

Abstract
The postgenomic era, as manifest, inter alia, by proteomics, offers unparalleled opportunities for the efficient discovery of safe, efficacious, and novel subunit vaccines targeting a tranche of modern major diseases. A negative corollary of this opportunity is the risk of becoming overwhelmed by this embarrassment of riches. Informatics techniques, working to address issues of both data management and through prediction to shortcut the experimental process, can be of enormous benefit in leveraging the proteomic revolution.In this disquisition, we evaluate proteomic approaches to the discovery of subunit vaccines, focussing on viral, bacterial, fungal, and parasite systems. We also adumbrate the impact that proteomic analysis of host-pathogen interactions can have. Finally, we review relevant methods to the prediction of immunome, with special emphasis on quantitative methods, and the subcellular localization of proteins within bacteria.
INTRODUCTION
Genomics has changed the world. Or at least, it has changed the intellectual landscape of the biosciences: its implications suggest that we should be able to gain access to information about biological function at a rate, and on a scale, previously beyond our wildest expectations. As ever, our hopes and dreams are yet to be fulfilled. What we can conceive of still far exceeds what can actually be done at the laboratory bench. Experimental science is playing catch up, developing so-called postgenomic strategies that seek to exploit the opportunities created by the information explosion implicit within genomics. Biology is at risk of being overcome by a bewildering deluge of new data on a hitherto unknown scale and of a hitherto unknown complexity. This is clearly both a blessing and a curse; the trick is to tease out useful information from the data with the hope that this will, in its turn, yield first knowledge, and then, ultimately, true understanding and the ability to efficiently manipulate biological systems.
Postgenomic approaches are legion. They include genomic sequencing, transcriptomics, proteomics, and the analysis of protein-protein interactions, as well as applied techniques, such as the high-throughput screening (HTS) for drug candidates, and integrated informatic strategies, including structure-function prediction. The key underlying factor here is parallelization: the ability to address specific questions not on an individual basis, through complex, intricate experiments, but en masse through elegantly conceived procedures that examine not a single biological object but hundreds, thousands, even hundreds of thousands. This is the area of functional genomics. Functional genomics relies implicitly on high-throughput techniques for measuring the mRNA (the transcriptome), protein (the proteome), and metabolite (the metabolome) components of cells, tissues, organs, and whole organisms.
We pause, momentarily, to examine some definitions. The word orismology, which, in English, dates to 1816, is the science of making and defining terms, especially scientific and technical ones. The need for constant and reliable definitions of terms in science is clear but is seldom realized. Orismology, the science of defining technical terms, seeks to address issues such as these. However, as in many areas of life, that which is apparently rigorous is often anything but risible. The meanings of words drift and vary with time and take on new (sub)discipline-dependent meanings. While this is natural and unavoidable, it can, at times, become obfuscatingly discombobulating. We have seen that with the rise of -ologies (psychology, sociology, even lipocalinology [1]) and, more recently, with the explosions of -omes and -omics. The now famous, or perhaps infamous, website http://www.genomicglossaries.com/content/omes.asp lists literally hundreds of different -omes and -omics. From the genome, countless other -omes have arisen, and within this -omic revolution there are many many conflicting definitions, some are useful, some are not. For the sake of completeness, we list a few of the most useful and the most germane to our present discussions
The genome is the DNA sequence of an organism. The number of sequenced genomes is now large and ever increasing. In the space of a few years the sequencing of a genome has gone from a transcendent achievement capable of stopping the scientific world in its tracks to the almost mundane, worthy of only a minor mention in a second line journal. In future times, genomic sequencing may simply become a workaday laboratory technique. Within a decade it may become the stuff of postgraduate students' theses; undergraduates might need to sequence a dozen to complete their final year projects.
Transcriptome is the complement of messenger RNAs (mRNAs) transcribed from a genome. This is a dynamic set of proteins, unlike the genome, which is constantly changing with time in response to the conditions experienced by the cell, hence the development of transcriptomics: the analysis, typically using MicroArrays, of mRNA expression profiles.
Proteome is the protein complement of a cell corresponding to the genome and transcriptome described above. Proteomics is the science that has developed to study the proteome. The proteome is, like the transcriptome, highly dynamic. Conceptually, the proteome is biology in a way that neither the genome nor transcriptome could ever be. Proteins make nature function. Genes, as nucleic acid memes (if one is to believe Richard Dawkins and his ilk), are the essence of inheritance, but it is only through the medium of the protein world, that they are able to propagate themselves.
Metabolome is the complement of all low molecular weight molecules present in a cell. As before, the state of the metabolome is highly dependent on the particular physiological or developmental state or the environmental challenge of the cell. We can usefully distinguish between primary and secondary metabolites. Primary metabolites are the intermediates (ATP, amino acids, membrane phospholipids, etc) in the key metabolic pathways of the cell. Secondary metabolites, at least in the context of microbial natural products, are compounds with no explicit role in the internal metabolic economy of the microbe that biosynthesized them. One argument predicates their existence within an evolutionary rationale: secondary metabolites enhance the survival of their producer organisms by binding specifically to macromolecular receptors in competing organisms with a concomitant physiological action. As a consequence of this intrinsic capacity for interaction with biological receptors, made manifest in their size and complexity, natural products will be generally predisposed to form macromolecular complexes. On this basis, and within a drug discovery context, one might expect that natural products would possess a high hit rate when screened and a good chance of high initial activity and selectivity.
Focussing on the size and complexity of the proteome, we continue by briefly looking at the vexed question of gene number. Pre-genome estimates of the size of the human genome have been revised down from an initial “best-guess” figure in excess of 100,000. As this review is being written, estimates of gene number are converging from a preliminary postgenomic estimate of 30,000–40,000 to a more realistic 65,000–70,000. This may also prove to be an underestimate. The proteome is, however, much larger, principally through the existence of splice variants [2], but also due to the existence of protein splicing elements (inteins) which catalyze their own excision from flanking amino acid sequences (exteins) thus creating new proteins in which the exteins are linked directly by a peptide bond [3]. Other mechanisms include posttranslational modifications, cleavage of precursors, and other types of proteolytic activation. Some estimates place the estimated number of proteins encoded by the human genomes to be two to three orders of magnitude higher than the number of genes. In certain senses at least, the proteome is, as we have said, also much more dynamic than the genome; it varies according to the cell type and the functional state of the cell. In addition, the proteome shows characteristic perturbations in response to disease and external stimuli. Proteomics, as a scientific discipline is relatively new, but is based upon rather older techniques, combining sophisticated analytical methods, such as 2D electrophoresis and mass spectrometry (MS), with bioinformatics. Thus proteomics is the study of gene expression at a functional level.
Returning once more to definitions, a comprehensive description of the proteome provides not only a catalogue of all proteins encoded by the genome but also data on protein expression under defined conditions, the occurrence of posttranslational modifications and, importantly, the distribution of specific proteins within the cell [4]. A forerunner to the current proteome paradigm was the concept adumbrated by Anderson and Anderson [5]: the “human protein index.” They wished to characterize all the proteins expressed by a cell using high-resolution two-dimensional electrophoresis (2DE). They thought that the human protein index would prove useful in clinical chemistry, pathology, and toxicology. In its proteomic form, this conceit has proved all too true.
The biome, and hence biomics, is an overall term encompassing all of these definitions and including informatic approaches as well. An oft-neglected part of the biome is the immunome: the set of antigenic peptides, or possibly immunogenic proteins, within a microorganism, be that virus, bacteria, fungus, or parasite [6, 7]. There are alternative definitions of the immunome that also include immunological receptors and accessory molecules, but in what follows we will restrict discussion to this initial definition. It is also possible to talk of the self-immunome, the set of potentially antigenic self-peptides. This is clearly important within the context of, for example, cancer (the cancer-immunone) and autoimmunity (the autoimmunome), which affect about 30% and 3% of the global population, respectively.
Many -omes are virtual, rather than literal, biological entities. For example, the recently christened chemome, or chemizome, may be defined as the set of all artificially created or natural products that interact with biological targets in the organism. In practice, this set is not bounded. It is not possible to ever derive or find all the molecules that are encompassed by this definition. In contrast, the immunone, at least for a particular pathogen, can be realized only in the context of a particular, defined host.
The nature of the immunome is clearly dependent upon the host as much as it is on what we will, for convenience, call the pathogen. This is implicit in the term antigenic or immunogenic. A peptide is not antigenic if the immune system does not respond to it. A good example of this is the major histocompatibility complex (MHC) restriction of T-cell responses. A particular MHC allele will have a peptide specificity that may, or may not, overlap, with other expressed alleles, but the total specificity of all individual alleles will not cover the whole possible sequence space of peptides. Thus peptides that do not bind to any of an individual's allelic MHC variants cannot be antigenic within a cellular context. The ability to define the specificity of different MHCs computationally, which we may call in silico immunomics or in silico immunological proteomics for want of a more succinct term, is an important, but eminently realizable goal of immuninformatics, the application of informatics techniques to immunological macromolecules, a newly emergent subdiscipline within bioinformatics. We will return to this key topic later.
From the perspective of human disease, a proper understanding of the immune system is vital. Indeed, the immune system has evolved to combat the threat of infectious disease. Disease is, arguably, the most significant cause of death worldwide, but it is also the greatest source of preventable human mortality, in that, and in contrast to other causes of death, it can be attacked systematically through the use of biological and chemical entities, such as vaccines and drugs, and through the efforts of surgeons and physicians, and through improvements in public health, drinking water, and sanitation. Although it may be argued quite cogently that the greatest benefit to man has come through improved public health, it is clear that drugs and vaccines have made a large contribution. In contrast, other than by dispensing of drugs and other therapies, the contribution made to public wellbeing by trained medics, though more direct, is also relatively small.
Immunology is also pivotal in other areas of human disease. Cancer is often a prey to immunological mechanisms, and the augmentation of the immune response to carcinomas and cancer antigens is a vital area for future development. Likewise, the inappropriate response of the immune system to self-proteins, as manifest in allergy and, more importantly, in autoimmune diseases, is an area where immunotherapy and immunomodulators can be effective. The discovery and development of vaccines is an important component of publically funded health-care programs throughout the developed and under-developed worlds. Most Western countries have a well developed or long standing centres devoted to its study. The Edward Jenner Institute for Vaccine Research is the United Kingdom's contribution to this worldwide movement.
From a wealth creation viewpoint, rather than from a purely humanitarian one, the world human vaccine market is currently only in the region of $5 billion. One must put this figure against the total worldwide annual sales for all human therapeutic drugs of about $350 billion and an annual global investment in R&D of around $30 billion. To put these large numbers into context, this $350-billion figure is comparable to the yearly gross national product of Taiwan, the Netherlands, or Los Angeles County. However, sales in the vaccine market are increasing at around 12% per annum compared to a yearly rate of about 5% for drugs. Likewise, increased concern by consumers regarding both chemical-free food and environmental and animal welfare has led to an increased interest in vaccines within the farm livestock and companion animal health markets, which worth $18 billion and $3 billion respectively [8]. In the aftermath of AIDS, antibiotic resistance, and the threat from bioterrorism, interest in vaccines has increased dramatically from, say, 10–15 years ago when the vaccine industry was floundering. In 1990, there were about 10 companies in the area worldwide, compared today to over 100, although the majority of current vaccine production is still in the hands of only four big players. The design of therapeutic vaccines (pharmaccines) is, then, an active area of research. Novel ways to rationalize and accelerate vaccine discovery are desperately needed however. Advances in molecular biology and computer science are now accelerating candidate vaccine antigens discovery rates.
To bring this introduction full circle, proteomics is poised to make a significant contribution to the elaboration of the immunome, and thus vaccinology. Proteomics is a pivotal discipline, or more accurately disciplines, within functional genomics. It is an umbrella term for the large-scale analysis of proteins. In fact, proteomics encompasses many different methods seeking to identify the protein complement of a cell or tissue at a given time. These include comparing apparent differences between treated and untreated or between normal and diseased samples, the determination of posttranslational modification (the most common of which are glycosylation and phosphorylation), and the large-scale identification of protein-protein interactions. We will begin with a review of experimental approaches to proteomic vaccinology and then we will present an analysis of computational approaches to vaccinology.
PROTEOMICS IN VACCINOLOGY
The discovery of vaccination is generally attributed to Edward Jenner (1749–1823). However, at the beginning of the 18th century, inoculation against smallpox had been brought to England by Lady Mary Wortley Montagu (1689–1762). Lady Mary, who is, perhaps, better known to history as a poet and witty correspondent, was born in London, the eldest child of Evelyn Pierrepont, Earl of Kingston. In 1716, after the accession of the first Hanoverian monarch George I (1660–1727) on the death of the last Stuart monarch Queen Anne (1665–1714), Lady Mary's husband was appointed Ambassador to Turkey. The Wortley Montagu's long and dangerous transcontinental journey, which was undertaken in the dead of winter was considered something of an achievement at the time. Constantinople was full of wonders which Lady Mary, unlike so many European wives, set out to explore and understand, immersing herself in all Turkish things, even learning the language. She visited the zenanas, meeting the upper class women secluded there, whom she came to admire, and absorbed Turkish customs. Her record of her experiences, Turkish Embassy Letters, is a primary source for historians of this period.
The Wortley Montagu's visit occurred during the reign of the sophisticated, cultured, and tulip-obsessed ottoman sultan Ahmed III (1667–1736). His reign marked something of a renaissance for the Ottoman Empire after its relative decline during the 17th century. Influenced by his son-in-law, or damut, vizier Ibrahim Pasha Kulliyesi, Ahmed III increasingly looked to the West, creating the first fire brigade and printing presses in Constantinople and also establishing the Empire's first foreign embassies. Ahmed III reigned from 1703 to 1730, the so-called Tulip Era, or lale devri, a period of rare hedonistic extravagance centering on the sultan's love for the tulip.
Wortley was recalled due to a change in English relations with Turkey, and the family appeared in London in the fall of 1718. Lady Mary discovered that the Turks inoculated healthy children with a weakened strain of smallpox in order to confer immunity from the more virulent strains of the disease, and determined to bring the practice to England. Lady Mary had her own son and daughter inoculated against smallpox, which had killed her brother and left her scarred by her 1715 bout, and thus introduced the custom to the nobility. However, Lady Mary struggled to interest the English medical establishment in inoculation. Their main objection seems to have been to being told by a woman what it was their business to know. While it has become fashionable among feminist revisionists to credit Lady Mary, rather than Jenner, with the discovery of vaccination, this is hardly accurate. While it is important to recognize her contribution, it is important as well to recall that protective immunity has been recognized for several millenia at least; in 430 BC Thucydides, principal historian of the Peloponnesian War, noted that during an Athenian plague only those who had recovered from the plague were able to nurse the sick without themselves falling ill. During the 15th century, both the Chinese and Turks deliberately induced immunity by inhaling dried crusts from smallpox pustules or by inserting the crusts into cuts in the skin.
After a period of first training in London and then working for a time as an army surgeon, Jenner, a native of Gloucestershire, spent his entire career working in the county as a country doctor. Jenner had noted that milkmaids who had contracted cowpox, a related virus, seemed to be immune to smallpox. On 14th May 1796, he introduced the fluid from a cowpox pustule he used to build protective immunity against smallpox in his gardener's 8-year old son. Jenner then infected him with smallpox. The boy did not become ill. Later, Louis Pasteur (1822–1895) adopted “Vaccination,” the word Jenner had invented for his treatment (from the Latin vacca, a cow), for immunization against any disease. Pasteur also made important empirical advances in vaccination, discovering that chickens injected with attenuated fowl cholera bacteria survived an infection with the virulent form. Later, Pasteur immunized sheep with attenuated anthrax bacillus and challenged them with virulent anthrax and showed that the attenuated anthrax protected the sheep from disease, and in 1885 Louis Pasteur saved the life of a boy bitten by a rabid dog by administering a rabies vaccine he had created. It is now generally accepted that mass vaccination, taking account, as it does, of the principal of herd immunity, is one of the most effective prophylactic approaches to the treatment, or rather, pretreatment, of infectious disease.
However, vaccination has, until relatively recently, been a highly empirical science, relying of poorly understood, nonmechanistic approaches to the development of new vaccines. As a consequence of this, relatively few effective vaccines have been developed and deployed during most of the two centuries that have elapsed since Jenner's work. This has been prompted by, amongst other things, worries over the emergence of antibiotic resistance and, latterly, bioterrorism.
Vaccinology is slowly evolving into immunovaccinology, a discipline that uses the rapid advances in immunological understanding extant within the last few decades to effect a paradigm shift in thinking within the discipline. Reverse immunogenetic approaches offer the tantalizing prospect of short cutting the process of vaccine discovery and also producing safer and more effective vaccines. Postgenomic approaches, of which proteomics is amongst the most prominent, are another broad tranche of techniques which offers much in this context.
Antigenicity or immunogenicity manifests itself within both humoral immunology (mediated primarily through the binding of whole antigens by antibodies) and cellular immunology (mediated by binding of proteolytically cleaved peptides). In the main, we will concentrate our attention on that part of the adaptive immune response that is mediated by T cells. Within the context of cellular immunology, the immunogenicity of peptides strongly depends on their ability to bind to MHC and to be recognized subsequently by T-cell receptors (TCR). Traditionally, T-cell epitopes, the small peptide fragments of whole proteins that cellular immunity recognizes, have been identified by examining the responses of T cells to sets of overlapping peptides generated from target antigens. This is adequate, if labour intensive, for the study of a single, small protein, but the experimental overhead becomes prohibitive for the study of proteomes from large viruses, bacteria, or parasites, which may contain thousands, if not tens of thousands, of gene products.
The application of proteomics, perhaps in combination with transcriptomic approaches, together with bioinformatics, should allow us to reduce the virtual set of open reading frames (ORFs) apparent within a genome. This set might number a few hundred for viruses, a few thousands for bacteria, or a few tens of thousands for parasitic microorganisms. Leverage of these technologies could reduce this to a manageably short list of candidate vaccines, perhaps numbering no more than a few dozens. Such candidates would then require channeling through a set of subsequent processes including recombinant expression, purification, and testing for immunogenicity and protective efficacy [9].
Hitherto, proteomics has been seen as a primarily analytical science, which combines multidimensional polyacrylamide gel electrophoretic techniques with sensitive biological MS, supported by rapidly growing protein and DNA databases, to effect the high-throughput identification of protein populations from different cell types or cells experiencing different environmental conditions. As we have said, the unambiguously identification of a protein is a prerequisite to their full functional investigation. This identification is usually effected through matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS), which is one of the current analytical methods for linking sequence databases to gel-separated proteins. There are at least two main MALDI-MS identification methods: peptide mass fingerprinting (PMF) and post-source decay analysis. PMF identifies proteins by comparison of experimentally and theoretically derived profiles of proteolytically digested peptides. Because both experimental data and sequence databases are limited, there usually remains some ambiguity with regard to posttranslational modification(s) and intrinsic sequence variation. Moreover, the role of electroblotting and Edman N-terminal sequencing as tools in protein identification should not be overlooked. As proteins derived from the same gene may be largely identical, and might differ only in limited yet functionally important details, the identification of proteins must not only pinpoint numerous proteins en masse but also differentiate between close relatives.
But obviously proteomics is more than a few techniques, however sophisticated. Indeed, it is a cohesive and overarching intellectual environment, replete with ideas, many now beginning to yield advanced, if less established, techniques. The cutting edge of proteomics has much to offer.
Other techniques, such as the yeast two-hybrid system [10], also cower under the proteomic umbrella, but are less relevant to vaccine discovery, and so are excluded from our discussion. Perhaps, the most exciting array of emergent proteomic techniques are the so-called protein arrays [11], where recombinant proteins can be arrayed to study protein-ligand and protein-protein interactions. Based on the rationale that altered abundance or a change in structure of proteins can lead to disease, and although protein arrays are currently more expensive and more technically difficult to produce than nucleotide arrays, protein and antibody arrays are now generating considerable excitement. Alternatively, arrays of protein-specific antibodies can quantitate protein levels, analogous to the detection of mRNA by microarrays [12]. It is to be hoped that as protein arrays become more sophisticated, they will impact on infectious disease research by profiling sera and body fluids to discover prognostic and diagnostic markers of particular infections.
The identification of antigenic or immunogenic proteins as putative whole protein subunit vaccines is a key goal of immunovaccinology. It offers the hope of eliciting significant responses from both humoral and cellular immune systems, far exceeding the efficacy of peptide vaccines, while avoiding potential toxicity problems associated with whole microbe vaccines [13]. Before we continue, however, we must raise a minor caveat: proteomics is, after all, only one part of a much larger postgenomic initiative. While our current focus will be on the role of proteomics in immunovaccinology, it is as well to note that it is a complement, rather than a replacement, for other like-minded technologies, such as genomics and transcriptomics. In order to maximize the potential didactic benefit of reading this review, it should be read in conjunction with other papers which cover other postgenomic techniques as applied to close related areas [12, 13, 14, 15, 16, 17, 18]. In what follows, we will concentrate, on viral, bacterial, fungal, and parasite proteomics, as well as host-pathogen interactions, largely, but not exclusively, within the context of vaccinology. However, we specifically omit discussion of autoimmune disease, cancer, and cancer antigen proteomics. These are primarily host-only proteomics that is clearly beyond our current scope.
Viral proteomics
As we will see in later sections, proteomics of bacterial systems is now well advanced, as cancer proteomics is, a subject we will adumbrate but not describe in great detail. By contrast, direct analysis of viruses has been rather limited. Mass spectrometry has long been used to increase our understanding of structure and function in viral proteins: being used to identify posttranslational modifications and mutants, and characterize individual capsid proteins. For example, VP6, the major structural protein of rotavirus, which makes up its inner capsid, has been studied recently using MALDI-TOF and electrospray ionization mass spectrometry [19]. Emslie et al were able to differentiate serovars of the virus and identify a number of posttranslational modifications, including the N-terminal acetylated methionine and deamidated ASP107. Other mass-based approaches, combined with time-resolved proteolysis (mass mapping), have revealed the dynamic nature of viral particles in solution [20]. More recently, Yao et al have used a novel isotope labelling approach, based on the differential incorporation of 18O, to investigate differences between the proteomes of two serotypes (Ad5 and Ad2) of adenovirus [21].
Most other proteomic studies have examined host-virus interactions. Currently, our understanding of the effects of virus infection on the proteomes of infected cells is poor. Toda et al profiled proliferative B-lymphoblastoid cell lines infected with Epstein-Barr virus using proteomic techniques and identified a spot, corresponding to the 16-kd protein phosphoprotein stathmin, that decreased significantly in immortalized cells [22]. Diaz and coworkers [23, 24] examined ribosomal modifications induced by herpes simplex virus type 1. Comparison of the highly basic ribosomal protein maps from infected and noninfected cells indicated that virus infection induces unusual phosphorylation of proteins of the small ribosomal subunit, including S2 and S3a, and the large subunits, including protein L30. Their most significant observation was the permanent phosphorylation of ribosomal protein S6, which plays an important role in controlling the translation of mRNAs that code for components of the translation apparatus.
In a proteomic analogue of transcriptomic analysis, Rodriguez et al [25] used electrophoresis to examine protein expression patterns in Vero infected with African swine fever virus (ASFV) attenuated strain BA71V and porcine alveolar macrophages cells treated with the ASFV virulent strain E70. The resultant data sets, for noninfected cells, included 177 basic and 818 acidic polypeptides from the macrophage and 1,127 acidic and 271 basic polypeptides from the Vero cell. Comparison of infected and noninfected proteomes indicated that ASFV infection shuts off protein synthesis for 65% of cellular proteins, while a small number of proteins—28 proteins (macrophages) and 48 proteins (Vero cells)—show a greater than 2-fold increase in expression.
Bacterial proteomics
In the last decade, rapid advancements in sequencing technology have lead to the completion of a whole tranche of bacterial genomes. Two main routes to bacterial genomics have been followed. The first was contingent upon the generation of a physical map using cloned genomic fragments in a phage or plasmid library, with the individual cloned fragments then being sequenced and aligned to the physical map. The genome sequence of Escherichia coli was determined in this way [26]. In the second, essentially random fragments of the genome were cloned in plasmid and phage libraries, with the inserts' terminal sequences then determined and the sequenced fragments assembled into the complete genome sequence. This methodology has been used to determine the genome sequences of many other bacteria including Haemophilus influenzae [27], Mycoplasma genitalium [28], Methanococcus jannaschii [29], and Helicobacter pylori [30]. The rest, of course, is history. Presently, the number of completed or partially completed sequencing projects is in the region of hundreds, rather than tens, of genomes [31].
Once determined, analysis of genome sequences using gene prediction programs has identified large numbers of ORFs, many were previously unknown. While, it proved possible to assign functions to proteins encoded by the majority of ORFs on the basis of their homology to extant sequences, a significant number of ORFs show no obvious similarity to genes of known function. As we have said, this has led to the development of many postgenomic strategies, such as proteomics, which seek to determine function. Bacteria have special features, generally lacking in other organisms, for proteomic analysis, that result from the abundance of information on their genomes, their low levels of functional redundancy, their relative simplicity of gene regulation, and their experimental tractability.
Within the context of vaccinology, one of the key goals of postgenomic research is to determine differences between two related microbes, or, more generally, cells, or between the same microbe or cell under different growth conditions. Proteomics approaches to this problem have been applied, with particular success, to bacteria. This work includes the determination of the proteomes for several bacterial species: Salmonella typhimurium [32], Bacillus subtilis [33], and Mycoplasma pneumoniae [34].
In another study, Chlamydia pneumoniae, an obligate intracellular human pathogen that causes acute and chronic respiratory tract diseases, was cultured in Hep-2 cells and proteins from its infectious elementary bodies were separated by two-dimensional gel electrophoresis [35]. Two hundred sixty-three protein spots were extracted in the pH range 3–11, these corresponded to 167 genes (about 15% of the genome) were identified. The proteins identified included 31 hypothetical proteins including several involved in the type III secretion apparatus, an important mediator of virulence amongst intracellular bacteria, and others involved in energy metabolism. In a related study, global gene expression in Chlamydia trachomatis serovars A, D, and L2, each is responsible for a different chlamydial disease, was investigated using proteomics [36]. Seven hundred protein spots were detected, from which 250 proteins, deriving from 144 genes, were identified, again from the elementary body. As well as again identifying proteins associated with the type III secretion system, 25 hypothetical ORFs and 5 polymorphic membrane proteins were also identified. Correlating protein expression with type of serovar suggests ways of tailoring the identification of specific antigens to particular disease states.
In another study on different, but closely related, bacteria, Piechaczek et al examined uropathogenic E coli strain 536 and some of its mutants [37]. Differences in proteins expressed by wild-type E coli as well as mutants 536delta102, 536-21, and 536R3, which differ in the presence or absence of different pathogenicity islands with their genome, were examined using two-dimensional polyacrylamide gel electrophoresis and MALDI-TOF mass spectrometry. The presence of 39 intracellular proteins with markedly different expression in the different strains was determined, of which 34 could be identified using MALDI-TOF-MS. Comparison of the different derivatives indicated that proteomics was an efficient approach to studying global gene expression and that the expression of various proteins including those encoded by many housekeeping genes is affected by the presence of different pathogenicity islands. Malhotra et al analyzed two strains, PAO1 and PD0300, of Pseudomonas aeruginosa to determine proteins that are differentially expressed as a consequence of mucoid conversion, a process implicated in chronic pulmonary infections in cystic fibrosis [38]. Using proteomic methods, they identified 6 proteins more abundant in mucoid strain PD0300, including 2 implicated in alginate biosynthesis (AlgA and AlgD), porin F, and DsbA (a disulfide bond isomerase).
We will now shift our emphasis and restrict our focus to two particular pathogens: Mycobacterium tuberculosis and H pylori. This pair of pathogens is chosen not only as a demonstration of what has been done but also as an example of what might easily be achieved for other bacterial pathogens.
Every day, over 5700 people will die from tuberculosis (TB), a chronic bacterial infection. It causes greater morbidity than any other infectious disease and is the only such disease to be declared a “global emergency” by the World Health Organization, yet it is over 40 years since a novel anti-TB drug was introduced. The intracellular pathogen M tuberculosis, the causative agent of TB, infects about one-third of the world's population, around 1.7 billion people. Although most infected people do not develop active TB, over 8 million people do develop the disease annually. The rapid spread of AIDS, especially in developing countries, has contributed to the recent sudden escalation in TB cases. This problem is exacerbated by the increased spread of antibiotic- or multidrug-resistant strains of M tuberculosis.
One approach to targeting TB is the development of novel antibiotics. For example, in the genomic era, a tranche of new drug targets, including mycobacterial cell wall components, which are vital for bacterial viability, and the metabolic pathways that biosynthesize them, have become available. Vaccines are another important research avenue. Only a few years ago, it was generally accepted that clinical trials of TB vaccines would not occur for at least a decade, yet the first trials are now beginning.
A number of studies, building on early work [39, 40], have begun to build a picture of the TB proteome, and how pathogenic and nonpathogenic strains of TB differ. For example, proteomic approaches can identify novel genes not apparent from automated gene hunting within genome sequences, as has been found for TB [41], where the existence of six ORFs was shown by electrophoresis and MS.
In a ground breaking study, proteomics was used to compare the proteome of two nonvirulent vaccine strains of attenuated Mycobacterium bovis Bacillus Calmette-Guerin (BCG) with two virulent strains of M tuberculosis. M tuberculosis usually resides within the host macrophage, but its mechanisms of survival are poorly understood. Whatever evidence exists suggests that M bovis BCG is both a deletion and regulatory mutant, yet retains the ability to live within the macrophage and is immunoprotective, albeit at a relative low efficacy. This leads to the identification of around 25 different proteins, which are either differentially expressed or modified, from a set of 2600 resolved protein spots out of the 3924 ORFs identified in the TB genome [42]. In a more recent study, the same group has identified a number of putative virulence factors and diagnostic markers of TB as well as interesting candidates for vaccination against tuberculosis [43]. About 1800 distinct protein spots were identified by electrophoresis, of which 56 spots were unique to virulent strains and 40 spots to the attenuated strains. Twelve spots specific for M tuberculosis were identified as proteins previously shown to be missing from M bovis BCG, while 20 M tuberculosis-specific spots were identified as genes not previously thought to be deleted in M bovis BCG.
Some of these differences seen in this last experiment may reflect differences in environment-dependent expression rather than differences between the complete proteome. In order to investigate this, a number of workers have examined the proteome of M tuberculosis and BCG under different conditions. In an early study, Wong et al [44] used proteomics to examine the effect of high and low extracellular iron concentration on the expression of genes in M tuberculosis. The expression of 15 proteins was induced, and the expression of 12 proteins was decreased under low-iron conditions. Mass spectrometry identified 10 proteins including fur and aconitase proteins, both of which are regulated by iron in certain bacterial systems. More recently, Monahan et al [45] have tried to define differences in gene expression during the interaction of BCG with macrophage cell line THP-1. They found that BCG resident within macrophages express different proteins than those expressed during growth in culture or under conditions of heat shock. In particular, they identified six abundant proteins with increased macrophage expression: Rv2623, InhA, GroEL-1, GroEL-2, alpha-crystalline, and elongation factor Tu. In a related study, Betts et al [46] have examined a laboratory model of the latent or “persistent” form of TB that may mimic its nongrowing, drug-resistant persistence in vivo. By using microarray and proteome analysis, they investigated the response of a nutrient-starved M tuberculosis and identified a number of interesting target proteins. In an earlier study, Betts and coworkers analyzed the recent clinical isolate CDC1551 M tuberculosis with laboratory strain H37Rv, which has been subject to in vitro passage, using standard proteomic techniques [47]. Although the two strains demonstrate different in vivo and in vitro phenotypes, visualization of 1750 protein spots indicated that their protein profiles were very similar. Of the 17 protein spot differences, 7 were unique to CDC 1551, 3 to H37Rv, and 2 showed increased expression in H37Rv.
Identification of proteins by a strategy that targets the differences between M tuberculosis and BCG, as well as strains grown under different conditions, will help elucidate the molecular basis of attenuation and the vaccine potential of BCG, as well as identifying TB-specific antigens, virulence factors, and diagnostic biomarkers that can distinguish vaccination by BCG from infection with M tuberculosis. Identification of potential subunit vaccines is greatly facilitated using this spot-the-difference technique or alternative proteomic approaches which focus on the identification of secreted proteins. In either case, it is often necessary to undertake old-style serial experiments where a set of potential antigens is expressed by hand and evaluated as a source of B-cell or T-cell epitopes. The work of Covert et al indicates a rapid, parallel, and facile postgenomic approach to this problem using proteomics to elucidate immunodominant T-cell antigens of pathogenic bacteria [48]. Subcellular protein fractions from M tuberculosis were resolved into 355 and 299 fractions of filtrate and cytosolic proteins. The reactions of splenocytes from C57Bl/6 mice infected with M tuberculosis were used to analyze dominant T-cell responses from these fractions, leading to the identification of 38 immunodominant fractions and 30 corresponding individual proteins. Many of these were previously known antigens, but 17 were novel T-cell antigens.
We now turn to a discussion of H pylori. The human stomach, on the basis of its low pH, has long been considered as an extremely hostile environment for the growth of bacteria. However, this view has changed dramatically with the discovery of the spiral microaerophilic bacterium H pylori from the human gastric mucosa. A report by Langenberg et al [49] began to unravel the mechanism of pathogenicity demonstrated by H pylori, by observing that it could produce large amounts of the virulence factor urease, thus explaining urease activity observed earlier in the mammalian stomach. This understanding, combined with evidence that H pylori causes chronic and acute gastritis, initiated interest into the prevalence and incidence of this bacterial infection. Epidemiological studies are consistent with the view that H pylori causes gastric infection in half the human population worldwide and over 80% of populations from developing countries. The prevalence of H pylori in gastric ulcer disease is greater than 90% and curing infection results in a cure for the gastric ulcer.
The definition of H pylori surface proteins is of particular importance in vaccine discovery. Two-dimensional electrophoresis combined with antibody detection and N-terminal sequencing was used to detect H pylori antigens [50, 51, 52]. Jungblut et al [53] studied H pylori whole cell proteins extensively by 2DE and 152 proteins were identified by MS. A single patient's serum was used to determine antibody reactivity. A small number of antigenic proteins were identified, leading the authors to suggest that several antigens may be minor components in whole cell lysates and therefore beyond detection in the absence of enrichment. Sample fractionation and enrichment of proteins using a chromatographic step prior to electrophoresis improves the identification of proteins at a low expression level. It may also improve the ratio of immunogenic versus nonimmunogenic proteins in a complex antigen preparation. In some studies of H pylori proteins, large pH gradients were used [51, 52, 53, 54] and basic proteins, common in H pylori, may have been poorly resolved. Isoelectric focusing using a more appropriate pH gradient allows greater resolution of proteins, and by 2DE immunoblotting it is possible to identify specific antigenic proteins as well as evaluate complex antigens. More precise identification of such immunogens will be necessary, in order to produce recombinant proteins, using either advanced MS-MS sequencing or more classical N-terminal microsequencing.
In a study designed to directly address the direct identification of vaccine targets, Chakravarti et al analyzed the H pylori genome [55] using both proteomic and genomic approaches. Two different approaches were taken for the identification of a set of potential candidate vaccines. In the first, proteins were identified from outer membrane preparations using proteomic technologies. An outer membrane fraction, purified from disrupted cells, was treated with Triton X-100, centrifuged, treated with detergent, centrifuged again, and then separated by 1D SDS-PAGE. Those proteins are reacting against monoclonal antibodies and are identified by mass fingerprinting. In the second approach, outer membrane proteins were separated by 2DE and transferred to PVDF membrane. Spots were trypsin-digested, and extracted peptides were analyzed by MALDI-TOF-MS. In a complementary study, Haas et al [56] compared the reactivity of sera from H pylori-infected patients, a control group with non- H pylori gastric illness, and patients with gastric cancer to electrophoretically separated proteins from H pylori strain HP 26695. Three hundred ten proteins were recognized by H pylori-positive sera. Notable amongst these were serine protease HtrA (HP1019), Cag3 (HP0522), and the predicted coding region HP0231. In an interesting variant study, McAtee et al examined protein differences between bacterial lysates from H pylori strain 26695, which is resistant to metronidazole (MTZ) due to a mutation in nitroreductases gene, rdxA, grown in the presence and absence of small quantity of MTZ [57]. The expression of a number of proteins decreased by twofold or more during growth with MTZ, yet the levels of various isoforms of alkylhydroperoxide reductase (AHP) (encoded by gene ahpC HP1563 and linked to oxygen toxicity resistance) increased.
Fungal, parasite, and cancer proteomics
In the following section, we briefly adumbrate several areas in eukaryotic proteome research. Two are emerging areas, fungal and parasite proteomics, while the third is relatively well developed, cancer proteomics. In examining the last of this triumvirate, it is difficult to disentangle it from host proteomics, which is clearly beyond the scope of this review. As a consequence, we will touch on the subject only briefly.
Currently, and in contrast to the application of genomic technologies, fungal proteomics is a ripe area for exploitation. Our present understanding of fungal virulence factors is somewhat limited and largely confined to fungi-plant interactions [58]. They may be classified as
(1) toxins and enzymes that degrade host defenses. These can be readily assessed via biochemical assays and were amongst the first virulence factors identified;
(2) elicitors that induce host defenses;
(3) transporters and signal transduction components that protect the fungus from host responses;
(4) signal transduction proteins that aid sensing of the host environment;
(5) penetration effectors, such as melanin or hydrophobins.
The group of fungal virulence factors is still small and is obviously incomplete given the complex lifestyle of pathogenic fungi [59, 60, 61]. Thus the aggressive use of proteomic methods, in conjunction with genome-wide comparisons coupled with transcriptomic expression profiling, will have much to contribute to studies of fungal pathogenesis.
A recent study by Lim et al [62] will illuminate what is possible. Two hundred twenty proteins associated with the cell envelope were extracted from active and quiescent mycelia of Trichoderma reesei. Of these, 56 spots were examined by MS and 20 spots were identified as known proteins on the basis of sequence, indicating that most fungal cell wall proteins are novel. Identified proteins included translation elongation factor beta, diphosphate kinase, disulfide isomerase, outer membrane porin, transaldolase, vacuolar protease A, enolase, and glyceraldehyde-3-phosphate dehydrogenase. However, the most abundant protein in active and quiescent mycelia was HEX1. This is the major protein in Woronin bodies which are only found in filamentous fungi. Future studies will identify genes that specifically determine fungal lifestyle and genes that distinguish between filamentous and single-cell growth. It will also allow genes and pathways involved in pathogenicity to be identified, leading to the identification of further virulence factors, and thus further candidate fungal vaccines.
Parasitic infections are a very common cause of serious disease, particularly in third world countries and amongst domesticated animal populations, engendering a greatly enhanced interest in developing prophylactic vaccines against them [63, 64]. Human vaccines against malaria and other parasites have not been overly successful. However, vaccines able to control the major parasites of livestock have proved more useful [8, 65], particularly those directed against major nematode and trematode infections. Apart from attenuated-live vaccines for the control of avian coccidiosis, toxplasmosis in sheep and anaplasmosis in cattle, vaccines have been developed against Haemonchus contortus, the pathogenic nematode of sheep and goats, and Fasciola hepatica, the liver fluke of sheep and cattle; Bm86 vaccine against Boophilus microplus; 45w and EG95 recombinant proteins against Taenia ovis and Echinococcus granulosis; and broad-spectrum gastrointestinal worm vaccines against Ostertagia and Trichostrongylus species. Vaccines in development include the cathepsin L vaccines against the liver fluke F hepatica, and the H11 vaccine against H contortus.
Jefferies et al [66] analyzed the excretory-secretory proteins from F hepatica using proteomics, identifying a number of proteins including cathepsin L proteases and other enzymes involved in protection from the host immune responses as part of a reactive oxygen detoxification system: superoxide dismutase, thioredoxin peroxidase, and glutathione S-transferases. Interestingly, host superoxide dismutase was the only such protein identified on the gel.
By comparison, molecular vaccines against protozoans are proving considerable more elusive in both animals and humans. This is no where more apparent than in the case of malaria. This disease, caused, in its most severe form, by the protozoan parasite Plasmodium falciparum, has plagued humanity throughout recorded history and results in the death of over 2 million people per year. Other parasitic diseases, such as leishmaniasis and schistosomiasis, are also important diseases in developing countries. Leishmaniasis, in its cutaneous (CL), mucocutaneous (MCL), and visceral (VL) forms, affects directly about 2 million people per year, with about 350 million individuals at risk worldwide. The 35-Mb genome of Leishmania, which should be sequenced late in 2002, contains about 8500 genes that will translate into more than 10000 proteins. Of all vaccines against human parasitic disease, those targeting malaria, leishmaniasis, and schistosomiasis are in the most advanced stages of development. However, despite the remarkable progress made in identifying protective antigens, at present there are no generally accepted vaccines against parasitic diseases. Vaccines for malaria and leishmaniasis have been taken to clinical trials while vaccines for schistosomiasis are in phase I/II trials. The control of leishmaniasis remains a problem and no vaccines exist for the VL, CL, or MCL forms of the disease.
While postgenomic approaches are being pursued actively for Leishmania [67], which combine MicroArray transcriptomics with random vaccine screening using cDNA libraries, relatively little has been done within the proteomic arena. Thiel and Bruchhaus [68] have analyzed the expression specific differences between the proteomes characterizing the promastigotes and amastigotes forms of Leishmania and also the transition between them. They mapped the Leishmania donovani proteome during distinct metamorphic stages, identifying stage-specific proteins and regulons, using isoelectric focusing compatible protocol. Around 400 proteins could be visualized and a significant decrease in protein synthesis during differentiation from promastigotes to amastigotes could be observed.
Toxoplasma gondii is another protozoan parasite that has been investigated using proteomic technology. There are two forms of T gondii associated with human hosts. The rapidly growing tachyzoites give rise to acute illness and the slowly dividing encysted bradyzoites can remain dormant within tissues for a lifetime. During infection, conversion occurs between the rapidly dividing tachyzoite stage (responsible for acute toxoplasmosis) and the much more slowly replicating bradyzoite, a process central to both pathogenesis and longevity of infection. Proteomics has helped identify several proteins specific to these different stages.
Cohen et al [69] analyzed proteins expressed during the tachyzoite stage of T gondii and separated over 1000 proteins in the pH ranges 4–7 and 6–11. Because the genome was not available in full, they were obliged to combine their proteomic approaches with searches of EST databases in order to identify proteins less equivocally. Many protein spots were encoded by the same gene, indicating that posttranslational modification and alternative splicing are common features of gene expression in T gondii. In a similar study, Dlugonska et al [70] analyzed a lysate of the tachyzoite stage of T gondii and separated 224 proteins. They could identify 14 proteins using mass fingerprinting including the excretory dense granule proteins GRA1-GRA8, S16/acid phosphatase, nucleoside triphosphate hydrolase, and the H4 protein, and two secreted antigens p36 and p40 were identified.
Proteomic analysis of host-pathogen interactions
The story of the proteomic analysis of host-pathogen interactions is the story of a series of dichotomies, which is to say that we can partition the subject into a bifurcating series of binary divisions. One division is between the nature of target cells (antigen presenting cells versus T cells), another is between the nature of stimulation used to engender changes in gene expression within target cells (bacterial or viral infection versus isolated immunomodulators, such as LPS). In the following section we will briefly review a number of studies addressing these issues. Each highlights a different theme or aspect relevant to the development of proteomic immunovaccinology. We begin with alterations apparent in gene expression within a small number of bacterial systems.
Fletcher et al investigated the effect of environmental factors on the expression and release of secreted or surface proteins, containing many virulence factors, from Actinobacillus actinomycetemcomitans, a bacteria implicated in periodontal diseases, where gum inflammation is associated with bone loss and gum recession leading to the formation of a so-called periodontal pocket [71]. Differences in expression of many proteins, including glycolytic enzyme triose phosphate isomerase, were observed for bacteria grown under varied conditions (anaerobic versus aerobic growth, biofilm versus planktonic growth, under iron depletion, or in the presence or absence of serum or blood), indicating its adaptability to changes within the periodontal microenvironment. Monahan et al analyzed changes in protein expression in attenuated vaccine strain M bovis BCG induced by host macrophage phagocytosis [72]. They used proteomics to show that BCG phagocytosed by the human macrophage cell line THP-1 expresses proteins not seen during heat shock or growth in culture media, and were able to identify six proteins showing increased expression: 16 kd alpha-crystalline (HspX), GroEL-1 and GroEL-2, a 31.7-kd hypothetical protein (Rv2623), InhA, and elongation factor Tu (Tuf).
We now turn to proteomic changes in antigen presenting cells and begin with the inverse experiment to that performed by Betts. Ragno et al combined transcriptomic and proteomic methods to evaluate changes in gene and protein expression in the leukaemic macrophage cell line THP-1 after infection with TB [73]. Initially, microarrays of 375 immunologically implicated human genes identified a set of early upregulated proteins that not unsurprisingly included a range of chemokines and cytokines, as well as other cell surface molecules. It was more difficult to detect changes using proteomics, although human IL-1beta and superoxide dismutase were shown to have increased expression after infection, and, in contrast, the heat-shock protein hsp27 was downregulated. In a similar study, Kovarova et al analyzed phagosome extracts from macrophages derived from host organisms resistant or susceptible to infection by Francisella tularensis LVS (live vaccine strain) [74]. They identified several proteins upregulated in susceptible macrophages including host proteins mitochondrial ATP synthase beta-chain and NADH-ubiquinone oxidoreductase as well as the bacterial 60-kd chaperonin GroEL and a hypothetical 23-kd protein, whose expression level correlate with susceptibility and F tularensis LVS pathogenicity. Pizarro-Cerda et al examined the molecular components that facilitate cellular uptake of Listeria monocytogenes into the phagosome in the human epithelial cell line LoVo using proteomics [75]. Their results confirmed literature precedents, with the exception of MSF, a member of the septin family of GTPases, which forms filaments that colocalize with the actin cytoskeleton in quiescent cells.
Moving now from cells presenting antigen to cells mediate immune recognition, we focus now on T cells. Truffa-Bachi et al used protemics to analyze the changes contingent on the removal of Concanavalin A or Cyclosporin A from cultures of activated murine T cells [76]. They found that a large number of proteins were strongly upregulated and downregulated after the immunosuppressive drugs were removed, indicating that T cells were programmed by Cyclosporin A to change expression levels without reactivation. In the context of developing a proteomic database of helper T cells, Nyman et al activated CD4+ T cells with anti-CD3+ anti-CD28 antibodies and visualized 2000 spots with autoradiography and 1500 spots using silver staining and identified 91 proteins using mass fingerprinting [77]. By using proteomics, Fratelli et al sought to identify T-cell proteins that undergo glutathionylation, the formation of mixed disulfides between glutathione and other proteins, under conditions of oxidative stress [78]. They observed several proteins not previously known to be glutathionylated, including enzymes, such as enolase (which is inhibited by glutathionylation); redox enzymes, such as peroxiredoxin 1 or cytochrome c oxidase; cytoskeletal proteins, such as vimentin, profilin, and actin; cyclophilin (which is not inhibited by glutathionylation); stress proteins, such as HSP60 and HSP70; and a number of miscellaneous proteins, such as galectin and fatty acid binding protein. The authors felt that their results supported the view that glutathionylation is a common global mechanism for the regulation of protein function.
INFORMATICS OF THE IMMUNOME
Informatic support for proteomics is now well established, and it would be futile to reiterate the content of many useful reviews on the subject (see [79, 80, 81, 82] and references therein). Equally software for the analysis and exploration of proteomics is now well developed and widely distributed. Indeed, online databases of proteomes or collections of proteomes have now proliferated. However, the informatic analysis of the immunome is currently less well developed. In many ways the informatic analysis of the imunome is the complement of the experimental analyzes described above. The immunome, the complement of short immunogenic peptides derived, by the complex, poorly understood molecular machinery of the immune system, from the proteome of some microbe is itself a subset of the peptidome. The peptidome is the set of all peptides, as opposed to proteins generated by the cell. It is composed of both genomic peptides, with a specific function, such as hormones or neuropeptides, and cleavage products generated by proteases. In some respects, it lies somewhere between the proteome and metabolome of small biosynthesized molecules and is highly compartmentalized within the cell. Bioscience is only now beginning to explore the peptidome. Because experimental methods do not address either the peptidome or immunome, informatic prediction has much to contribute here.
Approaches to predicting the immunone
A specialized type of immune cell mediates cellular immunity, the T cell, which constantly patrols the body searching out proteins that originate from a pathogenic organism, be that virus, bacterium, fungus, or parasite. The surface of T cells is, unsurprisingly, enriched in TCRs, which function by binding MHCs expressed on the surfaces of other cells. These proteins bind small peptide fragments derived from both host and pathogen proteins. It is the recognition of such complexes that lies at the heart of the cellular immune response. These short peptides are referred to as epitopes. The overall process leading to the presentation of antigen-derived epitopes on the surface of cells is a complicated, and not yet fully understood, process. There are many alternative processing pathways, but we will confine our attention to the two major types: class I and class II.
Class I MHCs are expressed by almost all cells in the body. They are recognized by T cells whose surfaces are rich in CD8 coreceptor protein. Class II MHCs are only expressed on the so-called “professional antigen presenting cells” and are recognized by T cells whose surfaces are rich in CD4 coreceptors. Class I peptides are ultimately derived from intracellular proteins, such as viruses. These proteins are targeted to the proteasome, which cuts them into short peptides of 8 to 11 amino acids in length. These peptides are then bound by the transmembrane peptide transporter TAP, which translocates them from the cell cytoplasm to the endoplasmic reticulum where they are bound by MHCs. Theoretical analyzes of proteasomal cleavage patterns have been conducted by several groups [83, 84], leading in turn to a number of prediction methods [85], some of which are available via the Internet [86, 87]. The amount of data studied remains relatively small, and the predictive power possessed by these different methods has yet to be evaluated objectively. Nonetheless, they represent useful contributions and important starting points for future study. Likewise, studies have also been conducted on the peptide substrate specificities of the TAP transporter [88], leading to the development of predictive models [89] for the determination of peptides that bind to TAP. Together, studies on proteasomal cleavage and TAP transport represent a good first attempt to produce useful, predictive tools for the processing aspect of class I restricted epitope presentation.
For class II, receptor-mediated ingestion of extracellular protein derived from a pathogen is targeted to an endosomal compartment, where the proteins are cleaved by cathepsins, a particular class of protease, to produce slightly longer peptides of 15–20 amino acids. Class II MHCs then bind these peptides. The peptide specificity of protein cleavage by cathepsins has also been investigated and simple cleavage motifs are well known [90]. However, more precise investigations are required before accurate predictive methods can be realized. The first attempts to computerize the identification of MHC binding peptides led to the development of motifs characterizing the peptide specificity of different MHC alleles. Such motifs—a concept with wide popularity amongst immunologists—characterize a short peptide in terms of dominant anchor positions with a strong preference for certain amino acids. Probably the first proper attempt to analyze MHC binding in terms of specific allele-dependant sequence motifs was undertaken by Sette et al [91]. They defined motifs for the mouse alleles I-Ad and I-Ed after measuring affinity for a large set of synthetic peptides originating from eukaryotic and prokaryotic organisms, as well as viruses; in addition they also assayed a set of overlapping peptides encompassing the entire staphylococcal nuclease molecule. Sette et al quote prediction rates at the 75% level for these two alleles. A large number of succeeding papers, both from this group and others, have extended this approach to many other human and mouse alleles.
As we have said, these motifs are usually expressed in terms of anchor residues: the presence of certain amino acids at particular positions that are thought to be essential for binding. For example, human class I allele HLA-A*0201, probably the best studied of all alleles, has anchor residues at peptide positions P2 and P9 for a nine amino acid peptide. At P2, acceptable amino acids would be L and M, and at the P9 anchor position would be amino acids V and L. Secondary anchors, residues that are favourable, but not essential for binding, can also be present. Moreover, sequencing of peptides, that are known to bind, show preferences for particular amino acids at particular positions, although whether this represents anything other than the inherent bias in protein sequences is seldom addressed. The method is admirably simple: it is easy to implement either by eye or more systematically using a computer to scan through protein sequences.
However, there are many problems with the motif approach. Although it is possible to score the relative contributions of primary and secondary anchors to produce a rough and ready measure of binding affinity [92, 93], the most significant problem with the motif approach is that it is, fundamentally, a deterministic method. A peptide is either a binder or is not a binder. Even a brief reading of the immunological literature shows that matches to motifs produce many false positives, and are, in all probability, producing an equal number of false negatives, though peptides predicted to be nonbinders are seldom screened.
While useful in themselves, binding motifs are, as we have said, very simplistic. They are not quantitative and their over-reliance on anchor positions can lead to unacceptable levels of false positives and false negatives. Alternative approaches abound and have different strengths and different weaknesses. The strategy adopted by many workers is to use data from binding experiments to generate matrices able to predict MHC binding. For want of a better term, we refer to these approaches as experimental matrix methods, as most such methods use their own measured data and relatively uncomplicated statistical treatments to produce their predictive models.
A step forward from deterministic motifs came with the work of Kenneth Parker [94]. This method, which is based on regression analysis, gives quantitative predictions in terms of half-lives for the dissociation of β2 -microglobulin from the MHC complex. It is founded on a series of important observations about peptide binding to MHC molecules [95, 96, 97, 98, 99, 100, 101, 102] and has been used in a number of applications [103, 104]. Moreover, apart from its intrinsic utility, one of the other important contributions of this approach is that it was the first to be made available on-line (http://bimas.dcrt.nih.gov/molbio/hla_bind). This method, often referred to as BIMAS, or occasionally, COMBIFORM, by immunologists, is, for this reason, widely used. Other empirical methods include EpiMatrix and EpiMer developed by DeGroot and coworkers and TEPITOPE developed by Hammer and colleagues.
A number of groups have used techniques from artificial intelligence research, such as artificial neural networks (ANNs) and hidden Markov models (HMMs), to tackle the problem of predicting peptide-MHC affinity. ANNs and HMMs are, for slightly different applications, the particular favourites when bioinformaticians look for tools to build predictive models. However, the development of ANNs is often complicated by several adjustable factors whose optimal values are seldom known initially. These can include, inter alia, the initial distribution of weights between neurons, the number of hidden neurons, the gradient of the neuron activation function, and the training tolerance. Other than chance effects, neural networks have, in their application, suffered from three kinds of limiting factors: overfitting, overtraining (or memorization), and interpretation. As new, more sophisticated neural network methods have been developed and statistics has been applied to their use, overfitting and overtraining have been largely overcome. Interpretation, however, remains an intractable problem; few, if any, can easily visualize or interpret the very complex weighting schemes used by neural networks.
Notwithstanding these potential problems, many workers have adopted an ANN strategy in seeking to solve the prediction of peptide-MHC binding. Bisset and Fierz [105] were amongst the first to use ANN in this context. They trained an ANN to relate binding to the class II allele HLA-DR1 to peptide structure and reported a correlation coefficient of 0.17 with a statistical significance of P = .0001. Amongst the best known names of those interested in the area of MHC binding prediction is Vladimir Brusic. Over many years, he and his coworkers have developed a range of artificial intelligence techniques, including, inter alia, ANN, HMMs, and evolutionary algorithms, aimed at solving problems of this kind [106, 107, 108, 109]. His work contains models of both class I and class II MHC alleles, as well as the TAP transporter [88, 89], and within the context of his own classification scheme [110], his models seem highly predictive.
A quite different approach to obtaining predictions of peptide is that MHC binding is based on atomistic molecular dynamic simulations. It attempts to calculate the free energy of binding for a given molecular system, which is closely related to experimentally observable quantities such as equilibrium constants or IC50s. It has the advantage that, in principal, there is no reliance on known binding data, as it attempts the de novo prediction of all relevant parameters given certain knowledge of the system. Essentially, all what is required is the experimentally determined structure, or a convincing homology model, of an MHC peptide complex.
DeLisi and coworkers were among the first to apply molecular dynamics to peptide, MHC binding, and have, subsequently, developed a series of different methods [111, 112, 113]. Part of this work has concentrated on accurate docking using molecular dynamics and another part on determining free energies from peptide MHC complexes. Didier Rognan has, over a long period, also made important contributions to this area [114, 115, 116]. In his work, dynamic properties of the solvated protein-peptide complexes, such as atomic fluctuations, solvent accessible surface areas, and hydrogen bonding patterns correlated well with available binding data. He has been able to discriminate between binders that remain tightly anchored to the MHC molecule and nonbinders that are significantly weaker. Other work in this area has come from two directions. The first direction is interested in using the methodology to analyze and predict features of peptide-MHC complexes. These methods have looked at both class I [117, 118] and class II [119]. The second direction is more interested in developing novel aspects of molecular dynamics (MD) methodology, including both simulation methodology [120] and solvation [121], and using the MHC peptide systems as a convenient example of binary molecular complex.
Quantitative approaches to predicting the immunone
In this section, we review quantitative approaches to the developing field of computational immunovaccinology. This includes our own contribution, including a discussion of our newly released JenPep database and two powerful new techniques for T-cell epitope prediction. The first is a 2D quantitative structure-activity relationships, or 2D-QSAR, approach which we have christened the “additive” method [122]. The other is a 3D-QSAR approach, based on comparative molecular similarity indices analysis (CoMSIA) [123, 124]. The methods were prototyped using the common class I allele, HLA-A∗0201, for which numerous binding data is available.
Virtual screening
A methodology closely related to MD, both being based, to a large degree, on molecular mechanics force fields, or, at least, drawing on analogies from pairwise atomistic potential energy functions, is a set of techniques grouped loosely under the name of “virtual screening.” There are two principal types of virtual screening methodology that have, thus far, been applied to the prediction of MHC binding. One derives from computational chemistry and the other from structural bioinformatics and the development of tools for fold prediction. Virtual screening is an expression derived from pharmaceutical research that is the use of predicted ligand-receptor interactions to rank or filter molecules as an alternative to high-throughput screening. Approaches to virtual screening cover a spectrum of methods which vary in complexity from molecular descriptors and QSAR variables, through simple scoring functions (such as Ludi, FlexX, Gold, or Dock), potentials of mean force (PMF) (such as Bleep), force field methods, QM/MM and linear response methods, to free energy perturbations. In this transition from, say, atom counts through to full molecular dynamics, we see a tremendous increase in required computer time. Virtual screening can be seen as seeking a pragmatic solution to the “accuracy gained” versus “time taken” equation. The point at which one stops on this spectrum is contingent upon the system being evaluated, the number of peptides being evaluated, and the computing resources available.
Didier Rognan has developed a virtual screening method called FRESNO and applied this algorithm, which relies on a simple physicochemical model of host-guest interaction, to the prediction of peptide binding to MHCs [125]. This model was trained on a combination of data and experimentally derived 3D structures from the alleles HLA-A∗0201 and H-2Kk. He found that lipophilic interactions contributed the most to HLA-A∗0201-peptide interactions, whereas H-bonding predominated in H-2Kk recognition. Cross-validated models were afterward used to predict the binding affinity of a test set of 26 peptides to HLA-A∗0204 (an allele closely related to HLA-A0201) and of a series of 16 peptides to H-2Kk. He concluded from their initial study that their scoring function was able to predict, with reasonable accuracy, binding free energies from 3D models. In a more comparative study [126], Rognan and colleagues found that for predicting the binding affinity of 26 peptides to the class I MHC molecule HLA-B∗2705, FRESNO outperformed six other available methods (Chemscore, Dock, FlexX, Gold, Pmf, and Score).
Turning now to bioinformatic-based approaches, others are using amino acid pair potentials, initially developed to predict the fold of a protein, to identify those peptides which will bind well to an MHC. Margalit and colleagues have proposed a number of virtual screening methodologies [127, 128], each is of increasing complexity. They used amino acid pair potentials, originally developed by Miyazawa and Jernigan [129], to evaluate the interprotein contact complementarity between peptide sequences and MHC binding site residues. They presented an analysis of peptide binding to four MHC alleles (HLA-A2, HLA-A68, HLA-B27, and H-2Kb), and were successful in predicting peptide binding to MHC molecules with hydrophobic binding pockets but not when MHC molecules with charged or hydrophilic pockets were investigated. Again focussing on class I alleles, a more recent study from this group [130] used an updated set of statistical pairwise potentials. These were developed from the Miyazawa and Jernigan potential by Betancourt and Thirumalai [131] and described the hydrophilic interactions more appropriately. This enables more accurate modelling of the threading of the candidate peptide sequence.
Because of the relative celerity of virtual screening methods compared with MD methods and its ability to tackle MHC alleles for which no known binding data is available, this method has considerable potential. While both MD and related methods hold out the greatest hope for such true de novo predictions of MHC binding, their present success rate is very much lower than that of data driven models.
Positional scanning peptide libraries
An alternative strategy is the use of positional scanning peptide libraries (PSPLs) to generate such matrices. A number of such studies have been conducted. Some are aimed at investigating the problem of MHC-peptide interaction [132, 133, 134], while others concern themselves with evaluating how variations in peptide sequence contribute to TCR recognition and T-cell activation [135, 136]. One of the most recent of these is also one of the most promising; Udaka et al [137] have used PSPLs to investigate the influence of positional sequence variation on binding to the mouse class I alleles Kb, Db, and Ld. From their analysis, a program that could score MHC-peptide interaction was developed and used to predict the experimental binding of an independent test set. Their results showed a good linear correlation but with substantial deviation. About 80% of peptides could be predicted within a log unit.
QSAR approaches
JenPep
Version 1.0 of JenPep [138] is composed of three subdatabases: (i) a compilation of quantitative affinity measures for 6000 peptides which bind class I and class II MHC; (ii) a compendium of 2300 dominant and subdominant T-cell epitopes; and (iii) a set of quantitative data for 400 peptide binding to the TAP peptide transporter. The database, and an HTML interface for searching, is freely available via the Internet. It can be found at http://www.jenner.ac.uk/JenPep. JenPep contains binding data on a wide variety of different MHC alleles; for class I MHC molecules, JenPep has data for 68 different restriction alleles with more than 50 genotype variations. For class II MHC molecules, there are over 40 restriction alleles with 52 genotype designations. Peptide lengths for class I MHC molecules are in the range of 7–16 residues and for class II MHC molecules are in the range of 9–35 residues. Measures of binding affinity include radiolabelled and fluorescent IC50 values, BL50, and half-lives. JenPep is the first database in immunology to concentrate on quantitative measurements, complementing existing systems. This compilation of binding data underlies our attempts to derive statistically sound QSAR tools for the accurate prediction of peptide binding to immunological molecules.
A 2D-QSAR method for binding affinity prediction
We have developed predictive techniques based on the so-called additivity concept, whereby each substituent makes an additive and constant contribution to the biological activity regardless of variation in the rest of the molecule. The IBS hypothesis, developed by Parker [94], is the immunological analogue of this idea. We extended this concept by adding additional terms that account for near neighbour side-chain interactions [122]. The binding affinity of a peptide will depend on contributions from each amino acid as well as interactions between adjacent and every second side-chain:
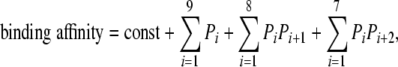 |
(1) |
where the const accounts, at least nominally, for the peptide backbone contribution, ∑9i=1 Pi is the sum of amino acids contributions at each position, ∑8i=1PiPi+1 is the sum of adjacent peptide side-chain interactions, and ∑7i=1 PiPi+2 is the sum of every second side-chain interactions.
Four hundred twenty IC50 values for 340 nonamer peptides were used in the development of the additive method. The peptide sequences and their binding affinities to the HLA-A∗0201 molecule were extracted from the JenPep database. More than one IC50 value was found for some of the peptides. As is common practice amongst QSAR practitioners, IC50 values were converted to P-units (negative decimal logarithm).
A program was developed to transform the nine amino acid peptide sequence into a row of a table. A term is equal to 1 when a certain amino acid at a certain position or a certain interaction exists, and equal to 0 when they are absent. Thus a matrix of 420 rows and 6120 columns was generated. One hundred eighty columns account for the contributions of the amino acids (20 amino acids × 9 positions), 3200 for the adjacent side-chains, or 1–2 interactions (20 × 20 × 8), and 2800 for the 1–3 side-chain interactions (20 × 20 × 7). To reduce the number of columns, the program omits columns that contain only zeros. The final matrix consists of 420 rows and 2158 columns.
As the columns are more numerous than the rows, the equations were solved using partial least square (PLS) method. The predictive power was assessed by the cross-validated q 2 (as generated by “leave-one-out” cross-validation [LOO-CV]), standard error of predictions (SEP), and residuals between the experimental and predicted by LOO-CV PIC50 values. A mean |residual| value and standard deviation for the set were also calculated. The non-cross-validated model was assessed by multiple linear regression (MLR) parameters: explained variance (r 2), standard error of estimate (SEE), and F ratio.
The final equation derived by the additive method consists of 1815 terms including the constant. It contains the contributions of the amino acids and the contributions of the significant side-chain interactions. There were 172 very well-predicted (residuals ≤ |0.5| log unit) peptides (50.5%), 128 well-predicted (|0.5| ≤ residuals ≤ |1.0| log unit) peptides (37.5%), and only 41 poorly predicted (residuals > |1.0| log unit) peptides (12.0%).
A 3D-QSAR method for binding affinity prediction
One of the most reliable methods for investigating the structure-activity trends within sets of biological molecules is 3D-QSAR. The explanatory power of 3D-QSAR methods is considerable, manifests not only in their ability to accurately predict binding affinities, but also in their capacity to display advantageous and disadvantageous interaction potential mapped onto the structures of molecules being investigated. We have applied the 3D-QSAR method (CoMSIA) to gain an understanding of the relationship between physicochemical properties (steric bulk, electrostatic potential, local hydrophobicity, hydrogen-bond donor, and hydrogen-bond acceptor abilities) and the affinities of peptides that bind to the MHC molecule HLA-A*0201 [123, 124].
Two hundred sixty-six nonamer peptides are included in the CoMSIA study. Their IC50 values were collected from the JenPep database and converted to P-units. All molecular modelling and QSAR calculations were performed using the sybyl 6.7 molecular modelling software. The X-ray structure of the nonameric viral peptide TLTSCNTSV was used as a starting conformation. The structures of the remaining peptides were built to this conformation. The partial atomic charges used in CoMSIA were computed using the AM1 semiempirical method, as available in MOPAC.
Five types of similarity index (steric, electrostatic, hydrophobic, and hydrogen-bond donor and acceptor) were calculated, using a common probe atom with 1 Å radius, charge +1, hydrophobicity +1, hydrogen-bond donor and acceptor properties +1. SEP, q 2, and residuals assessed the predictive power of the final model. The initial CV model had low q 2 and r 2 values. This result was not surprising, given the great diversity of peptides collected from a variety of sources. One hundred fifty-one were very well predicted, 83 were well-predicted peptides, and only 32 peptides were poorly predicted. The mean |residual| was 0.553. The model was improved by excluding a limited number of poorly predicted peptides in a stepwise manner, beginning with the peptide with the highest residual. The final CV model had significantly higher parameter values: q 2 = 0.683 at 7 components and r 2 = 0.891. This model was used to predict the binding affinities of the excluded peptides. The predictions were better for both the group of very well-predicted peptides and the group of poorly predicted peptides.
Comparison of the two methods in the context of peptide structure
It has long been known that all nine side-chains of the bound peptide contact the HLA-A∗0201 molecule and influence the energetics of binding. The antigen-binding groove has a 30-Å long surface accessible to a solvent probe. There are six pockets in the surface denoted by A through F. Some of them are nonpolar and can form hydrophobic contacts, while others contain polar atoms and can make hydrogen bonds with the side-chains. As statistical approaches, the additive method and CoMSIA seek to correlate relative differences in discriminating molecular descriptor values to a dependent property (eg, the binding affinity). In that respect, CoMSIA is a method able to map similarities or dissimilarities between molecules. The additive method is able to quantify the contributions made to the binding affinity by each amino acid, at each position, and by the interactions between them. Comparing the results of the additive method and CoMSIA, we have found a remarkable degree of congruence.
Positions within the peptide are defined as P1 to P9. CoMSIA suggests that hydrophobic steric bulk with negative potential is well tolerated at P1. Topologically, P1 corresponds to pocket A. The most suitable amino acids for this position seem to be Phe and Tyr. According to the additive method, Tyr is the favourite amino acid for P1. Phe and Lys also make positive contributions, while Arg, His, and Thr are not preferred. The steric map at P2 indicates that long side-chains such as Leu, Ile, and Met are well tolerated here. The additive method distinguishes two favourite amino acids for this position (Met and Leu). Ala, Cys, Gly, and Thr make negative contributions.
Hydrophobic volume with negative potential is preferred at P3. The side-chains of the amino acids at this position fall into pocket D. The hydrogen bonding ability map indicates that amino acids able to form hydrogen bonds will also be well accepted here. Tyr and Trp have the greatest positive contributions for this position, but Leu and Phe are also well accepted. Glu, Cys, His, Pro, and Ser negatively contribute. Short hydrophilic amino acids able to form hydrogen bonds are well tolerated at P4. Ser or Thr would be well tolerated here. According to the additive method, there is no favourite amino acid at P4. Gly, Pro, Ser, and Thr are well accepted here while Ile, Phe, Cys, and Met make negative contributions.
The maps indicate that amino acids with hydrophobic, branched or aromatic side-chains ending with small hydrophilic groups are well tolerated at P5. Favourite amino acids for P5 are Phe and Tyr. His, Leu, and Trp also positively contribute, while Arg should be avoided at this position. Amino acids with long hydrophobic side-chains are preferred at P6. Hydrogen-bond ability is an additional priority. Ile, Leu, Thr, and Tyr are well accepted here. Ala, Arg, Asp, Gln, His, and Lys negatively contribute. This side-chain falls into pocket C. This pocket is predominantly polar, which explains the acceptance of the hydrophilic Thr and Tyr, but it cannot explain the preference for the hydrophobic Ile and Leu. Short side-chains are favoured sterically at P7. The side-chain at P7 falls into pocket E. Pro is the favourite amino acid for this position according to the additive method, although His also makes a good contribution. Asn, Arg, Gln, Gly, Ser, and Thr are deleterious.
The side-chain at P8 should be short, with a hydrophobic core and an end capable of forming hydrogen bonds. Gln, Phe, Pro, and Ser are all well accepted here. The presence of Asp, Ile, His, Met, or Val is deleterious. Amino acids with hydrophobic, short side-chains are required for P9. Val is the favourite amino acid here. Interestingly, a small hydrophilic area, carrying negative potential, appears near P9, which is due to the Thr introduced here by the intermediate binder MLQDMAILT and the high binder YMLDLQPET. However, according to the additive method, Ser and Thr should be avoided.
Predicting subcellular location
There are obviously many other aspects to computational vaccine design other than the prediction of potential epitopes. Many of them are as yet only poorly developed. While we have seen that T-cell epitope prediction is now well developed, at least to the stage where it is beginning to become useful, the prediction of immunogenicity, particularly for subunit vaccines, which necessarily involves a deeper understanding of host responses, remains primitive. The prediction of antibody- or B-cell-mediated antigenicity is at an even more primitive stage [139, 140]. This relies on concepts of some antiquity [141, 142, 143] and quite simplistic software [144, 145]. However, some other techniques complementary to the prediction of host responses, such as the prediction of the subcellular location of potential antigen proteins, have reached a greater level of maturity. A prevailing hypothesis, amongst many, involved directly in the hunt for protective antigens is a belief that the majority of such immunogens will be secreted proteins. Proteomics can help in the systematic search for secreted proteins [146, 147]. This is also an area where computational techniques can produce direct results.
Consider a microbial genome or, more specifically, a bacterial genome. The total protein complement—say a few thousand gene products—is distributed between the inner and outer compartments of the bacteria. Some will reside in the cytoplasm, some will find their way to the periplasmic space, at least in Gram-negative bacteria, and others will be secreted from the cell. Some proteins will become integral membrane proteins located in the inner or outer membranes and some will become lipoproteins. An ability to predict these locations would be a great benefit when choosing which proteins to investigate as candidate vaccines; a secreted protein, for example, can be regarded, at least naively, to be a more likely target than, say, a cytoplasmic enzyme. A number of bioinformatic methods have been developed which address the prediction of subcellular location which has proved to be more complex than was originally envisaged.
In 1982, a strong link between amino acid composition (eg, Leu and Trp favoured, Pro disfavoured [148]) and cellular location was identified [149], but as the number of available protein structures increased, this relationship has become more blurred [150]. Despite the ambiguous relationship between amino acid composition and subcellular localization, many methods of increasing sophistication have been created that exploit this connection [151, 152, 153]. Nakashima and Nishikawa [154] describe a method where the average amino acid composition for a number of proteins, whose subcellular localization is known, was calculated. From these simply obtained results, trends in amino acid composition were observed such as intracellular proteins relatively rich in aliphatic residues. Just using basic rules like these, they were able to correctly identify 78% of the test set as being either intracellular or extracellular.
This idea was developed further by Andrade et al [155] who hypothesized that throughout evolution, each subcellular location has maintained a characteristic physio-chemical environment. The proteins in each location would have adapted to the environment and therefore each location would have proteins with signature structural characteristics. These characteristics are more likely to manifest at the surface (which is exposed to the environment) and therefore the surface residue composition is likely to give a very strong identification of the subcellular location. This method predicted 77% of protein locations accurately. Although amino acid composition is correlated with subcellular location, the former cannot be exclusively defined by the later. Neural networks have also been applied to this problem [156] and are the basis of the NNPSL web-based server. This provided an accuracy of 81% for prokaryotic prediction but only 66% for eukaryotic. This seems likely to be due to the persistent neural network shortcoming of overfitting to training data especially when the variables are complex.
The majority of methods for predicting localization are based on protein sorting signals [157]. These signals are normally represented as a short sequence with variable levels of conservation. Many are represented as well-defined motifs while others show vague sequence features that are undetectable by simple homology searching [158]. The most obvious protein sorting signal to investigate is the signal peptide. Looking at a simple bacterial model, if a protein has a signal peptide but no transmembrane domain, then it will be excreted through the inner membrane. If a protein with a signal peptide has a transmembrane domain, then it will become inserted into the membrane [159]. All signal peptides have a 3-region structure, the amino (N), the hydrophobic (H), and the carboxy (C) with a weak consensus pattern specifying the cleavage site [160]. Signal peptides are divided into classes on the basis of variation of structure of the N, H, and C regions, structure of the cleavage site, and different propensities for amino acids [161].
Many approaches have been taken to try and predict subcellular location from signal peptides and cleavage variations. The different amino acid propensities of N, H, and C regions for different classes can be identified by multivariate analysis of the individual amino acids [162]. A wide range of characteristics of amino acid properties has been determined, and the similarities/dissimilarities in the property profiles for different signal peptide classes were compared. Initially this method was applied just to E coli with some success but later expansion to Gram-positive bacteria was less successful and varied greatly from species to species [163]. Though, there were some factors such as charge, length, side-chain hydrophobicity, and volume that proved reasonably reliable factors that could be used as part of possible new techniques. The prediction of cleavage sites and inference of subcellular location has proved more fruitful than amino acid composition-based methods, with prediction as high as 96% [164, 165].
DISCUSSION
Prophylactic vaccination has made an essential contribution to the improvement of human health over the 20th century. However, we still lack efficient vaccines against major human diseases such as malaria or tuberculosis. Historically, at least in the area of parasite vaccinology, as in many other areas of the subject, one of the greatest problems has been the scarcity of relevant material and our concomitant inability to generate purified vaccine candidates using conventional protein chemistry. Proteomic and other postgenomic and molecular biology approaches, through the preparation of cDNA expression libraries, are now proving central to the identification of immunogenic proteins.
Preceding sections have addressed two approaches to the identification of the immunome: experimental proteomic analysis of microbial proteins and mechanistic informatic prediction. However, the present review is by no means exhaustive, nor does it pretend to be. We have not specifically addressed other important uses for proteomics within vaccine research, such as the systematic discovery of adjuvants and diagnostic and prognostic biomarkers. Rather, we have to suggest how computational strategies and experimental proteomic approaches are highly complimentary to the aim of identifying the immunome. In particular, proteomics will prove crucial in the correct identification of appropriate posttranslational modification and conformation, upon which the immunogenicity of many vaccines will depend. Various informatics strategies hold out the hope that they will be able to short cut some of the more intractable experimental procedures by quickly prioritizing candidate genes.
As we have shown, experimental proteomics can identify proteins that represent potential candidate vaccines. It can achieve this either by identification of highly expressed genes or proteins secreted from the cell. The discovery of potential virulence factors or antigens is achieved by comparing the proteomes of virulent and avirulent microbes, or microbes grown under different conditions, or changes apparent upon infection, or by identifying proteins that are coregulated with already known virulence genes. For example, identification of proteins by such strategies may help elucidate the molecular basis of the attenuation of BCG, and may provide antigens that distinguish infection with M tuberculosis from vaccination with BCG. Proteomics can also help trace out how pathogenic bacteria cope with the challenges imposed on them by therapy or host responses to infection. Generally, however, proteomics will only form part of large postgenomic strategies, incorporating many other techniques. Appropriate use of this technology should allow us to reduce the large number of protein products within the proteome down a much more manageable short list of candidate vaccines, perhaps numbering no more than a few dozens. Such candidates would then require subsequent channeling through recombinant expression, purification, and testing for immunogenicity and protective efficacy [55]. For example, the electroeluting of single protein spots, and the subsequent testing of eluted protein against an APC-T-cell clone system, for immunogenicity, is an interesting combination postgenomic approach which addresses the concept of whole protein antigenicity. Epitomics, the postgenomic identification of epitopes, is also an area falling under the proteomics revolution. Mass spectrometry is now being used routinely to sequence peptides eluted from MHC molecules [166, 167, 168].
The experimental and informatic techniques described above address the determination of immunogenicity, albeit parenthetically. Immunogenicity is one of the most widely used terms within immunobiology. Simply, immunogenicity is that property of a chemical moiety—be that protein, lipid, carbohydrate, or some combination thereof—that allows it to induce a significant response of the immune system. An exact definition might not be possible to formulate, being dependent on context. Put simply, a protein which is highly immunogenic within one species, within one population, or within one particular individual within a population is not necessarily immunogenic within another species, population, or individual. Immunogenicity is not the same as protective immunity although it is bound up with it, particularly from an immunovaccinology perspective. Protective immunity is, essentially, an enhanced immunity to reinfection, or to a first infection in the case of a successful vaccine. It is composed of an augmentation of preformed immune reactants, such as antigen-specific antibodies, and the formation of long lasting immune memory, which is mediated by memory B cells and memory T cells. Immunogenicity, per se, is an obvious requirement for protective immunity, yet while it is necessary, it is also clearly not sufficient. There are other factors—probably many other factors—as yet unknown, that mediate protection.
Although we cannot easily define immunogenicity, nonetheless, a fundamental understanding of immunological mechanisms operating, within this context, at the molecular level underlies most modern attempts to design vaccines rationally. The newly emergent discipline of immunovaccinology is bound up with the development of immunobiology as a postgenomic science. The sequences of genomes from both microbial pathogens and vertebrate hosts are now available, and the power of parallel approaches such as transcriptomics and proteomics is now being felt in the search for new vaccines. However, the manifestation of immunology at the whole animal level is an exceedingly complex phenomenon. It is only by investigating each of its individual stages, at the level of interacting molecules and cells, and in a physicochemical manner, that we can hope to formulate ways of modelling and manipulating the process effectively.
References
- Paine K, Flower D R. The lipocalin website. Biochim Biophys Acta. 2000;1482(1-2):351–352. doi: 10.1016/s0167-4838(00)00166-7. [DOI] [PubMed] [Google Scholar]
- Ji H, Zhou Q, Wen F, Xia H, Lu X, Li Y. AsMamDB: an alternative splice database of mammals. Nucleic Acids Res. 2001;29(1):260–263. doi: 10.1093/nar/29.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perler F B. InBase: the Intein Database. Nucleic Acids Res. 2002;30(1):383–384. doi: 10.1093/nar/30.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasinger V C, Cordwell S J, Cerpa-Poljak A, et al. Progress with gene-product mapping of the mollicutes: Mycoplasma genitalium . Electrophoresis. 1995;16(7):1090–1094. doi: 10.1002/elps.11501601185. [DOI] [PubMed] [Google Scholar]
- Anderson N G, Anderson L. The human protein index. Clin Chem. 1982;28(4 pt 2):739–748. [PubMed] [Google Scholar]
- Holtappels R, Grzimek N K, Thomas D, Reddehase M J. Early gene m18, a novel player in the immune response to murine cytomegalovirus. J Gen Virol. 2002;83(pt 2):311–316. doi: 10.1099/0022-1317-83-2-311. [DOI] [PubMed] [Google Scholar]
- Holtappels R, Thomas D, Podlech J, Reddehase M J. Two antigenic peptides from genes m123 and m164 of murine cytomegalovirus quantitatively dominate CD8 T-cell memory in the H-2d haplotype. J Virol. 2002;76(1):151–164. doi: 10.1128/JVI.76.1.151-164.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton J P, Mulcahy G. Parasite vaccines—a reality? Vet Parasitol. 2001;98(1-3):149–167. doi: 10.1016/s0304-4017(01)00430-7. [DOI] [PubMed] [Google Scholar]
- Chakravarti D N, Fiske M J, Fletcher L D, Zagursky R J. Mining genomes and mapping proteomes: identification and characterization of protein subunit vaccines. Dev Biol (Basel) 2000;103:81–90. [PubMed] [Google Scholar]
- Toby G G, Golemis E A. Using the yeast interaction trap and other two-hybrid-based approaches to study protein-protein interactions. Methods. 2001;24(3):201–217. doi: 10.1006/meth.2001.1182. [DOI] [PubMed] [Google Scholar]
- Templin M F, Stoll D, Schrenk M, Traub P C, Vohringer C F, Joos T O. Protein microarray technology. Trends Biotechnol. 2002;20(4):160–166. doi: 10.1016/s0167-7799(01)01910-2. [DOI] [PubMed] [Google Scholar]
- Walker J, Flower D, Rigley K. Microarrays in hematology. Curr Opin Hematol. 2002;9(1):23–29. doi: 10.1097/00062752-200201000-00005. [DOI] [PubMed] [Google Scholar]
- Grandi G. Antibacterial vaccine design using genomics and proteomics. Trends Biotechnol. 2001;19(5):181–188. doi: 10.1016/s0167-7799(01)01600-6. [DOI] [PubMed] [Google Scholar]
- Rathod P K, Ganesan K, Hayward R E, Bozdech Z, DeRisi J L. DNA microarrays for malaria. Trends Parasitol. 2002;18(1):39–45. doi: 10.1016/s1471-4922(01)02153-5. [DOI] [PubMed] [Google Scholar]
- Knox D P, Redmond D L, Skuce P J, Newlands G F. The contribution of molecular biology to the development of vaccines against nematode and trematode parasites of domestic ruminants. Vet Parasitol. 2001;101(3-4):311–335. doi: 10.1016/s0304-4017(01)00558-1. [DOI] [PubMed] [Google Scholar]
- Glynne R J, Watson S R. The immune system and gene expression microarrays—new answers to old questions. J Pathol. 2001;195(1):20–30. doi: 10.1002/1096-9896(200109)195:1<20::AID-PATH933>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Dhiman N, Bonilla R, O'Kane D J, Poland G A. Gene expression microarrays: a 21st century tool for directed vaccine design. Vaccine. 2001;20(1-2):22–30. doi: 10.1016/s0264-410x(01)00319-x. [DOI] [PubMed] [Google Scholar]
- de Veer M J, Holko M, Frevel M, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69(6):912–920. [PubMed] [Google Scholar]
- Emslie K R, Molloy M P, Barardi C R, et al. Serotype classification and characterisation of the rotavirus SA11 VP6 protein using mass spectrometry and two-dimensional gel electrophoresis. Funct Integr Genomics. 2000;1(1):12–24. doi: 10.1007/s101420000002. [DOI] [PubMed] [Google Scholar]
- Thomas J J, Bakhtiar R, Siuzdak G. Mass spectrometry in viral proteomics. Acc Chem Res. 2000;33(3):179–187. doi: 10.1021/ar9801200. [DOI] [PubMed] [Google Scholar]
- Yao X, Freas A, Ramirez J, Demirev P A, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73(13):2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- Toda T, Sugimoto M, Omori A, Matsuzaki T, Furuichi Y, Kimura N. Proteomic analysis of Epstein-Barr virus-transformed human B-lymphoblastoid cell lines before and after immortalization. Electrophoresis. 2000;21(9):1814–1822. doi: 10.1002/(SICI)1522-2683(20000501)21:9<1814::AID-ELPS1814>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Diaz J J, Giraud S, Greco A. Alteration of ribosomal protein maps in herpes simplex virus type 1 infection. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;771(1-2):237–249. doi: 10.1016/s1570-0232(02)00038-7. [DOI] [PubMed] [Google Scholar]
- Greco A, Bienvenut W, Sanchez J C, et al. Identification of ribosome-associated viral and cellular basic proteins during the course of infection with herpes simplex virus type 1. Proteomics. 2001;1(4):545–549. doi: 10.1002/1615-9861(200104)1:4<545::AID-PROT545>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Rodriguez J M, Salas M L, Santaren J F. African swine fever virus-induced polypeptides in porcine alveolar macrophages and in Vero cells: two-dimensional gel analysis. Proteomics. 2001;1(11):1447–1456. doi: 10.1002/1615-9861(200111)1:11<1447::AID-PROT1447>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Blattner F R, Plunkett G 3rd, Bloch C A, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277(5331):1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Fleischmann R D, Adams M D, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269(5223):496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Fraser C M, Gocayne J D, White O, et al. The minimal gene complement of Mycoplasma genitalium . Science. 1995;270(5235):397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- Bult C J, White O, Olsen G J, et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii . Science. 1996;273(5278):1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- Tomb J F, White O, Kerlavage A R, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori . Nature. 1997;388(6642):539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- Paine K, Flower D R. Bacterial bioinformatics: pathogenesis and the genome. J Mol Microbiol Biotechnol. 2002;4(4):357–365. [PubMed] [Google Scholar]
- O'Connor C D, Farris M, Fowler R, Qi S Y. The proteome of Salmonella enterica serovar Typhimurium: current progress on its determination and some applications. Electrophoresis. 1997;18(8):1483–1490. doi: 10.1002/elps.1150180823. [DOI] [PubMed] [Google Scholar]
- Hecker M, Engelmann S. Proteomics, DNA arrays and the analysis of still unknown regulons and unknown proteins of Bacillus subtilis and pathogenic gram-positive bacteria. Int J Med Microbiol. 2000;290(2):123–134. doi: 10.1016/S1438-4221(00)80080-6. [DOI] [PubMed] [Google Scholar]
- Regula J T, Ueberle B, Boguth G, et al. Towards a two-dimensional proteome map of Mycoplasma pneumoniae . Electrophoresis. 2000;21(17):3765–3780. doi: 10.1002/1522-2683(200011)21:17<3765::AID-ELPS3765>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Vandahl B B, Birkelund S, Demol H, et al. Proteome analysis of the Chlamydia pneumoniae elementary body. Electrophoresis. 2001;22(6):1204–1223. doi: 10.1002/1522-2683()22:6<1204::AID-ELPS1204>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Shaw A C, Gevaert K, Demol H, et al. Comparative proteome analysis of Chlamydia trachomatis serovar A, D, and L2. Proteomics. 2002;2(2):164–186. doi: 10.1002/1615-9861(200202)2:2<164::aid-prot164>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Piechaczek K, Dobrindt U, Schierhorn A, Fischer G S, Hecker M, Hacker J. Influence of pathogenicity islands and the minor leuX-encoded tRNA5Leu on the proteome pattern of the uropathogenic Escherichia coli strain 536. Int J Med Microbiol. 2000;290(1):75–84. doi: 10.1016/S1438-4221(00)80110-1. [DOI] [PubMed] [Google Scholar]
- Malhotra S, Silo-Suh L A, Mathee K, Ohman D E. Proteome analysis of the effect of mucoid conversion on global protein expression in Pseudomonas aeruginosa strain PAO1 shows induction of the disulfide bond isomerase, dsbA. J Bacteriol. 2000;182(24):6999–7006. doi: 10.1128/jb.182.24.6999-7006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkrands I, King A, Weldingh K, Moniatte M, Moertz E, Andersen P. Towards the proteome of Mycobacterium tuberculosis . Electrophoresis. 2000;21(17):3740–3756. doi: 10.1002/1522-2683(200011)21:17<3740::AID-ELPS3740>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Rosenkrands I, Weldingh K, Jacobsen S, et al. Mapping and identification of Mycobacterium tuberculosis proteins by two-dimensional gel electrophoresis, microsequencing and immunodetection. Electrophoresis. 2000;21(5):935–948. doi: 10.1002/(SICI)1522-2683(20000301)21:5<935::AID-ELPS935>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Jungblut P R, Muller E C, Mattow J, Kaufmann S H. Proteomics reveals open reading frames in Mycobacterium tuberculosis H37Rv not predicted by genomics. Infect Immun. 2001;69(9):5905–5907. doi: 10.1128/IAI.69.9.5905-5907.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungblut P R, Schaible U E, Mollenkopf H J, et al. Comparative proteome analysis of Mycobacterium tuberculosis and Mycobacterium bovis BCG strains: towards functional genomics of microbial pathogens. Mol Microbiol. 1999;33(6):1103–1117. doi: 10.1046/j.1365-2958.1999.01549.x. [DOI] [PubMed] [Google Scholar]
- Mattow J, Jungblut P R, Schaible U E, et al. Identification of proteins from Mycobacterium tuberculosis missing in attenuated Mycobacterium bovis BCG strains. Electrophoresis. 2001;22(14):2936–2946. doi: 10.1002/1522-2683(200108)22:14<2936::AID-ELPS2936>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Wong D K, Lee B Y, Horwitz M A, Gibson B W. Identification of fur, aconitase, and other proteins expressed by Mycobacterium tuberculosis under conditions of low and high concentrations of iron by combined two-dimensional gel electrophoresis and mass spectrometry. Infect Immun. 1999;67(1):327–336. doi: 10.1128/iai.67.1.327-336.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan I M, Betts J, Banerjee D K, Butcher P D. Differential expression of mycobacterial proteins following phagocytosis by macrophages. Microbiology. 2001;147(pt 2):459–471. doi: 10.1099/00221287-147-2-459. [DOI] [PubMed] [Google Scholar]
- Betts J C, Lukey P T, Robb L C, McAdam R A, Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002;43(3):717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- Betts J C, Dodson P, Quan S, et al. Comparison of the proteome of Mycobacterium tuberculosis strain H37Rv with clinical isolate CDC 1551. Microbiology. 2000;146(pt 12):3205–3216. doi: 10.1099/00221287-146-12-3205. [DOI] [PubMed] [Google Scholar]
- Covert B A, Spencer J S, Orme I M, Belisle J T. The application of proteomics in defining the T cell antigens of Mycobacterium tuberculosis . Proteomics. 2001;1(4):574–586. doi: 10.1002/1615-9861(200104)1:4<574::AID-PROT574>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Langenberg M-L, Tytgat G N.J, Schipper M E.I, Rietra P J.G.M, Zanen H C. Campylobacter-like organism in the stomach of patients and healthy individuals. Lancet. 1984;2(1348):. [PubMed] [Google Scholar]
- Utt M, Nilsson I, Ljungh A, Wadstrom T. Identification of novel immunogenic proteins of Helicobacter pylori by proteome technology. J Immunol Methods. 2002;259(1-2):1–10. doi: 10.1016/s0022-1759(01)00476-8. [DOI] [PubMed] [Google Scholar]
- McAtee C P, Lim M Y, Fung K, et al. Identification of potential diagnostic and vaccine candidates of Helicobacter pylori by two-dimensional gel electrophoresis, sequence analysis, and serum profiling. Clin Diagn Lab Immunol. 1998;5(4):537–542. doi: 10.1128/cdli.5.4.537-542.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel B, Bosserhoff A, Frank R, Gross R, Goebel W, Beier D. Identification of immunodominant antigens from Helicobacter pylori and evaluation of their reactivities with sera from patients with different gastroduodenal pathologies. Infect Immun. 2000;68(2):915–920. doi: 10.1128/iai.68.2.915-920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungblut P R, Bumann D, Haas G, et al. Comparative proteome analysis of Helicobacter pylori . Mol Microbiol. 2000;36(3):710–725. doi: 10.1046/j.1365-2958.2000.01896.x. [DOI] [PubMed] [Google Scholar]
- Nilsson I, Utt M, Nilsson H O, Ljungh A, Wadstrom T. Two-dimensional electrophoretic and immunoblot analysis of cell surface proteins of spiral-shaped and coccoid forms of Helicobacter pylori . Electrophoresis. 2000;21(13):2670–2677. doi: 10.1002/1522-2683(20000701)21:13<2670::AID-ELPS2670>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Chakravarti D N, Fiske M J, Fletcher L D, Zagursky R J. Application of genomics and proteomics for identification of bacterial gene products as potential vaccine candidates. Vaccine. 2000;19(6):601–612. doi: 10.1016/s0264-410x(00)00256-5. [DOI] [PubMed] [Google Scholar]
- Haas G, Karaali G, Ebermayer K, et al. Immunoproteomics of Helicobacter pylori infection and relation to gastric disease. Proteomics. 2002;2(3):313–324. doi: 10.1002/1615-9861(200203)2:3<313::aid-prot313>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- McAtee C P, Hoffman P S, Berg D E. Identification of differentially regulated proteins in metronidozole resistant Helicobacter pylori by proteome techniques. Proteomics. 2001;1(4):516–521. doi: 10.1002/1615-9861(200104)1:4<516::AID-PROT516>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Yoder O C, Turgeon B G. Fungal genomics and pathogenicity. Curr Opin Plant Biol. 2001;4(4):315–321. doi: 10.1016/s1369-5266(00)00179-5. [DOI] [PubMed] [Google Scholar]
- Yoder O C, Turgeon B G. Molecular genetic evaluation of fungal molecules for roles in pathogenesis to plants. J Genet. 1996;75:425–440. [Google Scholar]
- Oliver R, Osbourn A. Molecular dissection of fungal phytopathogenicity. Microbiology. 1995;141(pt 1):1–9. doi: 10.1099/00221287-141-1-1. [DOI] [PubMed] [Google Scholar]
- Hogan L H, Klein B S, Levitz S M. Virulence factors of medically important fungi. Clin Microbiol Rev. 1996;9(4):469–488. doi: 10.1128/cmr.9.4.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D, Hains P, Walsh B, Bergquist P, Nevalainen H. Proteins associated with the cell envelope of Trichoderma reesei: a proteomic approach. Proteomics. 2001;1(7):899–909. doi: 10.1002/1615-9861(200107)1:7<899::AID-PROT899>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Gutierrez J A. Genomics: from novel genes to new therapeutics in parasitology. Int J Parasitol. 2000;30(3):247–252. doi: 10.1016/s0020-7519(00)00005-9. [DOI] [PubMed] [Google Scholar]
- Colley D G. Parasitic diseases: opportunities and challenges in the 21st century. Mem Inst Oswaldo Cruz. 2000;95(suppl 1):79–87. doi: 10.1590/s0074-02762000000700015. [DOI] [PubMed] [Google Scholar]
- Knox D P, Redmond D L, Skuce P J, Newlands G F. The contribution of molecular biology to the development of vaccines against nematode and trematode parasites of domestic ruminants. Vet Parasitol. 2001;101(3-4):311–335. doi: 10.1016/s0304-4017(01)00558-1. [DOI] [PubMed] [Google Scholar]
- Jefferies J R, Campbell A M, van Rossum A J, Barrett J, Brophy P M. Proteomic analysis of Fasciola hepatica excretory-secretory products. Proteomics. 2001;1(9):1128–1132. doi: 10.1002/1615-9861(200109)1:9<1128::AID-PROT1128>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Almeida R, Norrish A, Levick M, et al. From genomes to vaccines: Leishmania as a model. Philos Trans R Soc Lond B Biol Sci. 2002;357(1417):5–11. doi: 10.1098/rstb.2001.0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel M, Bruchhaus I. Comparative proteome analysis of Leishmania donovani at different stages of transformation from promastigotes to amastigotes. Med Microbiol Immunol (Berl) 2001;190(1-2):33–36. doi: 10.1007/s004300100075. [DOI] [PubMed] [Google Scholar]
- Cohen A M, Rumpel K, Coombs G H, Wastling J M. Characterisation of global protein expression by two-dimensional electrophoresis and mass spectrometry: proteomics of Toxoplasma gondii . Int J Parasitol. 2002;32(1):39–51. doi: 10.1016/s0020-7519(01)00308-3. [DOI] [PubMed] [Google Scholar]
- Dlugonska H, Dytnerska K, Reichmann G, Stachelhaus S, Fischer H G. Towards the Toxoplasma gondii proteome: position of 13 parasite excretory antigens on a standardized map of two-dimensionally separated tachyzoite proteins. Parasitol Res. 2001;87(8):634–637. doi: 10.1007/s004360000367. [DOI] [PubMed] [Google Scholar]
- Fletcher J M, Nair S P, Ward J M, Henderson B, Wilson M. Analysis of the effect of changing environmental conditions on the expression patterns of exported surface-associated proteins of the oral pathogen Actinobacillus actinomycetemcomitans . Microb Pathog. 2001;30(6):359–368. doi: 10.1006/mpat.2000.0439. [DOI] [PubMed] [Google Scholar]
- Monahan I M, Betts J, Banerjee D K, Butcher P D. Differential expression of mycobacterial proteins following phagocytosis by macrophages. Microbiology. 2001;147(pt 2):459–471. doi: 10.1099/00221287-147-2-459. [DOI] [PubMed] [Google Scholar]
- Ragno S, Romano M, Howell S, Pappin D J, Jenner P J, Colston M J. Changes in gene expression in macrophages infected with Mycobacterium tuberculosis: a combined transcriptomic and proteomic approach. Immunology. 2001;104(1):99–108. doi: 10.1046/j.0019-2805.2001.01274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarova H, Halada P, Man P, et al. Proteome study of Francisella tularensis live vaccine strain-containing phagosome in Bcg/Nramp1 congenic macrophages: resistant allele contributes to permissive environment and susceptibility to infection. Proteomics. 2002;2(1):85–93. [PubMed] [Google Scholar]
- Pizarro-Cerda J, Jonquieres R, Gouin E, Vandekerckhove J, Garin J, Cossart P. Distinct protein patterns associated with Listeria monocytogenes InlA- or InlB-phagosomes. Cell Microbiol. 2002;4(2):101–115. doi: 10.1046/j.1462-5822.2002.00169.x. [DOI] [PubMed] [Google Scholar]
- Truffa-Bachi P, Lefkovits I, Frey J R. Proteomic analysis of T cell activation in the presence of cyclosporin A: immunosuppressor and activator removal induces de novo protein synthesis [Erratum in Mol Immunol. 2000;37(5):261] Mol Immunol. 2000;37(1-2):21–28. doi: 10.1016/s0161-5890(00)00054-7. [DOI] [PubMed] [Google Scholar]
- Nyman T A, Rosengren A, Syyrakki S, Pellinen T P, Rautajoki K, Lahesmaa R. A proteome database of human primary T helper cells. Electrophoresis. 2001;22(20):4375–4382. doi: 10.1002/1522-2683(200112)22:20<4375::AID-ELPS4375>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Fratelli M, Demol H, Puype M, et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci USA. 2002;99(6):3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A. Applications of computer software for the interpretation and management of mass spectrometry data in pharmaceutical science. Curr Top Med Chem. 2002;2(1):99–107. doi: 10.2174/1568026023394641. [DOI] [PubMed] [Google Scholar]
- Sidhu K S, Sangvanich P, Brancia F L, et al. Bioinformatic assessment of mass spectrometric chemical derivatisation techniques for proteome database searching. Proteomics. 2001;1(11):1368–1377. doi: 10.1002/1615-9861(200111)1:11<1368::AID-PROT1368>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Vihinen M. Bioinformatics in proteomics. Biomol Eng. 2001;18(5):241–248. doi: 10.1016/s1389-0344(01)00099-5. [DOI] [PubMed] [Google Scholar]
- Chakravarti D N, Chakravarti B, Moutsatsos I. Informatic tools for proteome profiling. Biotechniques. 2002;(suppl):4–15. [PubMed] [Google Scholar]
- Nussbaum A K, Dick T P, Keilholz W, et al. Cleavage motifs of the yeast 20S proteasome beta subunits deduced from digests of enolase 1. Proc Natl Acad Sci USA. 1998;95(21):12504–12509. doi: 10.1073/pnas.95.21.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzhutter H G, Frommel C, Kloetzel P M. A theoretical approach towards the identification of cleavage-determining amino acid motifs of the 20S proteasome. J Mol Biol. 1999;286(4):1251–1265. doi: 10.1006/jmbi.1998.2530. [DOI] [PubMed] [Google Scholar]
- Kuttler C, Nussbaum A K, Dick T P, Rammensee H G, Schild H, Hadeler K P. An algorithm for the prediction of proteasomal cleavages. J Mol Biol. 2000;298(3):417–429. doi: 10.1006/jmbi.2000.3683. [DOI] [PubMed] [Google Scholar]
- Nussbaum A K, Kuttler C, Hadeler K P, Rammensee H G, Schild H. PAProC: a prediction algorithm for proteasomal cleavages available on the WWW. Immunogenetics. 2001;53(2):87–94. doi: 10.1007/s002510100300. [DOI] [PubMed] [Google Scholar]
- Kesmir C, Nussbaum A K, Schild H, Detours V, Brunak S. Prediction of proteasome cleavage motifs by neural networks. Protein Eng. 2002;15(4):287–296. doi: 10.1093/protein/15.4.287. [DOI] [PubMed] [Google Scholar]
- Daniel S, Brusic V, Caillat-Zucman S, et al. Relationship between peptide selectivities of human transporters associated with antigen processing and HLA class I molecules. J Immunol. 1998;161(2):617–624. [PubMed] [Google Scholar]
- Brusic V, van Endert P, Zeleznikow J, Daniel S, Hammer J, Petrovsky N. A neural network model approach to the study of human TAP transporter. In Silico Biol. 1999;1(2):109–121. [PubMed] [Google Scholar]
- Chapman H A. Endosomal proteolysis and MHC class II function. Curr Opin Immunol. 1998;10(1):93–102. doi: 10.1016/s0952-7915(98)80038-1. [DOI] [PubMed] [Google Scholar]
- Sette A, Buus S, Appella E, et al. Prediction of major histocompatibility complex binding regions of protein antigens by sequence pattern analysis. Proc Natl Acad Sci USA. 1989;86(9):3296–3300. doi: 10.1073/pnas.86.9.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amaro J, Houbiers J G, Drijfhout J W, et al. A computer program for predicting possible cytotoxic T lymphocyte epitopes based on HLA class I peptide-binding motifs. Hum Immunol. 1995;43(1):13–18. doi: 10.1016/0198-8859(94)00153-h. [DOI] [PubMed] [Google Scholar]
- Rammensee H, Bachmann J, Emmerich N P, Bachor O A, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3-4):213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- Parker K C, Shields M, DiBrino M, Brooks A, Coligan J E. Peptide binding to MHC class I molecules: implications for antigenic peptide prediction. Immunol Res. 1995;14(1):34–57. doi: 10.1007/BF02918496. [DOI] [PubMed] [Google Scholar]
- Parker K C, DiBrino M, Hull L, Coligan J E. The beta 2-microglobulin dissociation rate is an accurate measure of the stability of MHC class I heterotrimers and depends on which peptide is bound. J Immunol. 1992;149(6):1896–1904. [PubMed] [Google Scholar]
- Parker K C, Bednarek M A, Coligan J E. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152(1):163–175. [PubMed] [Google Scholar]
- Parker K C, Bednarek M A, Hull L K, et al. Sequence motifs important for peptide binding to the human MHC class I molecule, HLA-A2. J Immunol. 1992;149(11):3580–3587. [PubMed] [Google Scholar]
- Parker K C, Carreno B M, Sestak L, Utz U, Biddison W E, Coligan J E. Peptide binding to HLA-A2 and HLA-B27 isolated from Escherichia coli. Reconstitution of HLA-A2 and HLA-B27 heavy chain/beta 2-microglobulin complexes requires specific peptides. J Biol Chem. 1992;267(8):5451–5459. [PubMed] [Google Scholar]
- DiBrino M, Parker K C, Shiloach J, et al. Endogenous peptides bound to HLA-A3 possess a specific combination of anchor residues that permit identification of potential antigenic peptides. Proc Natl Acad Sci USA. 1993;90(4):1508–1512. doi: 10.1073/pnas.90.4.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBrino M, Parker K C, Margulies D H, et al. The HLA-B14 peptide binding site can accommodate peptides with different combinations of anchor residues. J Biol Chem. 1994;269(51):32426–32434. [PubMed] [Google Scholar]
- Parker K C, Biddison W E, Coligan J E. Pocket mutations of HLA-B27 show that anchor residues act cumulatively to stabilize peptide binding. Biochemistry. 1994;33(24):7736–7743. doi: 10.1021/bi00190a029. [DOI] [PubMed] [Google Scholar]
- DiBrino M, Parker K C, Margulies D H, et al. Identification of the peptide binding motif for HLA-B44, one of the most common HLA-B alleles in the Caucasian population. Biochemistry. 1995;34(32):10130–10138. doi: 10.1021/bi00032a005. [DOI] [PubMed] [Google Scholar]
- DiBrino M, Tsuchida T, Turner R V, Parker K C, Coligan J E, Biddison W E. HLA-A1 and HLA-A3 T cell epitopes derived from influenza virus proteins predicted from peptide binding motifs. J Immunol. 1993;151(11):5930–5935. [PubMed] [Google Scholar]
- Honma K, Parker K C, Becker K G, McFarland H F, Coligan J E, Biddison W E. Identification of an epitope derived from human proteolipid protein that can induce autoreactive CD8+ cytotoxic T lymphocytes restricted by HLA-A3: evidence for cross-reactivity with an environmental microorganism. J Neuroimmunol. 1997;73(1-2):7–14. doi: 10.1016/s0165-5728(96)00161-0. [DOI] [PubMed] [Google Scholar]
- Bisset L R, Fierz W. Using a neural network to identify potential HLA-DR1 binding sites within proteins. J Mol Recognit. 1993;6(1):41–48. doi: 10.1002/jmr.300060105. [DOI] [PubMed] [Google Scholar]
- Brusic V, Schonbach C, Takiguchi M, Ciesielski V, Harrison L C. Application of genetic search in derivation of matrix models of peptide binding to MHC molecules. Proc Int Conf Intell Syst Mol Biol. 1997;5:75–83. [PubMed] [Google Scholar]
- Harrison L C, Honeyman M C, Trembleau S, et al. A peptide-binding motif for I-A(g7), the class II major histocompatibility complex (MHC) molecule of NOD and Biozzi AB/H mice. J Exp Med. 1997;185(6):1013–1021. doi: 10.1084/jem.185.6.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honeyman M C, Brusic V, Harrison L C. Strategies for identifying and predicting islet autoantigen T-cell epitopes in insulin-dependent diabetes mellitus. Ann Med. 1997;29(5):401–404. doi: 10.3109/07853899708999369. [DOI] [PubMed] [Google Scholar]
- Honeyman M C, Brusic V, Stone N L, Harrison L C. Neural network-based prediction of candidate T-cell epitopes. Nat Biotechnol. 1998;16(10):966–969. doi: 10.1038/nbt1098-966. [DOI] [PubMed] [Google Scholar]
- Brusic V, Rudy G, Harrison L C. MHCPEP, a database of MHC-binding peptides: update 1997. Nucleic Acids Res. 1998;26(1):368–371. doi: 10.1093/nar/26.1.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld R, Zheng Q, Vajda S, DeLisi C. Flexible docking of peptides to class I major-histocompatibility-complex receptors. Genet Anal. 1995;12(1):1–21. doi: 10.1016/1050-3862(95)00107-7. [DOI] [PubMed] [Google Scholar]
- Sezerman U, Vajda S, DeLisi C. Free energy mapping of class I MHC molecules and structural determination of bound peptides. Protein Sci. 1996;5(7):1272–1281. doi: 10.1002/pro.5560050706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasmatzis G, Zhang C, Cornette J L, DeLisi C. Computational determination of side chain specificity for pockets in class I MHC molecules. Mol Immunol. 1996;33(16):1231–1239. doi: 10.1016/s0161-5890(96)00090-9. [DOI] [PubMed] [Google Scholar]
- Rognan D, Reddehase M J, Koszinowski U H, Folkers G. Molecular modeling of an antigenic complex between a viral peptide and a class I major histocompatibility glycoprotein. Proteins. 1992;13(1):70–85. doi: 10.1002/prot.340130107. [DOI] [PubMed] [Google Scholar]
- Rognan D, Zimmermann N, Jung G, Folkers G. Molecular dynamics study of a complex between the human histocompatibility antigen HLA-A2 and the IMP58-66 nonapeptide from influenza virus matrix protein. Eur J Biochem. 1992;208(1):101–113. doi: 10.1111/j.1432-1033.1992.tb17163.x. [DOI] [PubMed] [Google Scholar]
- Rognan D, Scapozza L, Folkers G, Daser A. Molecular dynamics simulation of MHC-peptide complexes as a tool for predicting potential T cell epitopes. Biochemistry. 1994;33(38):11476–11485. doi: 10.1021/bi00204a009. [DOI] [PubMed] [Google Scholar]
- Caflisch A, Niederer P, Anliker M. Monte Carlo docking of oligopeptides to proteins. Proteins. 1992;13(3):223–230. doi: 10.1002/prot.340130305. [DOI] [PubMed] [Google Scholar]
- Lim J S, Kim S, Lee H G, Lee K Y, Kwon T J, Kim K. Selection of peptides that bind to the HLA-A2.1 molecule by molecular modelling. Mol Immunol. 1996;33(2):221–230. doi: 10.1016/0161-5890(95)00065-8. [DOI] [PubMed] [Google Scholar]
- Androulakis I P, Nayak N N, Ierapetritou M G, Monos D S, Floudas C A. A predictive method for the evaluation of peptide binding in pocket 1 of HLA-DRB1 via global minimization of energy interactions. Proteins. 1997;29(1):87–102. [PubMed] [Google Scholar]
- Froloff N, Windemuth A, Honig B. On the calculation of binding free energies using continuum methods: application to MHC class I protein-peptide interactions. Protein Sci. 1997;6(6):1293–1301. doi: 10.1002/pro.5560060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora N, Bashford D. Solvation energy density occlusion approximation for evaluation of desolvation penalties in biomolecular interactions. Proteins. 2001;43(1):12–27. doi: 10.1002/1097-0134(20010401)43:1<12::aid-prot1013>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Doytchinova I A, Blythe M J, Flower D R. Additive method for the prediction of protein-peptide binding affinity. Application to the MHC class I molecule HLA-A∗0201. J Proteome Res. 2002;1(3):263–272. doi: 10.1021/pr015513z. [DOI] [PubMed] [Google Scholar]
- Doytchinova I A, Flower D R. Toward the quantitative prediction of T-cell epitopes: CoMFA and CoMSIA studies of peptides with affinity to the class I MHC molecule HLA-A∗0201. J Med Chem. 2001;44(22):3572–3581. doi: 10.1021/jm010021j. [DOI] [PubMed] [Google Scholar]
- Doytchinova I A, Flower D R. Physicochemical explanation of peptide binding to HLA-A∗0201 major histocompatibility complex: a three-dimensional quantitative structure-activity relationship study. Proteins. 2002;48(3):505–518. doi: 10.1002/prot.10154. [DOI] [PubMed] [Google Scholar]
- Rognan D, Lauemoller S L, Holm A, Buus S, Tschinke V. Predicting binding affinities of protein ligands from three-dimensional models: application to peptide binding to class I major histocompatibility proteins. J Med Chem. 1999;42(22):4650–4658. doi: 10.1021/jm9910775. [DOI] [PubMed] [Google Scholar]
- Logean A, Sette A, Rognan D. Customized versus universal scoring functions: application to class I MHC-peptide binding free energy predictions. Bioorg Med Chem Lett. 2001;11(5):675–679. doi: 10.1016/s0960-894x(01)00021-x. [DOI] [PubMed] [Google Scholar]
- Altuvia Y, Schueler O, Margalit H. Ranking potential binding peptides to MHC molecules by a computational threading approach. J Mol Biol. 1995;249(2):244–250. doi: 10.1006/jmbi.1995.0293. [DOI] [PubMed] [Google Scholar]
- Altuvia Y, Sette A, Sidney J, Southwood S, Margalit H. A structure-based algorithm to predict potential binding peptides to MHC molecules with hydrophobic binding pockets. Hum Immunol. 1997;58(1):1–11. doi: 10.1016/s0198-8859(97)00210-3. [DOI] [PubMed] [Google Scholar]
- Miyazawa S, Jernigan R L. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J Mol Biol. 1996;256(3):623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- Schueler-Furman O, Altuvia Y, Sette A, Margalit H. Structure-based prediction of binding peptides to MHC class I molecules: application to a broad range of MHC alleles. Protein Sci. 2000;9(9):1838–1846. doi: 10.1110/ps.9.9.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt M R, Thirumalai D. Pair potentials for protein folding: choice of reference states and sensitivity of predicted native states to variations in the interaction schemes. Protein Sci. 1999;8(2):361–369. doi: 10.1110/ps.8.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryhn A, Pedersen L O, Romme T, Holm C B, Holm A, Buus S. Peptide binding specificity of major histocompatibility complex class I resolved into an array of apparently independent subspecificities: quantitation by peptide libraries and improved prediction of binding. Eur J Immunol. 1996;26(8):1911–1918. doi: 10.1002/eji.1830260836. [DOI] [PubMed] [Google Scholar]
- Stevens J, Wiesmuller K H, Walden P, Joly E. Peptide length preferences for rat and mouse MHC class I molecules using random peptide libraries. Eur J Immunol. 1998;28(4):1272–1279. doi: 10.1002/(SICI)1521-4141(199804)28:04<1272::AID-IMMU1272>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Stevens J, Wiesmuller K H, Barker P J, Walden P, Butcher G W, Joly E. Efficient generation of major histocompatibility complex class I-peptide complexes using synthetic peptide libraries. J Biol Chem. 1998;273(5):2874–2884. doi: 10.1074/jbc.273.5.2874. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Gran B, Pinilla C, et al. Combinatorial peptide libraries and biometric score matrices permit the quantitative analysis of specific and degenerate interactions between clonotypic TCR and MHC peptide ligands. J Immunol. 2001;167(4):2130–2141. doi: 10.4049/jimmunol.167.4.2130. [DOI] [PubMed] [Google Scholar]
- Pinilla C, Rubio-Godoy V, Dutoit V, et al. Combinatorial peptide libraries as an alternative approach to the identification of ligands for tumor-reactive cytolytic T lymphocytes. Cancer Res. 2001;61(13):5153–5160. [PubMed] [Google Scholar]
- Udaka K, Wiesmuller K H, Kienle S, et al. An automated prediction of MHC class I-binding peptides based on positional scanning with peptide libraries. Immunogenetics. 2000;51(10):816–828. doi: 10.1007/s002510000217. [DOI] [PubMed] [Google Scholar]
- Blythe M J, Doytchinova I A, Flower D R. JenPep: a database of quantitative functional peptide data for immunology. Bioinformatics. 2002;18(3):434–439. doi: 10.1093/bioinformatics/18.3.434. [DOI] [PubMed] [Google Scholar]
- Van Regenmortel M H, Daney de Marcillac G. An assessment of prediction methods for locating continuous epitopes in proteins. Immunol Lett. 1988;17(2):95–107. doi: 10.1016/0165-2478(88)90076-4. [DOI] [PubMed] [Google Scholar]
- Ferreira-da-Cruz Mde F, Giovanni-de-Simone S, Banic D M, Canto-Cavalheiro M, Camus D, Daniel-Ribeiro C T. Can software be used to predict antigenic regions in Plasmodium falciparum peptides? Parasite Immunol. 1996;18(3):159–161. doi: 10.1046/j.1365-3024.1996.d01-62.x. [DOI] [PubMed] [Google Scholar]
- Thornton J M, Edwards M S, Taylor W R, Barlow D J. Location of “continuous” antigenic determinants in the protruding regions of proteins. EMBO J. 1986;5(2):409–413. doi: 10.1002/j.1460-2075.1986.tb04226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow D J, Edwards M S, Thornton J M. Continuous and discontinuous protein antigenic determinants. Nature. 1986;322(6081):747–748. doi: 10.1038/322747a0. [DOI] [PubMed] [Google Scholar]
- Van Regenmortel M H, Pellequer J L. Predicting antigenic determinants in proteins: looking for unidimensional solutions to a three-dimensional problem? Pept Res. 1994;7(4):224–228. [PubMed] [Google Scholar]
- Alix A J. Predictive estimation of protein linear epitopes by using the program PEOPLE. Vaccine. 1999;18(3-4):311–314. doi: 10.1016/s0264-410x(99)00329-1. [DOI] [PubMed] [Google Scholar]
- Pellequer J L, Westhof E. PREDITOP: a program for antigenicity prediction. J Mol Graph. 1993;11(3):204–210. doi: 10.1016/0263-7855(93)80074-2. [DOI] [PubMed] [Google Scholar]
- Saleh M T, Fillon M, Brennan P J, Belisle J T. Identification of putative exported/secreted proteins in prokaryotic proteomes. Gene. 2001;269(1-2):195–204. doi: 10.1016/s0378-1119(01)00436-x. [DOI] [PubMed] [Google Scholar]
- Kumar A, Agarwal S, Heyman J A, et al. Subcellular localization of the yeast proteome. Genes Dev. 2002;16(6):707–719. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromiha M M. A simple method for predicting transmembrane alpha helices with better accuracy. Protein Eng. 1999;12(7):557–561. doi: 10.1093/protein/12.7.557. [DOI] [PubMed] [Google Scholar]
- Nishikawa K, Ooi T. Correlation of the amino acid composition of a protein to its structural and biological characters. J Biochem (Tokyo) 1982;91(5):1821–1824. doi: 10.1093/oxfordjournals.jbchem.a133877. [DOI] [PubMed] [Google Scholar]
- Eisenhaber F, Frőmmel C, Argos P. Prediction of secondary structural content of proteins from their amino acid composition alone. II. The paradox with secondary structural class. Proteins. 1996;25(2):169–179. doi: 10.1002/(SICI)1097-0134(199606)25:2<169::AID-PROT3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Chiapello H, Ollivier E, Landes-Devauchelle C, Nitschke P, Risler J L. Codon usage as a tool to predict the cellular location of eukaryotic ribosomal proteins and aminoacyl-tRNA synthetases. Nucleic Acids Res. 1999;27(14):2848–2851. doi: 10.1093/nar/27.14.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou K C, Elrod D W. Using discriminant function for prediction of subcellular location of prokaryotic proteins. Biochem Biophys Res Commun. 1998;252(1):63–68. doi: 10.1006/bbrc.1998.9498. [DOI] [PubMed] [Google Scholar]
- Cedano J, Aloy P, Perez-Pons J A, Querol E. Relation between amino acid composition and cellular location of proteins. J Mol Biol. 1997;266(3):594–600. doi: 10.1006/jmbi.1996.0804. [DOI] [PubMed] [Google Scholar]
- Nakashima H, Nishikawa K. Discrimination of intracellular and extracellular proteins using amino acid composition and residue-pair frequencies. J Mol Biol. 1994;238(1):54–61. doi: 10.1006/jmbi.1994.1267. [DOI] [PubMed] [Google Scholar]
- Andrade M A, O'Donoghue S I, Rost B. Adaptation of protein surfaces to subcellular location. J Mol Biol. 1998;276(2):517–525. doi: 10.1006/jmbi.1997.1498. [DOI] [PubMed] [Google Scholar]
- Reinhardt A, Hubbard T. Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res. 1998;26(9):2230–2236. doi: 10.1093/nar/26.9.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai K. Predicting various targeting signals in amino acid sequences. Bull Inst Chem Res Kyoto Univ. 1991;69:269–291. [Google Scholar]
- Nakai K. Protein sorting signals and prediction of subcellular localization. Adv Protein Chem. 2000;54:277–344. doi: 10.1016/s0065-3233(00)54009-1. [DOI] [PubMed] [Google Scholar]
- Briggs M S, Gierasch L M, Zlotnick A, Lear J D, DeGrado W F. In vivo function and membrane binding properties are correlated for Escherichia coli lamB signal peptides. Science. 1985;228(4703):1096–1099. doi: 10.1126/science.3158076. [DOI] [PubMed] [Google Scholar]
- von Heijne J Mol BiolG. Signal sequences. The limits of variation. J Mol Biol. 1985;184(1):99–105. doi: 10.1016/0022-2836(85)90046-4. [DOI] [PubMed] [Google Scholar]
- Martoglio B, Dobberstein B. Signal sequences: more than just greasy peptides. Trends Cell Biol. 1998;8(10):410–415. doi: 10.1016/s0962-8924(98)01360-9. [DOI] [PubMed] [Google Scholar]
- Sjőstrőm M, Wold S, Wieslander A, Rilfors L. Signal peptide amino acid sequences in Escherichia coli contain information related to final protein localization. A multivariate data analysis. EMBO J. 1987;6(3):823–831. doi: 10.1002/j.1460-2075.1987.tb04825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edman M, Jarhede T, Sjőstrőm M, Wieslander A. Different sequence patterns in signal peptides from mycoplasmas, other gram-positive bacteria, and Escherichia coli: a multivariate data analysis. Proteins. 1999;35(2):195–205. [PubMed] [Google Scholar]
- Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10(1):1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- Jagla B, Schuchhardt J. Adaptive encoding neural networks for the recognition of human signal peptide cleavage sites. Bioinformatics. 2000;16(3):245–250. doi: 10.1093/bioinformatics/16.3.245. [DOI] [PubMed] [Google Scholar]
- Mouritsen S, Dalum I, Engel A M, et al. MHC class II-bound self-peptides can be effectively separated by isoelectric focusing and bind optimally to their MHC class II restriction elements around pH 5.0. Immunology. 1994;82(4):529–534. [PMC free article] [PubMed] [Google Scholar]
- Dongre A R, Kovats S, deRoos P, et al. In vivo MHC class II presentation of cytosolic proteins revealed by rapid automated tandem mass spectrometry and functional analyses. Eur J Immunol. 2001;31(5):1485–1494. doi: 10.1002/1521-4141(200105)31:5<1485::AID-IMMU1485>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Purcell A W, Gorman J J. The use of post-source decay in matrix-assisted laser desorption/ionisation mass spectrometry to delineate T cell determinants. J Immunol Methods. 2001;249(1-2):17–31. doi: 10.1016/s0022-1759(00)00361-6. [DOI] [PubMed] [Google Scholar]