1).
Trypsin Digestion and Reconstitution
Matched uncrosslinked controls and crosslinked erythrocyte membranes were electrophoresed into an SDS gel until the tracking dye migrated 0.5 cm. The gel was then fixed, stained with colloidal Coomassie blue, the entire region from the top of the gel to the dye front was excised and digested with trypsin using an in-gel digestion protocol, as described previously (Harper et al., 2013; Speicher et al., 2000). The tryptic digests were dried using a SpeedVac followed by reconstitution in 0.1% formic acid and 3% acetonitrile at a total peptide concentration of 2.0 μg/μL, based on the amount of protein applied to the gel lane.
LC-MS/MS Data Collection
Control and crosslinked membranes were analyzed using a Thermo Scientific Q Exactive Plus mass spectrometer interfaced with a Waters nanoACQUITY UPLC system. Buffer A was 0.1% formic acid in water, buffer B was 0.1% formic acid in acetonitrile, and a 2-hr gradient with a total runtime of 125 min, which proceeds as follows: the starting conditions were 95% buffer A, 5% buffer B at a flow rate of 0.2 μL/min. At 105 min, the solvent ratios were altered to 70% buffer A, 30% buffer B. At 110 min, the ratios were further altered to 20% buffer A, 80% buffer B. These conditions were held until 115.5 min, when the flow rate was increased to 0.3 μL/min. At 121 min, the solvent ratios were returned to 95% buffer A, 5% buffer B, where they remained until the conclusion of the run. The mass spectrometer was operated in positive ion mode, with an m/z peak isolation width of 1.5 m/z, an approximate injection size of 1 μg total peptides, an MS automatic gain control (AGC) target of 3 × 106 ions, an MS/MS AGC target of 1 × 106 ions, and a Top-20 data-dependent acquisition method (a full scan at a resolution of 70,000, followed by MS/MS analysis of the 20 most intense ions using a resolution of 17,500). The maximum MS fill time was 30 ms, the maximum MS/MS fill time was 120 ms, and the peptide match setting was set on preferred. Dynamic exclusion was set at 45 ms. Precursors with charge states less than +3 were excluded from MS/MS analyses. All other instrument parameters were used in their default settings.
MS/MS Data Analysis
Thermo RAW files from the control and crosslinked samples were converted to mzXML format (ProteoWizard MSConvert) (Kessner et al., 2008). The control and crosslinked sample MS scans were de-isotoped, and precursors were compared using a label-free comparison module in ZXMiner with a mass error tolerance of 10 ppm over the retention time range where peptides eluted, which was 15–110 min. Only precursors present in one or more cross-linked samples and absent in the uncrosslinked control were considered further as putative crosslinked peptides. Putative crosslinked peptides were identified using ZXMiner (Sriswasdi et al., 2014b). We used a forward database consisting of the 57 most abundant erythrocyte membrane proteins, as estimated by MaxQuant (Cox and Mann, 2008) quantitation of an LC-MS/MS analysis of a purified erythrocyte membrane sample. Proteins were selected as part of the database if ten or more unique MS/MS spectra were identified in the MaxQuant analysis of the control erythrocyte membrane sample. A reversed Escherichia coli protein database was appended to the erythrocyte membrane database to estimate confidence of crosslinked peptide assignments. A precursor specific to crosslinked samples was considered to be a putative crosslinked peptide if it matched a theoretical crosslinked sequence with a precursor mass tolerance of ≤10 ppm, and some of its MS/MS spectra met all of the following conditions: geometric mean (GM) score ≥0.2, ion coverage score ≥0.1, and ΔGM score ≥0.05 (the difference in GM score between the top-scoring match and the next best match). When putative crosslinks were compared with crystallographic structures, Cα-Cα distances between identified crosslinked residues were expected to be within 12 Å for well-ordered regions of known crystal structures. This distance included the lengths of the amino acid side chains and several angstroms of molecular flexibility. However, regions with substantial conformational flexibility such as surface loops and inter-chain interfaces were sometimes observed to have crosslinkable sites with larger Cα-Cα distances as described previously (Rivera-Santiago et al., 2015a, 2015b; Sriswasdi et al., 2014b).
Increasing Confidence in Crosslinked Peptides
All putative crosslinked peptides were subsequently further evaluated to verify the assignments. First, only matches where the peak coverage score was >0.5 (more than 50% of the masses in the MS/MS spectra matched predicted fragments) were further considered. The next step was to view the match between the MS/MS spectra and assigned fragment ions using the XLinkInspector graphical module in the ZXMiner software suite (Rivera-Santiago et al., 2015b). Putative matches that did not have good fragment ion coverage for both peptides in the crosslinked complex or where any of the three most intense MS/MS peaks were not assigned to predicted fragment ions were dropped from further consideration. Peptides meeting all of these criteria were considered “verified crosslinks.” Verified crosslinked peptides containing multiple alternative internal crosslink sites were further evaluated based on alternative scores and with the assistance of graphical interfaces in the ZXMiner program to determine the highest probability linkage between the two peptides.
Immunoprecipitation of Crosslinked AE1
In selected experiments, AE1 was immunoprecipitated using a polyclonal AE1 antibody (Abcam, product no. ab78067) that was bound to Protein G Dynabeads (Life Technologies) and covalently coupled as described by the vendor. Crosslinked and control membranes were solubilized in 50 mM Tris, 150 mM NaCl, 1 mM EDTA, and 0.5% SDS (pH 7.4) with mild sonication followed by addition of an equal volume of the same buffer where the SDS was replaced with 2% IGEPAL CA-630 and 1% sodium deoxycholate. After centrifugation to remove insoluble material, the supernatant was incubated with the immobilized antibody for 1 hr at room temperature with mild agitation, unbound proteins were removed, the resin was washed and bound proteins were eluted using 50 mM glycine, 150 mM NaCl, and 1% IGEPAL CA-630 (pH 2.5). Eluents were neutralized with 1 M Tris-HCl (pH 8.5).
Homology Modeling
The AE1 protein sequence, and the 1HYN and 4KY9 crystal structures for the AE1 N terminus (Shnitsar et al., 2013; Zhang et al., 2000), and the 4YZF crystal structure for the AE1 C terminus (Arakawa et al., 2015) were submitted to MODELLER 9v14 (Sali and Blundell, 1993) to generate and refine full-length human AE1 solution structures. All modeling experiments were run as 50-model trials using the “very slow” refinement algorithm and discrete optimized protein energy (DOPE) score as an output. Homology modeling and refinement were performed simultaneously by including known intra-subunit-validated crosslinks as distance restraints between α carbons imposed at 11.0 ± 0.1 Å. Each model was subject to 1,000 iterations and 10 optimization repeats. The completed models were then analyzed according to their DOPE score, and the highest-scoring model under this criterion was chosen for further analysis. Molecular graphics were illustrated using Open-Source PyMOL version 1.7 (Schrödinger), which also was used to calculate distances between α carbons of crosslinked glutamate, aspartate, and lysine. No crosslinks involving the N and C termini of the protein were detected.
Supplementary Material
Highlights.
Zero-length crosslinking of intact cell membranes was used to probe AE1 structure
A full-length AE1 model was deduced using crosslinks and known partial structures
Previously unresolved loops form extensive contact between the two major domains
Protein-protein interaction sites are more extensive than reported previously
Acknowledgments
The authors gratefully acknowledge the assistance of the Wistar Institute Proteomics and Metabolomics Core Facility. This work was supported by the US NIH grants R01HL038794 and R01DK084188 (to D.W.S.), P30CA010815 (NCI core grant to the Wistar Institute), and T32GM008275 as well as a Philadelphia Health Care Trust Fellowship (to R.R.S.).
Footnotes
Supplemental Information includes four figures, one movie, and one data file and can be found with this article online at http://dx.doi.org/10.1016/j.str.2016.11.017.
AUTHOR CONTRIBUTIONS
S.S. wrote and optimized the ZXMiner software and assisted with data interpretation. S.H. purified membranes, performed crosslinking reactions, and assisted with the data interpretation. P.H. performed crosslinking reactions, trypsin digestions, and immunoprecipitation experiments. R.R.S. optimized LC-MS/MS conditions, performed LC-MS experiments, data analysis, homology modeling, and data interpretation. R.R.S. and D.W.S. conceived and designed the study and wrote the manuscript, and all authors reviewed and edited the final manuscript.
References
- Alper SL. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J Exp Biol. 2009;212:1672–1683. doi: 10.1242/jeb.029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa T, Kobayashi-Yurugi T, Alguel Y, Iwanari H, Hatae H, Iwata M, Abe Y, Hino T, Ikeda-Suno C, Kuma H, et al. Crystal structure of the anion exchanger domain of human erythrocyte band 3. Science. 2015;350:680–684. doi: 10.1126/science.aaa4335. [DOI] [PubMed] [Google Scholar]
- Back JW, de Jong L, Muijsers AO, de Koster CG. Chemical cross-linking and mass spectrometry for protein structural modeling. J Mol Biol. 2003;331:303–313. doi: 10.1016/s0022-2836(03)00721-6. [DOI] [PubMed] [Google Scholar]
- Bonar PT, Casey JR. Plasma membrane Cl(−)/HCO(3)(−) exchangers: structure, mechanism and physiology. Channels (Austin) 2008;2:337–345. doi: 10.4161/chan.2.5.6899. [DOI] [PubMed] [Google Scholar]
- Bumpus NN, Hollenberg PF. Cross-linking of human cytochrome P450 2B6 to NADPH-cytochrome P450 reductase: identification of a potential site of interaction. J Inorg Biochem. 2010;104:485–488. doi: 10.1016/j.jinorgbio.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton NM, Bruce LJ. Modelling the structure of the red cell membrane. Biochem Cell Biol. 2011;89:200–215. doi: 10.1139/o10-154. [DOI] [PubMed] [Google Scholar]
- Bustos SP, Reithmeier RA. Protein 4.2 interaction with hereditary spherocytosis mutants of the cytoplasmic domain of human anion exchanger 1. Biochem J. 2011;433:313–322. doi: 10.1042/BJ20101375. [DOI] [PubMed] [Google Scholar]
- Che A, Cherry RJ. Loss of rotational mobility of band 3 proteins in human erythrocyte membranes induced by antibodies to glycophorin A. Biophys J. 1995;68:1881–1887. doi: 10.1016/S0006-3495(95)80365-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I. SLC4A transporters. Curr Top Membr. 2012;70:77–103. doi: 10.1016/B978-0-12-394316-3.00003-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Woods N, Sawasdee N, Guizouarn H, Pellissier B, Borgese F, Yenchitsomanus PT, Gowrishankar M, Cordat E. Band 3 Edmonton I, a novel mutant of the anion exchanger 1 causing spherocytosis and distal renal tubular acidosis. Biochem J. 2010;426:379–388. doi: 10.1042/BJ20091525. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p. p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Escobar L, Mejia N, Gil H, Santos F. Distal renal tubular acidosis: a hereditary disease with an inadequate urinary H(+) excretion. Nefrologia. 2013;33:289–296. doi: 10.3265/Nefrologia.pre2012.Oct.11592. [DOI] [PubMed] [Google Scholar]
- Gaubitz C, Oliveira TM, Prouteau M, Leitner A, Karuppasamy M, Konstantinidou G, Rispal D, Eltschinger S, Robinson GC, Thore S, et al. Molecular basis of the rapamycin insensitivity of target of rapamycin complex 2. Mol Cell. 2015;58:977–988. doi: 10.1016/j.molcel.2015.04.031. [DOI] [PubMed] [Google Scholar]
- Greber BJ, Boehringer D, Leibundgut M, Bieri P, Leitner A, Schmitz N, Aebersold R, Ban N. The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014;515:283–286. doi: 10.1038/nature13895. [DOI] [PubMed] [Google Scholar]
- Grey JL, Kodippili GC, Simon K, Low PS. Identification of contact sites between ankyrin and band 3 in the human erythrocyte membrane. Biochemistry (Mosc) 2012;51:6838–6846. doi: 10.1021/bi300693k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves JD, Tanner MJ. The effects of glycophorin A on the expression of the human red cell anion transporter (band 3) in Xenopus oocytes. J Membr Biol. 1994;140:81–88. doi: 10.1007/BF00234488. [DOI] [PubMed] [Google Scholar]
- Harper SL, Sriswasdi S, Tang HY, Gaetani M, Gallagher PG, Speicher DW. The common hereditary elliptocytosis-associated alpha-spectrin L260P mutation perturbs erythrocyte membranes by stabilizing spectrin in the closed dimer conformation. Blood. 2013;122:3045–3053. doi: 10.1182/blood-2013-02-487702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarolim P, Palek J, Amato D, Hassan K, Sapak P, Nurse GT, Rubin HL, Zhai S, Sahr KE, Liu SC. Deletion in erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proc Natl Acad Sci USA. 1991;88:11022–11026. doi: 10.1073/pnas.88.24.11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings ML, Nicknish JS. Localization of a site of intermolecular cross-linking in human red blood cell band 3 protein. J Biol Chem. 1985;260:5472–5479. [PubMed] [Google Scholar]
- Jons T, Drenckhahn D. Identification of the binding interface involved in linkage of cytoskeletal protein 4.1 to the erythrocyte anion exchanger. EMBO J. 1992;11:2863–2867. doi: 10.1002/j.1460-2075.1992.tb05354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkhof S, Ihling C, Mechtler K, Sinz A. Chemical cross-linking and high-performance Fourier transform ion cyclotron resonance mass spectrometry for protein interaction analysis: application to a calmodulin/target peptide complex. Anal Chem. 2005;77:495–503. doi: 10.1021/ac0487294. [DOI] [PubMed] [Google Scholar]
- Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles DW, Chasis JA, Evans EA, Mohandas N. Cooperative action between band 3 and glycophorin A in human erythrocytes: immobilization of band 3 induced by antibodies to glycophorin A. Biophys J. 1994;66:1726–1732. doi: 10.1016/S0006-3495(94)80965-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci USA. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A, Walzthoeni T, Kahraman A, Herzog F, Rinner O, Beck M, Aebersold R. Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol Cell Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Tang HY, Speicher DW. A structural model of the erythrocyte spectrin heterodimer initiation site determined using homology modeling and chemical cross-linking. J Biol Chem. 2008;283:1553–1562. doi: 10.1074/jbc.M706981200. [DOI] [PubMed] [Google Scholar]
- Li D, Harper SL, Tang HY, Maksimova Y, Gallagher PG, Speicher DW. A comprehensive model of the spectrin divalent tetramer binding region deduced using homology modeling and chemical cross-linking of a mini-spectrin. J Biol Chem. 2010;285:29535–29545. doi: 10.1074/jbc.M110.145573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marekov LN. Determination of protein contacts by chemical cross-linking with EDC and mass spectrometry. Curr Protoc Protein Sci. 2007;Chapter 19(Unit 19.16) doi: 10.1002/0471140864.ps1916s47. [DOI] [PubMed] [Google Scholar]
- Michaely P, Tomchick DR, Machius M, Anderson RG. Crystal structure of a 12 ANK repeat stack from human ankyrinR. EMBO J. 2002;21:6387–6396. doi: 10.1093/emboj/cdf651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao R, Suzuki T, Okumura A, Niikura A, Iwai M, Dohmae N, Tomo T, Shen JR, Ikeuchi M, Enami I. Topological analysis of the extrinsic PsbO, PsbP and PsbQ proteins in a green algal PSII complex by cross-linking with a water-soluble carbodiimide. Plant Cell Physiol. 2010;51:718–727. doi: 10.1093/pcp/pcq042. [DOI] [PubMed] [Google Scholar]
- Navare AT, Chavez JD, Zheng C, Weisbrod CR, Eng JK, Siehnel R, Singh PK, Manoil C, Bruce JE. Probing the protein interaction network of Pseudomonas aeruginosa cells by chemical cross-linking mass spectrometry. Structure. 2015;23:762–773. doi: 10.1016/j.str.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Bron C, Girardet M, Cherry RJ. Band 3-glycophorin A association in erythrocyte membrane demonstrated by combining protein diffusion measurements with antibody-induced cross-linking. Biochemistry (Mosc) 1980;19:1887–1893. doi: 10.1021/bi00550a024. [DOI] [PubMed] [Google Scholar]
- Olson AL, Tucker AT, Bobay BG, Soderblom EJ, Moseley MA, Thompson RJ, Cavanagh J. Structure and DNA-binding traits of the transition state regulator AbrB. Structure. 2014;22:1650–1656. doi: 10.1016/j.str.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramelle D, Miralles G, Subra G, Martinez J. Chemical cross-linkers for protein structure studies by mass spectrometry. Proteomics. 2013;13:438–456. doi: 10.1002/pmic.201200305. [DOI] [PubMed] [Google Scholar]
- Pasini EM, Kirkegaard M, Mortensen P, Lutz HU, Thomas AW, Mann M. In-depth analysis of the membrane and cytosolic proteome of red blood cells. Blood. 2006;108:791–801. doi: 10.1182/blood-2005-11-007799. [DOI] [PubMed] [Google Scholar]
- Rappsilber J. The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J Struct Biol. 2011;173:530–540. doi: 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh B, Karp JM, Sparks S, Dutta K, Rout MP, Sali A, Cowburn D. Slide-and-exchange mechanism for rapid and selective transport through the nuclear pore complex. Proc Natl Acad Sci USA. 2016;113:E2489–E2497. doi: 10.1073/pnas.1522663113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Santiago RF, Harper SL, Zhou S, Sriswasdi S, Feinstein SI, Fisher AB, Speicher DW. Solution structure of the reduced form of human peroxiredoxin-6 elucidated using zero-length chemical cross-linking and homology modelling. Biochem J. 2015a;468:87–98. doi: 10.1042/BJ20141463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Santiago RF, Sriswasdi S, Harper SL, Speicher DW. Probing structures of large protein complexes using zero-length cross-linking. Methods. 2015b;89:99–111. doi: 10.1016/j.ymeth.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero MF, Fulton CM, Boron WF. The SLC4 family of HCO 3-transporters. Pflugers Arch. 2004;447:495–509. doi: 10.1007/s00424-003-1180-2. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Kalkhof S, Ihling C, Cooper DM, Sinz A. Mapping protein interfaces by chemical cross-linking and Fourier transform ion cyclotron resonance mass spectrometry: application to a calmodulin/adenylyl cyclase 8 peptide complex. Eur J Mass Spectrom (Chichester, Eng) 2005;11:525–534. doi: 10.1255/ejms.748. [DOI] [PubMed] [Google Scholar]
- Shnitsar V, Li J, Li X, Calmettes C, Basu A, Casey JR, Moraes TF, Reithmeier RA. A substrate access tunnel in the cytosolic domain is not an essential feature of the solute carrier 4 (SLC4) family of bicarbonate transporters. J Biol Chem. 2013;288:33848–33860. doi: 10.1074/jbc.M113.511865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinz A. Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- Speicher DW, Weglarz L, DeSilva TM. Properties of human red cell spectrin heterodimer (side-to-side) assembly and identification of an essential nucleation site. J Biol Chem. 1992;267:14775–14782. [PubMed] [Google Scholar]
- Speicher KD, Kolbas O, Harper S, Speicher DW. Systematic analysis of peptide recoveries from in-gel digestions for protein identifications in proteome studies. J Biomol Tech. 2000;11:74–86. [PMC free article] [PubMed] [Google Scholar]
- Sriswasdi S, Harper SL, Tang HY, Gallagher PG, Speicher DW. Probing large conformational rearrangements in wild-type and mutant spectrin using structural mass spectrometry. Proc Natl Acad Sci USA. 2014a;111:1801–1806. doi: 10.1073/pnas.1317620111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriswasdi S, Harper SL, Tang HY, Speicher DW. Enhanced identification of zero-length chemical cross-links using label-free quantitation and high-resolution fragment ion spectra. J Proteome Res. 2014b;13:898–914. doi: 10.1021/pr400953w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck TL. Cross-linking the major proteins of the isolated erythrocyte membrane. J Mol Biol. 1972;66:295–305. doi: 10.1016/0022-2836(72)90481-0. [DOI] [PubMed] [Google Scholar]
- Telen MJ, Chasis JA. Relationship of the human erythrocyte Wrb antigen to an interaction between glycophorin A and band 3. Blood. 1990;76:842–848. [PubMed] [Google Scholar]
- van den Akker E, Satchwell TJ, Williamson RC, Toye AM. Band 3 multiprotein complexes in the red cell membrane; of mice and men. Blood Cells Mol Dis. 2010;45:1–8. doi: 10.1016/j.bcmd.2010.02.019. [DOI] [PubMed] [Google Scholar]
- Young MT, Beckmann R, Toye AM, Tanner MJ. Red-cell glycophorin A-band 3 interactions associated with the movement of band 3 to the cell surface. Biochem J. 2000;350:53–60. [PMC free article] [PubMed] [Google Scholar]
- Young MT, Tanner MJ. Distinct regions of human glycophorin A enhance human red cell anion exchanger (band 3; AE1) transport function and surface trafficking. J Biol Chem. 2003;278:32954–32961. doi: 10.1074/jbc.M302527200. [DOI] [PubMed] [Google Scholar]
- Zhang D, Kiyatkin A, Bolin JT, Low PS. Crystallographic structure and functional interpretation of the cytoplasmic domain of erythrocyte membrane band 3. Blood. 2000;96:2925–2933. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.