

ABSTRACT
The majority of patients with classical myeloproliferative neoplasms (MPN) of polycythemia vera, essential thrombocythemia, and primary myelofibrosis harbor distinct disease-driving mutations within the JAK2, CALR, or MPL genes. The term triple-negative has been recently applied to those MPN without evidence of these consistent mutations, prompting whole or targeted exome sequencing approaches to determine the driver mutational status of this subgroup. These strategies have identified numerous novel mutations that occur in alternative exons of both JAK2 and MPL, the majority of which result in functional activation. Current molecular diagnostic approaches may possess insufficient coverage to detect these alternative mutations, prompting further consideration of targeted exon sequencing into routine diagnostic practice. How to incorporate these illuminating findings into the expanding molecular diagnostic algorithm for MPN requires continual attention.
KEYWORDS: CALR, JAK2, myeloproliferative neoplasms, MPL, molecular diagnostics
The classical Philadelphia chromosome-negative myeloproliferative neoplasms (MPN) comprise polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF), all characterized by over production of mature haematopoietic cells, extra-medullary hematopoiesis and clinically by a tendency for thrombosis and/or hemorrhage and the potential to transform into acute leukemia. Diagnosis of MPN is multidisciplinary, requiring consideration of the presenting clinical features, morphological assessment of the peripheral blood and bone marrow aspirate and biopsy, standard laboratory parameters and ever increasingly, the underlying acquired mutation status. The landmark discovery of the constitutively activating, JAK2 exon 14 V617F mutation in nearly all PV patients and in approximately half of ET and PMF patients more than a decade ago prompted a renewed interest in the molecular pathogenesis of these diseases. In addition to the JAK2 V617F, other driver mutations such as those within JAK2 exon 12 in PV, MPL exon 10 and CALR exon 9 in ET and PMF have subsequently been identified and which have been demonstrated to directly contribute to MPN pathogenesis.1 MPN driver mutations all appear to contribute to the myeloproliferative phenotype through convergent activation of intracellular JAK-STAT signaling and their downstream effectors: an obvious target for therapeutic intervention. Supplementary “passenger” mutations may be acquired that can influence the initiation and evolution of the disease, however these mutations are not specific for MPN as they are also present in other myeloid malignancies such as myelodysplastic syndromes and acute myeloid leukemia. From a molecular diagnostic perspective, the importance of JAK2, MPL and CALR mutation analysis in differentiating between a reactive and a clonal, neoplastic process is paramount with a variety of approaches adopted that either screen for, or specifically detect and/or quantitate, the common JAK2 V617F, MPL W515L/K and CALR exon 9 insertion/deletion (indel) mutations. Each methodological approach has its own analytical specificity and sensitivity with selection largely dependent on resource and the clinical requirements of the service provider.2 Moreover, demonstration of any of these 3 mutation types will be a major diagnostic criterion in the forthcoming revision of the World Health Organization classification of MPN.3
The term “triple-negative” MPN has recently been coined and applied to those 10-15% of ET and PMF patients without evidence of the 3 major mutation types of JAK2 V617F, MPL W515L/K and CALR exon 9 indels (Fig. 1). This particular signature has been reported to be associated with a relatively adverse survival, particularly in PMF patients.4 The understandable question that arises is what genetic events, if any, drive the myeloproliferation in triple-negative ET and PMF? Rare, alternative, somatic mutations in both JAK2 exon 14 and MPL exon 10, often at the same codon as the more common mutations, have been previously noted but may account for only a minority of triple-negative MPN.5-9 In order to address this issue, several groups have recently applied either whole exome sequencing and/or targeted exon sequencing to triple negative MPN, particularly ET cases, and which have informed further understanding of the molecular pathogenesis of this group of patients.10-12 In addition to sporadic mutations in epigenetic modifying (ASXL1, TET2), spliceosome (SF3B1, SRSF2), and regulators of cytokine signaling (CBL, SH2B3) genes, a significant number of mutations were detected in both the JAK2 and MPL genes, strikingly in exons not normally mutated in MPN (Fig. 2).10-12 All novel MPL mutations and 2 novel JAK2 mutations detected by one group resulted in a gain of function by inducing ligand-independent JAK2-STAT5 signaling when analyzed in functional assays.10 These findings were confirmed by demonstration of thrombopoietin hypersensitivity and independence of 2 such novel MPL mutations in a further study.11 The diversity of MPL mutations occurring in the extracellular domain-encoding exons of triple negative MPN has been further expanded.12
Figure 1.
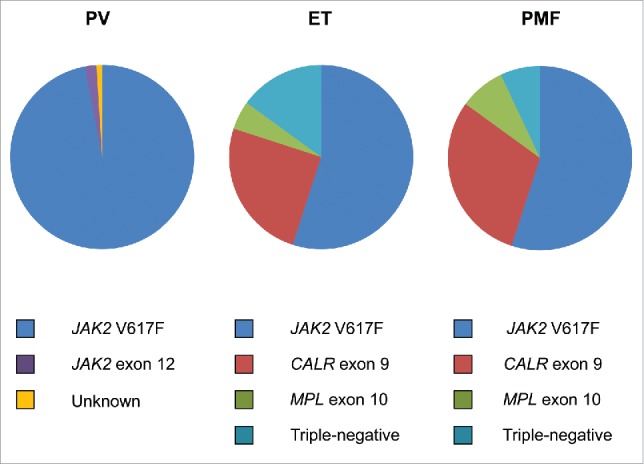
Distribution of driver mutations in polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF).
Figure 2.
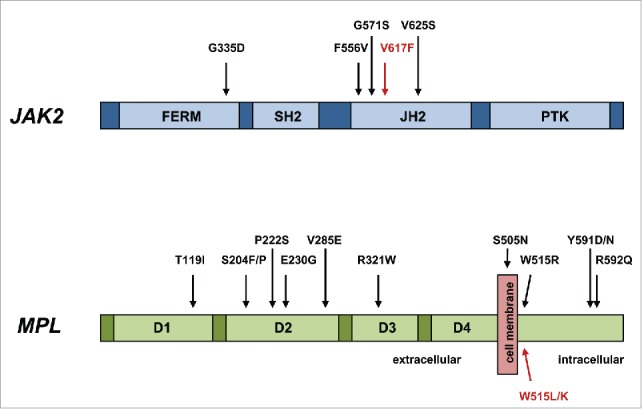
Novel activating mutations of JAK2 and MPL in triple-negative MPN. Those mutations indicated in red are the most frequently observed in MPN.
Clearly these findings have important implications for molecular diagnostics which aims to distinguish true MPN from reactive causes. Those diagnostic approaches that target specific mutations such as allele-specific PCR will not identify these, admittedly rare, variants. Furthermore, in the light of these findings, mutation screening of specific exons or amplicons with techniques such as high resolution melt curve analysis now appears insufficient. The ability of next-generation sequencing (NGS) approaches, able to rapidly sequence multiple amplicons simultaneously, expedites the adoption of these techniques into routine practice. Both commercially available and institutional targeted NGS panels have been developed and are becoming increasingly established in diagnostic testing algorithms which display variable coverage of mutational hotspots and selected exons.13-15 Whether such NGS gene panels should now cover all coding exons of JAK2 and MPL is perhaps worthy of consideration. In addition to exonic sequences, mutational analysis might also be extended to include the 5' untranslated regions of genes known to be altered in rare cases of sporadic and hereditary MPN.16-17
It must be noted that a significant number of MPN patients classified as triple negative have no detectable alternative JAK2 or MPL mutations, nor any evidence of clonal hematopoiesis by X chromosome inactivation patterns.10-12 Whether these cases, analogous to a larger proportion of pediatric patients classified as MPN, truly have a clonal malignancy is questionable.18 Re-consideration of the role of haematopoietic clonality assessment by X-chromosome inactivation patterns in triple-negative MPN may be necessary, however these approaches apply only to female patients, require considerable interpretation, and have been further confounded by the recent description of polyclonal hematopoiesis in some female MPN patients.19 Furthermore, the pathogenesis of MPN may not be entirely explained by the detection of driver mutations: other complex processes, such as inflammation, are likely to play a key role in the development of these diseases.20 The continual challenge remains in how to incorporate these discoveries into the ever evolving MPN molecular diagnostic algorithm.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
- [1].Nangalia J, Grinfeld J, Green AR. Pathogenesis of myeloproliferative disorders. Annu Rev Pathol 2016; 11:101-26; PMID:27193452; http://dx.doi.org/ 10.1146/annurev-pathol-012615-044454 [DOI] [PubMed] [Google Scholar]
- [2].Langabeer SE, Andrikovics H, Asp J, Bellosillo B, Carillo S, Haslam K, Kjaer L, Lippert E, Mansier O, Oppliger Leibundgut E, et al.. Molecular diagnostics of myeloproliferative neoplasms. Eur J Haematol 2015; 95(4):270-9; PMID:25951317; http://dx.doi.org/ 10.1111/ejh.12578 [DOI] [PubMed] [Google Scholar]
- [3].Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127(20):2391-405; PMID:27069254; http://dx.doi.org/ 10.1182/blood-2016-03-643544 [DOI] [PubMed] [Google Scholar]
- [4].Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, Gangat N, Fjerza R, Belachew AA, Lasho TL, et al.. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014; 124(16):2507-13; PMID:25037629; http://dx.doi.org/ 10.1182/blood-2014-05-579136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schnittger S, Bacher U, Kern W, Schröder M, Haferlach T, Schoch C. Report on two novel nucleotide exchanges in the JAK2 pseudokinase domain: D620E and E627E. Leukemia 2006; 20(12):2195-7; PMID:16871281; http://dx.doi.org/ 10.1038/sj.leu.2404325 [DOI] [PubMed] [Google Scholar]
- [6].Cleyrat C, Jelinek J, Girodon F, Boissinot M, Ponge T, Harousseau JL, Issa JP, Hermouet S. JAK2 mutation and disease phenotype: a double L611V/V617F in cis mutation of JAK2 is associated with isolated erythrocytosis and increased activation of AKT and ERK1/2. Leukemia 2010; 24(5):1069-73; PMID:20182460; http://dx.doi.org/ 10.1038/leu.2010.23 [DOI] [PubMed] [Google Scholar]
- [7].Chaligné R, Tonetti C, Besancenot R, Roy L, Marty C, Mossuz P, Kiladjian JJ, Socié G, Bordessoule D, Le Bousse-Kerdilès MC, et al.. New mutations of MPL in primitive myelofibrosis: only the MPL W515 mutations promote a G1/S-phase transition. Leukemia 2008; 22(8):1557-66; http://dx.doi.org/ 10.1038/leu.2008.137 [DOI] [PubMed] [Google Scholar]
- [8].Schnittger S, Bacher U, Haferlach C, Beelen D, Bojko P, Bürkle D, Dengler R, Distelrath A, Eckart M, Eckert R, et al.. Characterization of 35 new cases with four different MPL W515 mutations and essential thrombocythemia or primary myelofibrosis. Haematologica 2009; 94(1):141-4; PMID:19029146; http://dx.doi.org/ 10.3324/haematol.13224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gong JZ, Cook JR, Greiner TC, Hedvat C, Hill CE, Lim MS, Longtine JA, Sabath D, Wang YL. Laboratory practice guidelines for detecting and reporting JAK2 and MPL mutations in myeloproliferative neoplasms: a report of the association for molecular pathology. J Mol Diagn 2013; 15(6):733-44; PMID:23978506; http://dx.doi.org/ 10.1016/j.jmoldx.2013.07.002 [DOI] [PubMed] [Google Scholar]
- [10].Milosevic Feenstra JD, Nivarthi H, Gisslinger H, Leroy E, Rumi E, Chachoua I, Bagienski K, Kubesova B, Pietra D, Gisslinger B, et al.. Whole exome sequencing identifies novel MPL and JAK2 mutations in triple negative myeloproliferative neoplasms. Blood 2016; 127(3):325-32; PMID:26423830; http://dx.doi.org/ 10.1182/blood-2015-07-661835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cabagnols X, Favale F, Pasquier F, Messaoudi K, Defour JP, Ianotto JC, Marzac C, Le Couédiec JP, Droin N, Chachoua I, et al.. Presence of atypical thrombopoietin receptor (MPL) mutations in triple negative essential thrombocythemia patients. Blood 2016; 127(3):333-42; PMID:26450985; http://dx.doi.org/ 10.1182/blood-2015-07-661983 [DOI] [PubMed] [Google Scholar]
- [12].Angona A, Fernández-Rodríguez C, Alvarez-Larrán A, Camacho L, Longarón R, Torres E, Pairet S, Besses C, Bellosillo B. Molecular characterisation of triple negative essential thrombocythaemia patients by platelet analysis and targeted sequencing. Blood Cancer J 2016; 6(8):e463; PMID:27564461; http://dx.doi.org/ 10.1038/bcj.2016.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kirschner MM, Schemionek M, Schubert C, Chatain N, Sontag S, Isfort S, Ortiz-Brüchle N, Schmitt K, Krüger K, Zerres K, et al.. Dissecting genomic aberrations in myeloproliferative neoplasms by multiplex-PCR and next generation sequencing. PLoS One 2015; 10(4):e0123476; PMID:25894969; http://dx.doi.org/ 10.1371/journal.pone.0123476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Magor GW, Tallack MR, Klose NM, Taylor D, Korbie D, Mollee P, Trau M, Perkins AC. Rapid molecular profiling of myeloproliferative neoplasms using targeted exon resequencing of 86 genes involved in JAK-STAT signalling and epigenetic regulation. J Mol Diagn 2016; 18(5):707-18; PMID:27449473; http://dx.doi.org/ 10.1016/j.jmoldx.2016.05.006 [DOI] [PubMed] [Google Scholar]
- [15].Delic S, Rose D, Kern W, Nadarajah N, Haferlach C, Haferlach T, Meggendorfer M. Application of an NGS-based 28-gene panel in myeloproliferative neoplasms reveals distinct patterns in essential thrombocythaemia, primary myelofibrosis and polycythaemia vera. Br J Haematol 2016; 175(3):419-26; PMID:27447873 [DOI] [PubMed] [Google Scholar]
- [16].Hussein K, Percy M, McMullin MF, Schwarz J, Schnittger S, Porret N, Martinez-Aviles L, Bellosillo-Paricio B, Giraudier S, Skoda R, et al.. Clinical utility gene card for: hereditary thrombocythemia. Eur J Hum Genet 2014; 22(2):e1-e5; PMID:23736217; http://dx.doi.org/ 10.1038/ejhg.2013.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nelson ND, Marcogliese A, Bergstrom K, Scheurer M, Mahoney D, Bertuch AA. Thrombopoietin measurement as a key component in the evaluation of pediatric thrombocytosis. Pediatr Blood Cancer 2016; 63(8):1484-7; PMID:27100794; http://dx.doi.org/ 10.1002/pbc.26032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Randi ML, Geranio G, Bertozzi I, Micalizzi C, Ramenghi U, Tucci F, Notarangelo LD, Ladogana S, Menna G, Giordano P, et al.. Are all cases of paediatric essential thrombocythaemia really myeloproliferative neoplasms? Br J Haematol 2015; 169(4):584-9; PMID:25716342; http://dx.doi.org/ 10.1111/bjh.13329 [DOI] [PubMed] [Google Scholar]
- [19].Swierczek S, Lima LT, Tashi T, Gregg XT, Prchal JT. Presence of polyclonal hematopoiesis in females with Ph-negative myeloproliferative neoplasms. Leukemia 2015; 29(12):2432-4; PMID:26369983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hermouet S, Bigot-Corbel E, Gardie B. Pathogenesis of myeloproliferative neoplasms: role and mechanisms of chronic inflammation. Mediators Inflamm 2015; 2015:145293; PMID:26538820 [DOI] [PMC free article] [PubMed] [Google Scholar]