

Abstract
This manuscript describes the systematic development of pyridine-type ligands, which promote the Pd catalyzed, non-directed amination of benzene in combination with novel, hydroxylamine-based electrophilic amination reagents. DFT calculations and mechanistic experiments provide insights into the factors influencing the arene C–H amination protocol.
Introduction
Ligand design has the potential to access more active and selective catalysts and is thus situated to address core challenges in transition metal catalysis.1 However, despite their importance, the role of ligands in promoting kinetically challenging, non-directed C–H bond functionalizations2,3 in particular with Pd catalysts, is poorly understood. Consequently, the majority of such reaction protocols relies on screening as the major tool for identifying suitable ligands.4
During the last decade, several types of ligands have been shown to promote Pd catalyzed C–H oxidations of simple arenes.4 Among these ligands, pyridines have found use in non-directed, aromatic C–H bond oxygenations,5,6 olefinations,7,8 and arylations.9–11 In contrast, non-directed aromatic C–H aminations through organometallic intermediates are less developed, even though directed C–H aminations are well-known.12 Notably, the majority of non-directed C–H aminations proceed through radical13–15 or cationic16 mechanisms, whose selectivities are mostly controlled by substrate preferences; in other cases, the role of the transition metal catalyst is not well understood and several mechanisms are possible.17,18 Among the known methods are systems that employ metal-free conditions,19 photoredox catalysis,14 Cu catalysis,18,20 or a combination of Ag and Pd catalysis13 to achieve the desirable transformation. Based on the myriad of other C–H functionalizations of simple arenes which can be catalyzed by Pd-based systems,5–11 it is surprising that only one Pd-catalyzed method is known to affect the non-directed C–H amination of arenes (Scheme 1A).21
Scheme 1.

Previously known, only example of Pd catalyzed, non-directed C–H amination of simple arenes (1A) and herein described approach (1B).
This only known example suffers from significant background reactivity forming the amination product in the absence of Pd catalyst; furthermore, other Pd catalyzed side reactions under the established conditions produce large amounts of oxygenated byproduct (C–N to C–O product ratios up to 1 : 2). As demonstrated by this example, a major challenge in oxidative C–H aminations is the identification of suitable oxidant/amine combinations for organometallic C–H functionalization pathways, as amines are typically not stable in the presence of oxidants; an additional problem is that oxidants can undergo unwanted, catalyzed or non-catalyzed side reactions.15,21 Most importantly, due to such competing pathways, obtaining experimental mechanistic information on catalytic, non-directed C–H aminations through organometallic intermediates is difficult, which complicates the rational design of more efficient protocols. In order to address this gap in the knowledge we report herein the systematic development of Pd catalysts for non-directed C–H aminations employing atom-economic oxidants that combine amine and oxidant in one reagent (Scheme 1B).¶22–24
Results and discussion
Establishing C–H amination reactivity with hydroxylamine-based reagents
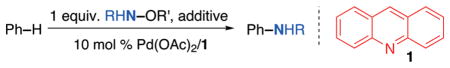
Our initial efforts towards this goal focused on designing and synthesizing hydroxylamine-derived amination reagents, which combine oxidant and amine source in one reagent. We hypothesized that these reagents could deliver the amine source directly to Pd upon oxidation. In comparison to previous trimolecular strategies involving reactions between catalyst, oxidant, and amine source,15,21 such a bimolecular strategy can be expected to suppress non-chemoselective reactivity between the catalyst and the oxidant in the absence of amine source, thus likely resulting in increased chemoselectivity of the reaction. Several such reagents (Table 1) were synthesized and evaluated for their ability to react in the C–H amination of benzene using a previously established C–H acetoxylation/ amination catalyst system (Pd(OAc)2/acridine 1 : 1).5,15
Table 1.
Reaction optimizationa
| ||||
|---|---|---|---|---|
| Entry | Additive | OR′ | R | Yield [%] |
| 1 | — | OAc | Ac | 11 |
| 2 | — | OAc | COCF3 | 21 |
| 3 | — | OAc | Ts | 13 |
| 4 | — | OTs | Ts | 10 |
| 5 | — | O2CCF3 | COCF3 | 22 |
| 6 | 1 equiv. AcOH | OAc | Ac | 29 |
| 7 | 5 mol% AgOAc | OAc | Ac | 27 |
| 8 | 1 equiv. AcOH | O2CCF3 | COCF3 | 17 |
| 9 | 5 mol% AgOAc | O2CCF3 | COCF3 | 22 |
| 10 | 1 equiv. AcOHb | OAc | Ac | 0 |
Conditions: benzene (0.50 mL, 5.6 mmol, 40 equiv.), Pd(OAc)2 (3.1 mg, 14 μmol, 10 mol%), 1 (2.5 mg, 14 μmol, 10 mol%), RNH–OR′ (140 μmol, 1.0 equiv.), 100 °C, 24 h.
No Pd catalyst.
Interestingly, in the absence of any additives, both reagents with trifluoroacetyl protecting groups on the N atom performed best (21% and 22%, respectively; entries 2/5). In contrast, in the presence of AgOAc or AcOH as additives, AcNH–OAc outperformed the fluorinated amination reagents (entries 6/7 vs. 8/9). Remarkably, the formation of PhOAc was limited to less than 1% in all reactions, suggesting that C–N bond forming reductive elimination outcompetes C–O bond formation under these reaction conditions. No C–N or C–O bond formation was observed in the absence of Pd(OAc)2 (entry 10), suggesting that the novel amination reagents do not react with benzene in the absence of catalyst. Other Pd sources or phosphine ligands (see Table S1†) only resulted in low yields of product (<10%). All reaction mixtures using Pd(OAc)2 as catalyst also showed formation of Pd black even after reaction times of less than 1 h and GCMS analysis of the reaction mixtures revealed the presence of biphenyl. This suggests that bimolecular catalyst decomposition25 is faster under these conditions than the reaction with the electrophilic amination reagent to form the desired aniline product. We reasoned that rational ligand development might be able to address these issues and aid in understanding why the unusual ligand acridine (1) provided substantially higher yields than all other ligands tested (Table S1†).
Exploring the source of the “acridine effect”: DFT calculations
Experimental and DFT work by others6,26 has suggested that the active species for C–H activation in Pd(OAc)2/pyridine catalyst systems for C–H oxygenation [through a Pd(II/IV) cycle] and C–H olefinations [through a Pd(0/II) cycle] is the monomeric species [Pd(OAc)2L] 2, which is in equilibrium with dimeric Pd2(OAc)4(L)2 (3) species, which are off-cycle resting states. These dimers principally can occur as two geometrical isomers, syn-3 or anti-3. To quantify the influence of different ligands on the equilibrium between monomer [Pd(OAc)2(L)] and the two possible dimers syn-3 or anti-3, we calculated the free enthalpies and geometries of this series of complexes at 100 °C, using the solvent correction for AcOH to mimic catalytic conditions closely (see e.g. Table 1, entry 6). The resulting differences between the free enthalpies of the monomers and dimers are the free reaction enthalpies associated with dimerization of 2, ΔGanti and ΔGsyn.
Interestingly, all analogous monomers 2 [Pd(OAc)2(L)] (L = 1, 4, 5) show a similar geometry (Fig. 1), in which one acetate ligand occupies two coordination sites in a κ2 fashion; the other acetate ligand binds in a κ1 fashion. Binding of the L-type ligands (acridine 1, quinoline 4, or pyridine 5) is in a κ1-fashion as expected from a monodentate L-type ligand and the only notable feature is that the more sterically bulky ligands show a large dihedral angle between the plane of the aromatic rings and the plane defined by the square planar Pd complex. In order to calculate the reaction enthalpies associated with dimerization of 2, ΔGanti and ΔGsyn, the geometries and energies of anti-3 and syn-3 were determined. For the purpose of this discussion, the free energy of the corresponding monomers 2 are defined as 0 kcal mol−1.
Fig. 1.

Graphic representations of DFT structures for 2 [Pd(OAc)2L] (L = 1, 4, 5). Geometry in analogous projections (Gaussian 09, B3LYP, 6-311+G(2d,p), LANL2Dz for Pd, AcOH, 100 °C). Ball-and stick model with H omitted for clarity.
Interestingly, the energies of syn-3 and anti-3 are distinctly different for each ligand, with the bulkier ligands 1 and 4 favoring the syn-dimers over the anti-dimers. In contrast, for simple pyridine (5) as ligand the anti-dimer is favored over the syn-dimer by 3.7 kcal mol−1. This change in preference can be explained by the fact that the steric bulk of ligands 4 and 1 force the corresponding anti-dimers to exhibit a chair conformation (see Fig. 2), while the syn-dimers take on a boat conformation,27 which is the typically observed conformation for Pd dimers bridged by acetate ligands.28
Fig. 2.

Graphic representations of DFT structures for the syn- and anti-dimers of [Pd(OAc)2(4)]2. Geometry in analogous projections (Gaussian 09, B3LYP, 6-311+G(2d,p), LANL2Dz for Pd, AcOH, 100 °C). Ball-and stick model with H omitted for clarity.
Overall, the resulting computed values for ΔGanti and ΔGsyn (Scheme 2) that are calculated on the basis of the energy differences between monomers and each dimer (see ESI† for details) show that the more sterically bulky ligands 1 and 4 strongly favor the formation of the monomeric species Pd(OAc)2(L) (2) by 12 to 8 kcal mol−1. In the parent system with pyridine (5) as ligand, the dimer anti-3 is energetically comparable to the monomeric species 2 (ΔGanti = +0.7 kcal mol−1). This suggests that a higher concentration of the catalytically active monomeric species can be expected with more sterically bulky ligands, which might be one reason why ligand 5 (pyridine) does not perform as well as the annulated ligands 1 and 4 in non-directed C–H amination (see ESI,† Table S1).
Scheme 2.

Free energies ΔG of monomeric Pd(OAc)2(L) (2) reacting to give dimeric Pd2(OAc)4(L)2 species (3) in AcOH at 373 K (100 °C; Gaussian 09, B3LYP, 6-311+G(2d,p), LANL2Dz for Pd). a The free energy of the monomer is defined as 0 kcal mol−1 in all calculations. The values for the most thermodynamically favourable species are shown in bold.
Exploring the source of the “acridine effect”: probing ligand influence on C–H activation activity by H/D exchange
To experimentally evaluate if the predicted preference for the active species 2 would translate into higher C–H activation activity under catalytically relevant conditions, H/D exchange experiments in AcOH-D4 were performed using pyridine (5), quinoline (4), and acridine (1) in combination with Pd(OAc)2 as catalysts (Scheme 3).
Scheme 3.

H/D exchange with ligands 1, 4 and 5 in AcOH-D4. TON = turnover number.
Remarkably, the turnover numbers (TONs) after 2 h for ligands 5, 4, and 1 follow the trend predicted by DFT calculations, with acridine (1) being the most activating and pyridine (5) the least activating ligand. This suggests that initial C–H activation reactivity in these systems is correlated to the relative concentration of active catalytic species 2 in solution. In contrast, long-term H/D exchange reactivity as described by the TONs after 24 h does not match the trend observed for short-term activity. As Pd black is observed in all 24 h reactions, we conclude from these data that catalytic systems with acridine (1) are more prone to catalyst decomposition than the other two systems, but that decomposition occurs regardless in all cases.
Tricylic ligands: synthesis and activity in H/D exchange
Due to the acceleration of the initial activity observed in H/D exchange with acridine (1) as the ligand, we synthesized a series of more electron-donating, tricyclic ligands 6–9, which would be expected to vary the steric bulk around the N ligation site (Scheme 4). Initial evaluations of these ligands were performed by H/D exchange in AcOH-D4 to gain insight into their C–H activation reactivity. All ligands showed 2 h TONs comparable to pyridine (5) and quinoline (4); however, ligands 6 and 9 did not result in significantly improved yields after 24 h. These low activities suggest fast catalyst decomposition, which is also accompanied by the observation of Pd black after only 30 min in these H/D exchange reaction mixtures.
Scheme 4.

H/D exchange with ligands 6–9 in AcOH-D4.
In order to rationalize the low activity of the very bulky ligand 9, we considered the DFT-derived geometry of the putative active catalyst [Pd(OAc)2(9)] (Fig. 3). Both the space- filling model and the ball-and stick model of this lowest energy configuration suggest significant steric interactions between 9 and the acetate ligands, which might accelerate the equilibrium dissociation of 9 from [Pd(OAc)2(9)]. Such dissociation would most likely lead to catalyst decomposition and reduced long-term activity, which provides a rationalization for both the very small increase in H/D exchange activity after 2 h (4 vs. 6 TONs) and the observation of fast Pd black formation even after 2 h.
Fig. 3.

Graphic representations of [Pd(OAc)2(9)] geometry in analogous projections (Gaussian 09, B3LYP, 6-311+G(2d,p), LANL2Dz for Pd, AcOH, 100 °C). Left to right: Space-filling model, ball-and-stick model, ball-and stick with H omitted for clarity.
In order to understand potential side reactions occurring with the other ligands, we investigated each reaction mixture after 24 h by GCMS. Interestingly, no traces of any modifications were observed for ligand 7; however, reaction mixtures containing 6 and 8 showed significant amounts of dehydrogenated ligands (Fig. 4) suggesting that ligand modification might result in lower activities.
Fig. 4.

Oxidative decomposition products of ligands 6 and 8.
C–H amination with new tricyclic ligands
Having established that C–H activation is relatively slow, but possible with the annulated ligands 6–9, we evaluated their potential to promote the C–H amination of benzene, using either AcNH–OAc or F3CC(O)NH–OAc as amination reagents (Scheme 5). Remarkably, all new ligands performed better than acridine (1) under all tested conditions, suggesting that electron-rich alkyl substituents contribute to C–H amination reactivity. This trend is in contrast to the diminished C–H activation behaviour observed in H/D exchange (see Scheme 4 above) and implies that catalytic steps beyond C–H activation are influenced by these ligands. As the novel ligands are more electron-rich than acridine (1), they can be expected to promote oxidation of the Pd-aryl intermediate formed through C–H activation. These data overall suggest that C–H activation might not be the rate-determining step in the established C–H amination protocol.
Scheme 5.

Ligand effects on C–H amination of benzene with different additives.
Both AcOH and AgOAc as additives were beneficial in the presence of 6–9, in analogy to previous observations with 1 as ligand (see Table 1 above). Reactions employing 5 mol% AgOAc consistently provided better yields than reactions in the presence of AcOH or higher/lower AgOAc loadings (up to 47% with AcONHAc and up to 52% with AcO–NHCOCF3).
A slight excess of ligand (15 mol% instead of 10 mol%) provided another raise in the yields, achieving up to 50% and 53% of C–H amination product, respectively. Notably, very little formation of PhOAc was observed in all of these reactions (<5%), suggesting that C–N bond formation is preferred over C–O bond formation under the established conditions. All optimal yields in this study were obtained with ligand 7. Thus, we conclude that oxidative stability combined with moderate steric bulk around the ligation site and the electron-donating features of four alkyl substituents is most beneficial for C–H amination.
Amination reagents and arene substrate scope
A study of the amination reagent scope (see Scheme 6) revealed that the reaction is generally possible with other protecting groups on N. However, sterically more bulky protecting groups than Ac (e.g. in iPrC(=O)NH–OAc) diminish the obtained yields significantly. In these reactions the yields of the C–C coupling product Ph–Ph are substantially higher than observed with either F3CCONH–OAc or AcNH–OAc as reagents, which suggests that reaction of the amination reagent with the Pd-aryl intermediate is slowed by steric bulk on N, which leads to preferential biaryl coupling (see mechanistic proposal, Scheme 8 wide infra).
Scheme 6.

Amination reagent scope. Conditions: benzene (60 equiv.), Pd(OAc)2 (15 mol%), 7 (18 mol%), RNH–OAc (140 μmol, 1.0 equiv.), 100 °C, 24 h. Yield were determined by calibrated NMR or calibrated GC analysis.
Scheme 8.

Proposed mechanism of Pd catalyzed C–H amination with hydroxylamine-derived amination reagents. Potential ligand binding sites at Pd with unknown ligands are shown as ligands X.
Additional studies employed various simple arenes as substrates (for details see ESI,† Table S12). Interestingly, electron-rich arenes such as anisol and veratrol formed preferentially the biaryl coupling product under standard conditions (see Scheme 5 above) with only traces of C–H amination product (<1%). This suggests that in these cases with relatively fast C–H activation3 the concentration of Pd-aryl species is high, favouring the bimetallic mechanism of C–C bond formation. In contrast, relatively electron-poor substrates such as PhCO2Et and PhBr formed significant amounts of the C–H amination product (14% (o : m : p = 1 : 2 : 7) and 12% (o : m : p = 1 : 3 : 7), respectively); in these cases, the biaryl coupling product was not detected. However, the highest yield achieved in this substrate scope study was 21% for toluene as substrate (o : m : p = 1 : 3 : 7), which suggested that further mechanistic studies are needed to work towards a rational design of more active and selective C–H amination catalysts and reagent.
Product inhibition study
In order to gain insight into reasons for the relatively low yields (<55%) in the established non-directed C–H amination, we tested the catalytic system for its sensitivity to product inhibition: C–H amination yields were determined in the absence and presence of 50% added PhNHAc (Scheme 7). Remarkably, all tested ligand/additive systems showed a strongly decreased yield of newly formed PhNHAc in the presence of PhNHAc. In addition to the observed formation of biphenyl representing product decomposition, this observation suggests that the product interacts with the active catalyst species and thus inhibits turnover.
Scheme 7.

Product inhibition study.
Proposed mechanism of C–H amination
Based on the above described studies, we propose the following reaction mechanism for the established C–H amination (Scheme 8): (i) sterically bulky ligands promote catalyst initiation by favoring the formation of the monomeric complex [Pd(OAc)2L] 2. According to literature studies,6,26,29,30 this is the catalytically active species for C–H activation of benzene. (ii) Catalyst inhibition in the presence of product can occur, possibly through coordination of product to form an off-cycle species such as [Pd(OAc)2(L)(PhNHR)]. (iii) Electron-rich arene substrates favour the formation of biaryl products instead of C–H amination products, but electron-poor arenes favour formation of the C–H amination product. This suggests that C–H amination products and biaryls are formed through a common intermediate, most likely a Pd-aryl species 10. Fast C–H activation (such as in electron-rich arenes) would lead to accumulation of 10, favouring bimetallic biaryl formation as described previously.25 (iv) The best results for C–H amination are obtained with F3CNH–OAc, the most oxidizing amination reagent. In combination with the observation that steric bulk of the protecting group favors biaryl formation at the expense of C–H amination, this suggests that the amination reagent directly interacts with the Pd-aryl intermediate. (v) The alkyl substituted ligands 6–9 promote C–H amination better than acridine (1), but are less active in H/D exchange. In combination with the observed effects of different amination reagents, these ligand effects suggest that oxidation of the Pd-aryl intermediate is rate-determining under the established conditions, which is promoted by the more electron-donating ligands 6–9. Scheme 8 visually summarizes all these results into one mechanistic proposal.||31
Mechanistic studies
In order to investigate if C–H bond cleavage occurs in the reaction, we determined the kinetic isotope effect (KIE) under intermolecular competition conditions (Scheme 9).32 To this end, a 1 : 1 mixture of benzene and benzene-D6 was employed and the KIE was determined by GCMS analysis of the product peak. Under all conditions, a primary KIE (>2) was observed, suggesting that C–H activation occurs in the catalytic cycle leading to C–H amination. However, this experiment does not require that C–H cleavage is the rate determining step.32
Scheme 9.

Competition kinetic isotope effect with Pd(OAc)2/7.
Other data discussed further above (H/D exchange dependence on ligands; reactivity dependence on amination reagents) indeed suggests that the rate-determining step of the developed protocol is the oxidation of the Pd-aryl intermediate formed by C–H activation. As such, the C–H cleavage process could be reversible. We hypothesized that quantifying the deuterium (D) incorporation into benzene recovered from the reaction in the presence of AcOH-D4 should provide insight into the reversibility of C–H activation. If C–H cleavage is irreversible, we would expect no D incorporation into the recovered starting material, as all Pd-aryl species would go on to either form biaryl or C–H aminated products. In contrast, if C–H cleavage is a reversible reaction, followed by irreversible oxidation of the Pd-aryl species, significant D incorporation into recovered benzene can be expected. In this case, if the Pd-aryl intermediate (10; see Scheme 8 above) formed by C–H activation can readily undergo exchange of ligated AcOH with AcOH-D4, then D incorporation would occur through the microscopic reverse of C–H activation, i.e. by protonation of the aryl ligand by bound AcOH-D4. Thus, in order to elucidate if C–H activation is indeed reversible under the reaction conditions, a C–H amination reaction was performed in the presence of 0.60 equivalent (84 μmol) of AcOH-D4 (Scheme 10).
Scheme 10.

Deuterium incorporation into benzene recovered from reaction mixture with Pd(OAc)2/7 catalyst system.
The resulting isotope distribution of recovered benzene was calculated by deconvoluting fragmentation data obtained by GCMS analysis (see ESI† for details). Notably, this analysis showed that 0.30 equiv. (42 μmol) of benzene molecules had been transformed into C6H5D, suggesting that C–H activation is reversible under the established C–H amination conditions and that the rate-determining step takes place after C–H cleavage.
In an effort to better understand if the proposed Pd-aryl complex 10 (Scheme 8 above) is a feasible intermediate of C–H amination, we synthesized organometallic complex 11 (Scheme 11). We considered 11 to be a good model for 10, as it contains a non-cyclometalated aryl ligand, an acetate ligand (similar to the proposed intermediate 10) as well as two L-type ligands (AcOH/X in 10) which are combined into the chelating ligand 4,4′-di-tert-butyl-2,2′-bipyridyl (tBu2bipy). We further hypothesized that if 11 is a good model complex for 10, it should allow the observation of several pathways shown in the proposed mechanism above (Scheme 8), such as protonation by AcOH (the microscopic reverse of C–H activation), biaryl formation, as well as C–N bond formation. Furthermore, we expected to gain initial insights into the effect of catalytic additives (e.g. AcOH, AgOAc) and the chemoselectivity of C–N vs. C–O bond formation.
Scheme 11.

Reactivity of isolated Pd-aryl complex 11 with amination reagent AcNH–OAc in the presence of different catalytically beneficial additives.
To evaluate 11 for reactivity in the presence of hydroxyl-based amination reagents, a 1 : 1 mixture of 11 and AcNH–OAc was heated to 85 °C in C6D6 (Scheme 11). In the absence of any additives, bitolyl 15 (32%) was the major product along with small amounts of toluene (13; 5%). Interestingly, the presence of AcOH, a promoter of C–H amination reactivity (see Table 1 and Scheme 5 above), considerably depressed the formation of the dimerization product 15 to 7%, albeit again in the absence of any other functionalized products. This suggests that the role of AcOH in the catalytic C–H amination reaction mixture may be to suppress biaryl formation.
Excitingly, in the presence of AgOAc, the most efficient additive in catalytic C–H aminations, 7% of C–N bond formation product 12 was obtained; even higher yields of 12 (14%) were observed in the presence of pentafluoropyridine (C6F5N), which had shown promise as a catalytic additive in early optimizations with acridine (1) (see ESI,† Table S1). These data suggest that both AgOAc or C6F5N promote C–N bond formation over biaryl formation, potentially through providing an additional ligand for a high-oxidation state Pd species. Notably, less than 1% of oxygenation product 14 was obtained under any of the reaction conditions shown in Scheme 11, suggesting that C–N bond formation outperforms C–O bond formation with model complex 11.
Overall, the described mechanistic studies suggest that C–H bond cleavage in the established C–H amination protocol occurs before reaction of the Pd catalyst with the amination reagent and that C–H activation is reversible. Furthermore, stoichiometric studies provide insight into the roles of catalytic additives and point towards a mechanism inherently favouring C–N over C–O bond formation.
Conclusions
In conclusion, we describe herein a catalytic system that enables the chemoselective synthesis of aniline derivatives by Pd catalyzed C–H amination of benzene, using hydroxylamine derivatives as concurrent oxidant and amine source. Preliminary mechanistic investigations suggest that the steric bulk of tricyclic pyridine-type ligands promotes C–H activation reactivity by favouring the reactive, monomeric catalyst [Pd(OAc)2L] (2) over the corresponding dimeric Pd species. Based on this result, a series of electron-donating, alkyl substituted tricyclic ligands (6–9) was developed, which enhance the activity of the resulting catalysts towards C–H amination. KIE studies indicate that concerted C–H bond cleavage occurs in the catalytic cycle; however, the observation of D incorporation in recovered starting material (benzene) suggests that C–H activation is reversible under C–H amination conditions and that a step after C–H bond activation is rate determining. The documented product inhibition provides another reason for why non-directed C–H aminations are less developed than non-directed C–H oxygenations or olefinations, as the products of C–H amination catalysis may deactivate the catalyst by coordination. Moreover, stoichiometric studies provide initial insights into the effects of additives on C–H amination catalysts.
Supplementary Material
Footnotes
Electronic supplementary information (ESI) available: Detailed procedures for ligand preparation and catalytic experiments, results of catalytic experiments including standard deviations, details of DFT calculations. See DOI: 10.1039/ c6cy00457a
Similar reagents have previously been employed by others directed, Pd catalyzed and ferrocene catalyzed aromatic C–H aminations; see ref. 22–24.
As an intramolecular C–H amination reaction with hydroxylamine-derived reagents has been documented to likely proceed through a Pd(0/II) mechanism, a Pd(0/II) catalytic cycle is an alternative possibility for the herein discussed reactivity. However, the stoichiometric reactions detailed in Scheme 11 are more consistent with oxidation of a Pd(II)-aryl species by the amination reagents.
Notes and references
- 1.Engle KM, Yu JQ. J Org Chem. 2013;78:8927–8955. doi: 10.1021/jo400159y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emmert MH, Cook AK, Xie YJ, Sanford MS. Angew Chem, Int Ed. 2011;50:9409–9412. doi: 10.1002/anie.201103327. [DOI] [PubMed] [Google Scholar]
- 3.Emmert MH, Gary JB, Villalobos JM, Sanford MS. Angew Chem, Int Ed. 2010;49:5884–5886. doi: 10.1002/anie.201002351. [DOI] [PubMed] [Google Scholar]
- 4.Kuhl N, Hopkinson MN, Wencel-Delord J, Glorius F. Angew Chem, Int Ed. 2012;51:10236–10254. doi: 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]
- 5.Cook AK, Emmert MH, Sanford MS. Org Lett. 2013;15:5428–5431. doi: 10.1021/ol4024248. [DOI] [PubMed] [Google Scholar]
- 6.Cook AK, Sanford MS. J Am Chem Soc. 2015;137:3109–3118. doi: 10.1021/jacs.5b00238. [DOI] [PubMed] [Google Scholar]
- 7.Zhang YH, Shi BF, Yu JQ. J Am Chem Soc. 2009;131:5072–5074. doi: 10.1021/ja900327e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kubota A, Emmert MH, Sanford MS. Org Lett. 2012;14:1760–1763. doi: 10.1021/ol300281p. [DOI] [PubMed] [Google Scholar]
- 9.Stuart DR, Fagnou K. Science. 2007;316:1172–1175. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]
- 10.Izawa Y, Stahl SS. Adv Synth Catal. 2010;352:3223–3229. doi: 10.1002/adsc.201000771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hickman AJ, Sanford MS. ACS Catal. 2011;1:170–174. [Google Scholar]
- 12.Shang M, Sun SZ, Dai HX, Yu JQ. J Am Chem Soc. 2014;136:3354–3357. doi: 10.1021/ja412880r. [DOI] [PubMed] [Google Scholar]; Thirunavukkarasu VS, Kozhushkov SI, Ackermann L. Chem Commun. 2014;50:29–39. doi: 10.1039/c3cc47028h. [DOI] [PubMed] [Google Scholar]; Zhu D, Yang G, He J, Chu L, Chen G, Gong W, Chen K, Eastgate MD, Yu JQ. Angew Chem, Int Ed. 2015;54:2497–2500. doi: 10.1002/anie.201408651. [DOI] [PubMed] [Google Scholar]; Shin K, Kim H, Chang S. Acc Chem Res. 2015;48:1040–1052. doi: 10.1021/acs.accounts.5b00020. [DOI] [PubMed] [Google Scholar]
- 13.Boursalian GB, Ngai MY, Hojczyk KN, Ritter T. J Am Chem Soc. 2013;135:13278–13281. doi: 10.1021/ja4064926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen LJ, Cabrera PJ, Lee M, Sanford MS. J Am Chem Soc. 2014;136:5607–5610. doi: 10.1021/ja501906x. [DOI] [PMC free article] [PubMed] [Google Scholar]; Romero NA, Margrey KA, Tay NE, Nicewicz DA. Science. 2015;349:1326–1330. doi: 10.1126/science.aac9895. [DOI] [PubMed] [Google Scholar]
- 15.Bandara HMD, Sosin MH, McKeogh BJ, Emmert MH. GSTF J Chem Sci. 2013;1:17–31. [Google Scholar]
- 16.Kantak AA, Potavathri S, Barham RA, Romano KM, DeBoef B. J Am Chem Soc. 2011;133:19960–19965. doi: 10.1021/ja2087085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foo K, Sella E, Thome I, Eastgate MD, Baran PS. J Am Chem Soc. 2014;136:5279–5282. doi: 10.1021/ja501879c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawakami T, Murakami K, Itami K. J Am Chem Soc. 2015;137:2460–2463. doi: 10.1021/ja5130012. [DOI] [PubMed] [Google Scholar]
- 19.Kim HJ, Kim J, Cho SH, Chang S. J Am Chem Soc. 2011;133:16382–16385. doi: 10.1021/ja207296y. [DOI] [PubMed] [Google Scholar]; Antonchick AP, Samanta R, Kulikov K, Lategahn J. Angew Chem, Int Ed. 2011;50:8605–8608. doi: 10.1002/anie.201102984. [DOI] [PubMed] [Google Scholar]
- 20.John A, Byun J, Nicholas KM. Chem Commun. 2013;49:10965–10967. doi: 10.1039/c3cc46412a. [DOI] [PubMed] [Google Scholar]; Wang S, Ni Z, Huang X, Wang J, Pan Y. Org Lett. 2014;16:5648–5651. doi: 10.1021/ol502724u. [DOI] [PubMed] [Google Scholar]
- 21.Shrestha R, Mukherjee P, Tan Y, Litman ZC, Hartwig JF. J Am Chem Soc. 2013;135:8480–8483. doi: 10.1021/ja4032677. [DOI] [PubMed] [Google Scholar]
- 22.Zhu D, Yang G, He J, Chu L, Chen G, Gong W, Chen K, Eastgate MD, Yu JQ. Angew Chem, Int Ed. 2015;54:2497–2500. doi: 10.1002/anie.201408651. [DOI] [PubMed] [Google Scholar]
- 23.Dong Z, Dong G. J Am Chem Soc. 2013;135:18350–18353. doi: 10.1021/ja410823e. [DOI] [PubMed] [Google Scholar]
- 24.Foo K, Sella E, Thome I, Eastgate MD, Baran PS. J Am Chem Soc. 2014;136:5279–5282. doi: 10.1021/ja501879c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yagyu T, Hamada M, Osakada K, Yamamoto T. Organometallics. 2001;20:1087–1101. [Google Scholar]
- 26.Zhang S, Shi L, Ding Y. J Am Chem Soc. 2011;133:20218–20229. doi: 10.1021/ja205294y. [DOI] [PubMed] [Google Scholar]
- 27.Powell J, Jack T. Inorg Chem. 1972;11:1039–1048. [Google Scholar]
- 28.O’Keefe BJ, Steel PJ. Organometallics. 1998;17:3621–3623. [Google Scholar]
- 29.Ke Z, Cundari TR. Organometallics. 2010;29:821–834. [Google Scholar]
- 30.Dick AR, Remy MS, Kampf JW, Sanford MS. Organometallics. 2007;26:1365–1370. [Google Scholar]
- 31.Tan Y, Hartwig JF. J Am Chem Soc. 2010;132(11):3676–3677. doi: 10.1021/ja100676r. [DOI] [PubMed] [Google Scholar]
- 32.Simmons EM, Hartwig JF. Angew Chem, Int Ed. 2012;51:3066–3072. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.