

Abstract
Manufacturers of insulin products for diabetes therapy have long sought ways to modify the absorption rate of exogenously administered insulins in an effort to better reproduce the naturally occurring pharmacokinetics of endogenous insulin secretion. Several mechanisms of protraction have been used in pursuit of a basal insulin, for which a low injection frequency would provide tolerable and reproducible glucose control; these mechanisms have met with varying degrees of success. Before the advent of recombinant DNA technology, development focused on modifications to the formulation that increased insulin self‐association, such as supplementation with zinc or the development of preformed precipitates using protamine. Indeed, NPH insulin remains widely used today despite a frequent need for a twice‐daily dosing and a relatively high incidence of hypoglycaemia. The early insulin analogues used post‐injection precipitation (insulin glargine U100) or dimerization and albumin binding (insulin detemir) as methods of increasing therapeutic duration. These products approached a 24‐hour glucose‐lowering effect with decreased variability in insulin action. Newer basal insulin analogues have used up‐concentration in addition to precipitation (insulin glargine U300), and multihexamer formation in addition to albumin binding (insulin degludec), to further increase duration of action and/or decrease the day‐to‐day variability of the glucose‐lowering profile. Clinically, the major advantage of these recent analogues has been a reduction in hypoglycaemia with similar glycated haemoglobin control when compared with earlier products. Future therapies may bring clinical benefits through hepato‐preferential insulin receptor binding or very long durations of action, perhaps enabling once‐weekly administration and the potential for further clinical benefits.
Keywords: basal insulin, pharmacodynamics, pharmacokinetics, hypoglycaemia
1. INTRODUCTION
Endogenous insulin is stored as hexamers (formed by 3 dimers combining with 2 zinc ions), together with cargo ions, molecules and proteins in β‐cell vesicles of the pancreas. Once released into the bloodstream, hexamers rapidly dissociate into biologically active monomers.1 Secretion of endogenous insulin is dynamic in response to physiological need, but there is typically a constant “basal” level of insulin secretion upon which rapidly produced prandial peaks of secretion in response to food intake are superimposed. The absorption kinetics of unmodified human insulin after subcutaneous (s.c.) injection, however, match neither the physiological basal nor the prandial secretion fully because of the rate at which the injected hexamers dissociate into the smaller dimers and monomers, which in turn are able to penetrate capillary membranes. Manufacturers have therefore sought ways to modify the absorption kinetics of exogenously administered insulin to reproduce the dynamic insulin profile of normal physiology more accurately. Consequently, fast‐acting insulin products are available that are given at mealtimes to suppress postprandial blood glucose excursions, while long‐acting basal insulin products provide a constant suppression of hepatic glucose release between mealtimes and overnight.1
In the case of developing basal insulins, the absorption rate needs to be slowed as much as possible, thereby permitting a low injection frequency that produces a steady‐state profile with a low peak:trough ratio.2 The major challenge in developing such a basal insulin is to achieve stable release profiles, thereby allowing reproducible glucose control.
Various approaches have been made to modify the native insulin molecule and/or its formulation to develop safe and efficacious basal insulin products. These include re‐formulation (e.g. cobalt substitution of zinc, higher zinc concentration), precipitation, protein binding (namely albumin), up‐concentration of formulations (including the up‐concentration of insulins with additional protraction mechanisms), the formation of higher‐order structures after injection (e.g. multihexamer chains and precipitates), PEGylation, and conjugation to non‐glycosylated human Fc. The impact of these different modes of protraction on the resulting pharmacokinetic (PK)/pharmacodynamic (PD) profiles and, ultimately, clinical outcomes is reviewed in the present paper, along with possible future strategies.
2. INSULINS AND FORMULATIONS ENGINEERED FOR INCREASED SELF‐ASSOCIATION
2.1. NPH insulin
Precipitation, as a mechanism of protraction of insulin action, was first successfully achieved by the addition to the formulation of protamine, together with zinc.3 The resulting intermediate‐acting insulin, NPH, is protracted by being injected as a suspension of preformed protein–insulin conglomerates, formed from insulin and protamine in a 5 : 1 molar ratio.4
On injection, the solvent from NPH insulin suspensions diffuses freely into the s.c. tissue, much as soluble insulin would do after injection, but the insulin crystals are retained in “heaps” at the injection depot (Figure 1).5, 6 These “heaps” dissolve slowly, hence NPH action is protracted; however, the conglomerates of protein and insulin under the skin vary in shape and size; therefore, so too do the absorption kinetics, from one injection to another, according to the depot environment and injection technique.6 Invading macrophages and protamine‐splitting enzymes in the s.c. tissue are thought to be responsible for dissolution of NPH insulin heaps and are one example of a variable present in the injection environment.6
Figure 1.
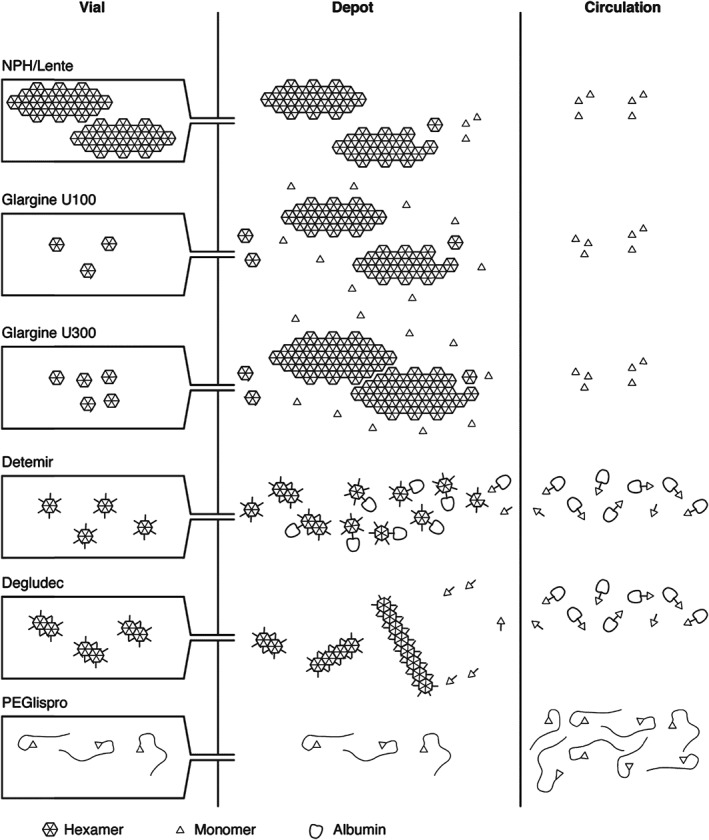
Summary of the different mechanisms of protraction. NPH insulin is injected as a pre‐formed protein–insulin conglomerate. On injection, the solvent from NPH insulin suspensions diffuses freely into the subcutaneous tissue but the crystals are retained in “heaps” at the injection depot. IGlar U100 is soluble in acidic formulation but, on subcutaneous injection and reaching physiological pH, it forms crystals. IGlar U300 also precipitates at physiological pH but these precipitates are much more compact compared with those of IGlar U100, so the surface area from which absorption can occur is reduced, thereby further slowing absorption. Acylation of IDet with a fatty acid side chain facilitates self‐association of IDet at the injection depot as dihexamers and reversible binding to albumin, both in the depot and in circulation, thereby slowing its absorption. IDeg also has a fatty acid side chain, which facilitates dihexamer formation in the vial and albumin binding in the circulation. However, protraction of absorption is primarily achieved via multihexamer chain formation in the depot. Subsequent dissociation of zinc causes the terminal hexamers to break down. The large hydrodynamic size of PEGlispro prolongs its action by slowing absorption and reducing clearance, effectively producing a circulating depot. The PEGlispro clinical trial programme was terminated in 2015.
Another major source of variability in the PK/PD profile of NPH arises because the product requires adequate resuspension (e.g. by rolling the vial 20 times) before s.c. injection. Failure to achieve resuspension can alter the PK/PD profile in different ways, depending on the angle at which the injection device was stored.7, 8
Although the duration of action of NPH is protracted compared with regular soluble insulin, it is typically only 12 to 14 hours in total, and this relatively short duration is associated with a peak effect at 4 to 6 hours, gradually declining thereafter; therefore, NPH usually requires at least twice‐daily dosing to ensure that basal insulin levels are sustained across 24 hours.9, 10 Variability in both resuspension technique and the formation and dissolution of the precipitate contribute to the substantial injection‐to‐injection variability in PD profiles observed for individuals treated with NPH.8, 11 Consequently, hypoglycaemia – particularly nocturnal hypoglycaemia, resulting from unpredictability in the nocturnal insulin peak after evening administration – is a common issue for patients treated with NPH.12 Nevertheless, NPH insulin is a low‐cost product that remains widely used today.13
2.2. Lente insulins
Hallas‐Møller and colleagues first developed the Lente insulin suspensions in the 1950s by complexing animal‐derived insulin suspensions with small amounts of zinc. Insulin action was protracted because of the slow dissolution of crystals, as reviewed by Owens et al.14 Advantage was taken of the different solubilities of bovine and porcine insulin at a neutral pH to create a trilogy of zinc–insulin suspensions: Lente (3 : 7 mixture of amorphous porcine insulin and bovine crystalline particles with an “intermediate” duration of action similar to that of NPH), Semilente (amorphous insulin particles) and Ultralente (large bovine crystalline particles providing the first “long”‐acting insulin preparation).15 The Lente insulins were reformulated to use recombinant human insulin after the advent of commercial production of human insulin in the 1980s, but they remained limited by their ≤24‐hour duration of action,9 the need for resuspension, and their highly variable action profile. Another limitation, more so before the advent of the insulin pen device, was that Lente insulins could not be mixed with human soluble insulin. This is in contrast to NPH, the absorption kinetics of which are unaffected when mixed with human insulin.16
2.3. Co(III) insulin
A subsequent strategy, which did not make it to clinical practice, was to strengthen insulin hexamers by substituting the zinc ions with cobalt (Co).17 While Co(III) insulin did behave as predicted, with the hexamers being slower to dissociate, it offered no real pharmacological advantages over NPH insulin.18 Instead, alternative strategies were undertaken, including modifying the amino acid sequence of the insulin itself to produce insulin analogues.
3. PRECIPITATION AFTER INJECTION
In pursuit of a more stable and protracted release formulation, research focused on shifting the iso‐electric point of insulin by altering the insulin molecule itself to enable precipitation at physiological pH after injection. This approach enables the insulin to be injected as a solute (in a slightly acidic formulation) that forms precipitates (of various sizes) in the neutral environment of the subcutis (Figure 1). The process therefore avoids the problems associated with resuspension.
3.1. NovoSol Basal
NovoSol Basal was the first insulin analogue to employ this mechanism of protraction by virtue of 2 amino acid substitutions (B27Arg, A21Gly) and amidation of the C‐terminal of the human insulin B chain (Figure 2).19 After injection, crystals <5 µm in size form and slow absorption follows, such that the T50% of NovoSol Basal (namely, the time elapsed until 50% of injected radio‐labelled insulin has disappeared from the depot) is significantly longer than that of Ultratard HM, a long‐acting human insulin (35.3 hours vs 25.5 hours; P < .001).20 NovoSol Basal was also shown to result in less within‐patient variation versus Ultratard HM, but between‐patient variation remained high. As bioavailability was markedly reduced with NovoSol Basal versus Ultratard HM, necessitating high doses, and as NovoSol Basal was also associated with local inflammatory reactions, it was withdrawn from further studies.21, 22, 23
Figure 2.
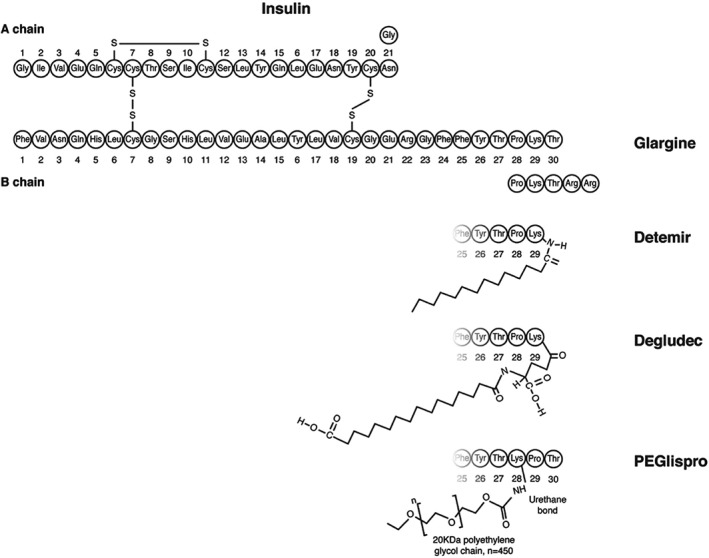
Molecular structure of insulin analogues. Molecular modifications made to the human insulin molecule in order to protract action are shown. The isoelectric point of IGlar U100 was raised by substituting glycine 21 on the A chain (A21) of human insulin for asparagine, and adding 2 asparagine molecules to the amino terminal of the B chain. IDet is an analogue in which threonine B30 has been removed and lysine B29 is acylated with a 14‐carbon myristoyl fatty acid. Threonine B30 is also removed in IDeg but lysine B29 is attached to a 16‐carbon fatty diacid via a glutamic acid spacer. In PEGlispro, the order of proline and lysine is reversed such that proline 29 follows lysine 28, which is attached to a polyethylene glycol chain via a urethane bond.
3.2. Insulin glargine U100
Insulin glargine 100 units/mL (IGlar U100) was the first basal insulin analogue approved for clinical use. It is engineered to have an iso‐electric point of pH 6.7, achieved by substituting glycine A21 on the A chain of human insulin for asparagine, and adding 2 asparagine molecules to the amino terminal of the B chain (Figure 2).24, 25 The post‐injection precipitation of IGlar U100 results in a longer, flatter time–action profile versus NPH.25, 26 The mean duration of IGlar U100 action, namely, the time between injection and blood glucose levels rising again in clamp studies to reach 8.3 mmol/L (150 mg/dL),26 has been reported to be 22 to 24 hours under single‐dose conditions,9, 10, 11 and 24 to 25.6 hours under steady‐state conditions,27, 28 with a gentle rise and fall in the PK/PD profile across this interval.26, 28 Consequently, once‐daily dosing is effective in most patients, although not all.26, 29 There is evidence of a waning of effect over 24 hours with once‐daily IGlar U100,30 and clinical studies have confirmed that glycaemic control can be further improved in some patients with type 1 diabetes with twice‐daily dosing of IGlar U100.29, 31
Precipitation itself is inherently variable6 and so, although IGlar U100 is associated with less within‐subject variability than NPH in clinical practice, where patient adherence to resuspension protocol varies,7 the problem of a variable PK/PD profile from injection to injection is not completely eliminated.11 A repeat‐clamp study in which IGlar U100 was compared with NPH (given after controlled resuspension) did suggest reduced variability in the injection‐to‐injection PD profile with IGlar U100 versus NPH, although this was not statistically tested.11 In clinical studies, once‐daily dosing of IGlar U100 resulted in lower risks of nocturnal hypoglycaemia versus once‐ or twice‐daily dosing of NPH in patients with type 132 and type 2 diabetes,33, 34, 35 and IGlar U100 is now the most widely used basal insulin worldwide. However, while the mean PK/PD and variability profiles of IGlar U100 represent welcome improvements on those of NPH that translate into clinical advantages, the within‐patient injection‐to‐injection variability of IGlar U100 remains relatively high compared with more recently developed basal insulins such as insulin detemir (IDet),11 insulin degludec (IDeg)36 and PEGylated insulin lispro.37
4. PROTEIN‐BOUND INSULIN ANALOGUE: INSULIN DETEMIR
There are several properties unique to IDet compared with other insulins, although the possible connection between these properties and IDet's mechanism of protraction has yet to be fully elucidated. IDet is a pH‐neutral, soluble insulin analogue that is acylated at residue B29‐lysine with a 14‐carbon myristoyl fatty acid. This facilitates self‐association of IDet molecules at the injection depot as dihexamers, and reversible binding to albumin, thereby slowing its systemic absorption (Figure 1).38, 39 The mean duration of action for IDet (using the same definition as for IGlar U100 above) was calculated to be 21.5 hours in people with type 1 diabetes, hence slightly shorter than for IGlar U100, based on data for a 0.4 U/kg dose level in a single‐dose clamp study10, 26; however, a head‐to‐head, double‐blind clamp study showed no appreciable difference in duration of effect at clinically relevant doses in people with type 2 diabetes who still had some (although low) endogenous insulin secretion capacity.27
The observational study PREDICTIVE showed that, similarly to IGlar U100, once‐daily dosing of IDet is possible for some patients with type 1 diabetes, but many require twice‐daily dosing.40 Compared with IGlar U100 and NPH, IDet produces significantly lower (between 2‐ and 4‐fold lower) within‐patient variability in the glucose‐lowering response from injection to injection, as shown in a large‐scale, repeated clamp study11 and a crossover trial involving 32 children or adolescents with type 1 diabetes.41 This may be partly attributable to the reversible binding of IDet to albumin in the circulation, buffering changes in absorption rate caused by changes in local blood flow at the depot.18, 27, 38
Insulin detemir therefore provides more predictable glycaemic control with consistent risk reductions for hypoglycaemia and less weight gain versus NPH in clinical trials.26, 39 The majority of studies comparing IDet and IGlar U100 show similar rates of hypoglycaemia,42, 43, 44, 45, 46 with the exception of Pieber et al.47 who showed a benefit of twice‐daily IDet in nocturnal hypoglycaemia compared with once‐daily IGlar U100. Less weight gain for IDet versus IGlar U100 is consistently observed, ranging from 25% up to 50% less weight gain with IDet, with this advantage being greatest when IDet is dosed once daily. The reasons for this relative reduction in weight gain are not yet understood, but might be related to a slight hepato‐preferential effect48 or satiety effects on the central nervous system.49
Insulin detemir is formulated at 4 times the concentration of human insulin because it has a lower molar potency than human insulin and other insulin analogues.50 It has been proposed that this might reflect the reduced affinity of IDet for the insulin receptor18; however, a reduced molar potency in glucose‐lowering effect is generally not seen with other insulin analogues that have a reduced receptor‐binding affinity, nor in other albumin‐binding analogues. The reasons for the reduced molar potency of IDet have therefore yet to be elucidated. From a clinical perspective, however, the important point is that one unit of IDet is defined as 24 nmol, as opposed to 6 nmol for other insulins, but this has a similar total blood glucose‐lowering effect to one unit of other insulins.18 The increased molar concentration of IDet therefore facilitates unit‐for‐unit switches between basal insulins, and should not be confused with the up‐concentration of other basal insulins (described below) that has been used as a method of increasing duration or reducing injection volume.
5. MULTIHEXAMER CHAIN FORMATION: INSULIN DEGLUDEC
Insulin degludec is an analogue in which threonine has been removed at B30, and B29 has been acetylated with a 16‐carbon fatty diacid via a glutamic acid spacer. This change confers a slower rate of absorption to IDeg by enabling formation of high‐molecular‐weight complexes, and albumin‐binding after s.c. injection. IDeg forms highly stable dihexamers (closed configuration) in phenol‐ and zinc‐containing formulation as a result of an interaction between one of the fatty diacid side chains of one hexamer and a zinc atom of another (Figure 3).51, 52, 53 In the absence of phenol, as will occur after injection, the IDeg dihexamers change to adopt an open configuration allowing the multihexamer chains to form, as shown by size‐exclusion chromatography.2 The dihexamers link up to form these multihexamer chains, again by the interaction of fatty diacid chains and zinc atoms between adjacent hexamers.54 Subsequent diffusion of zinc from each terminal of the chain causes the terminal hexamers to break apart into dimers, which then dissociate into monomers. This process results in a steady and gradual release of monomers, which are absorbed into the systemic circulation.2, 55
Figure 3.
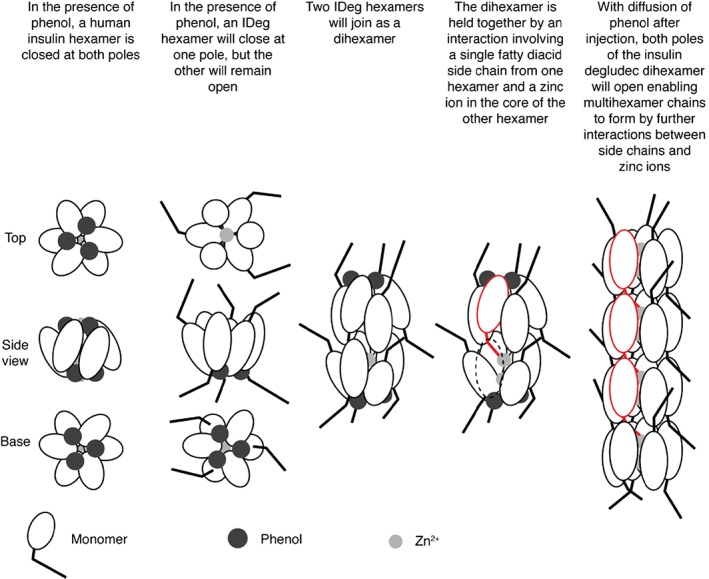
IDeg dihexamer formation.51, 52, 53 Insulin hexamers are arranged such that they have 2 poles, each formed by 3 of the constituent monomers, and these poles can be “open” (to expose the zinc‐containing core of the hexamer) or “closed” (shielding the core). In the presence of phenol or phenolic derivatives, which bind to hydrophobic pockets of the hexamers, the poles are closed.51, 52, 53 Two IDeg hexamers link together to form stable dihexamers, by the interaction of a single fatty diacid chain from one hexamer with a zinc atom of a neighbouring hexamer. On subcutaneous injection, these dihexamers link up to form multihexamer chains in the same manner because depletion of phenol after injection causes the closed poles to open, thereby exposing the second zinc ion. Ultimately, diffusion of zinc causes the terminal hexamers of these chains to break down into dimers, which then dissociate into monomers. Figure adapted from Jonassen et al.2 Republished with permission from Springer New York LLC; permission conveyed through Copyright Clearance Center, Inc.
In a head‐to‐head, 42‐hour glucose clamp study in patients with type 1 diabetes, the mean half‐life of IDeg 100 units/mL (U100) was 25.4 hours versus 12.1 hours for IGlar U100, with at least a 42‐hour duration of IDeg U100 action at steady state using once‐daily administration of 0.4 U/kg.56 The coefficient of variation of glucose‐lowering effect of IDeg U100 was 4 times lower than for IGlar U100 at the same dose (20% vs 82%), with a more even distribution of the glucose‐lowering effect over 24 hours.36, 57
Importantly, once‐daily dosing of the same dose of an insulin analogue with a duration of action >24 hours will not result in excessive accumulation or “stacking” in the patient. Rather, the serum insulin concentration will accumulate slowly until a very flat, steady‐state profile is reached in 3 to 5 days. It is therefore important to note that patients should allow 3 days before performing dose adjustments.58
IDeg U100 provided glycaemic control with a reduced risk of nocturnal hypoglycaemia versus IGlar U100 for patients with type 1 diabetes and patients with type 2 diabetes in the phase III clinical trial programme59 and in subsequent 64‐week, double‐blind, crossover trials (SWITCH 160 and SWITCH 261). A prespecified meta‐analysis of patient‐level data revealed a significantly lower risk of overall confirmed (17% lower) and nocturnal confirmed hypoglycaemic events (32% lower) for patients with type 2 diabetes receiving IDeg U100 versus IGlar U100.59 The rate of nocturnal confirmed hypoglycaemic events was also significantly (25%) lower for IDeg U100 versus IGlar U100 in the type 1 diabetes population (where rapid‐acting mealtime insulins are routinely used), whereas the rate of overall confirmed hypoglycaemia was trending higher, although it did not reach statistical significance. In both populations, treatment differences were more pronounced after 16 weeks of IDeg U100 initiation, when the insulin doses had reached stable levels.59 A separate meta‐analysis of additional endpoints in the clinical trial programme showed that, in both the pooled basal–bolus‐treated type 1 diabetes and pooled insulin‐naïve type 2 diabetes trial populations, the lower rates of hypoglycaemia were achieved with a significantly lower insulin dose versus IGlar U100: 12% and 10% lower, respectively.62 Similar risk reductions were observed in subsequent 64‐week, double‐blind, crossover trials in which IDeg U100 resulted in significantly lower hypoglycaemia (overall, severe and nocturnal) throughout the trial period versus IGlar U100 in patients with type 1 diabetes60 and in patients with type 2 diabetes61 who were at high risk of hypoglycaemia.
Also of clinical importance is the suitability of IDeg U100 for dosing any time of day, by virtue of its long duration of action and flat, predictable PD profile. This capability has been shown in 2 26‐week, open‐label, treat‐to‐target trials in type 1 and type 2 diabetes trial populations in which IDeg U100 given in a “forced‐flexible” schedule (with minimum 8, maximum 40 hours between doses) was compared with IDeg U100 or IGlar U100 given at the same time daily.63, 64
The formation of stable dihexamers in formulation also offers the potential for IDeg U100 to be co‐formulated with the rapid‐acting analogue insulin aspart without hybrid hexamers forming,65 or with liraglutide, a glucagon‐like peptide‐1 receptor agonist. The clinical utility of these fixed‐combination products has been demonstrated in several cohorts of patients with type 2 diabetes.66, 67
6. IMPACT ON THE PHARMACOKINETIC/PHARMACODYNAMIC PROFILE OF UP‐CONCENTRATED FORMULATIONS
The impact of up‐concentration on insulin absorption has long been observed with regular mealtime insulins and has since been employed to further protract the absorption of basal insulin analogues. For example, Humulin R U‐100 has a significantly shorter duration of action and a higher peak effect compared with its up‐concentrated formulation, Humulin R U‐500,68 which, at least at very high doses, has a blood glucose‐lowering effect lasting 21 hours. Onset of action, however, remains similar such that dosing 30 minutes before mealtime is suitable for both concentrations.68 This phenomenon, whereby up‐concentration of regular insulin results in a longer action profile, is explained by the observation that more compact conglomerates are formed: absorption is slowed because the surface area from which absorption can occur is reduced and there is a greater distance from this surface to capillaries.69 The availability of up‐concentrated, lower‐volume basal insulin formulations in devices capable of delivering >80 units per dose offers the possibility of fewer injections for patients requiring high doses of basal insulin; however, because of the alterations in the PD profile and potential changes in bioavailability, studies have investigated whether switching to the up‐concentrated formulation may require adjustments to dose and regimen.
6.1. Insulin glargine U300
When concentrating IGlar U100 to 300 U/mL (IGlar U300) for the purpose of being able to administer higher insulin doses in smaller volumes, the expected protraction in action profile was observed. After 1 week at steady state (0.4 units/kg/day), the half‐life was 19.0 hours for IGlar U300 versus 13.5 hours for IGlar U100.70 Importantly, the PK/PD profile was flatter than that of IGlar U100 and the duration of action extended upwards to 32 hours in patients with type 1 diabetes.70 A duration of action that extends beyond 24 hours is advantageous as it means that with once‐daily dosing, circulating insulin concentrations will rise over a few days until a steady‐state profile with a low peak:trough ratio is reached. This reduces waxing and waning of effect, thereby reducing the risk of hypoglycaemia.58 It is also important to note, however, that a significant 27% relative reduction in the biopotency of IGlar U300 at steady state was observed in this study versus IGar U100.70 IGlar U100 and U300 are therefore neither bioequivalent nor directly interchangeable, and a patient switching to the up‐concentrated formulation will need to adjust dose accordingly.71 The definition of a unit of IGlar U300 does not take the lower biopotency of IGlar U300 versus IGlar U100 into account, and therefore higher doses of IGlar U300 versus IGlar U100 would be expected. This was found to be the case in clinical trials, with a 12% higher dose of IGlar U300 required after 6 months,72 and a 44% higher dose increase than with IGlar U300 over 1 year.73
Results from a post hoc meta‐analysis of EDITION I to III, in which the safety and efficacy of IGlar U100 and IGlar U300 were compared in patients with type 2 diabetes, reported that non‐inferiority of IGlar U300 compared with IGlar U100 regarding fasting plasma glucose and glycated haemoglobin (HbA1c) reduction was achieved within 12 weeks. Superiority of IGlar U300 over IGlar U100 in terms of hypoglycaemia was confirmed in the meta‐analysis, with a significantly lower relative risk of overall [confirmed (≤3.9 mmol/L or ≤70 mg/dL) or severe] hypoglycaemia [relative risk 0.86, 95% confidence interval (CI) 0.74; 0.97] for the total duration of the trial. The study's titration phase was defined as ending at week 9; however, insulin dose did not stabilize until week 16.72 Nevertheless, while IGlar U300 showed a significant reduction in the rate of confirmed overall and nocturnal hypoglycaemia during the titration phase in a post hoc analysis, only the difference in nocturnal hypoglycaemia risk reduction reached statistical significance in the period from week 9 to end of study (predefined study endpoint). Thus, once the doses of IGlar U300 and IGlar U100 are equipotent, the cumulative mean number of hypoglycaemic events run in parallel to one another. This pattern is in contrast to the trials of IDeg U100 versus IGlar U100, in which the relative risks for hypoglycaemia did not tend to diverge until after 16 weeks of treatment, when the doses had been titrated to near‐final levels.72
6.2. Insulin degludec U200
An up‐concentrated formulation, IDeg U200 (200 U/mL), is also available. Interestingly, and in contrast to IGlar, the 2 IDeg concentrations (100 and 200 U/mL) are similar in terms of PK and PD characteristics.74 In a post hoc analysis, the 90% CIs for area under the serum IDeg U100 and U200 concentration–time curves and maximum IDeg U100 and U200 concentrations, at steady state during a dosing interval, were within the limits for bioequivalence (0.80‐1.25).74 The glucose‐lowering effects of IDeg U100 and IDeg U200 were both evenly distributed between the first and second 12‐hour periods post‐dosing. Therefore, one can switch from IDeg U100 to IDeg U200 and maintain glycaemic control without changing the dose administered or the regimen used.
A 26‐week, open‐label, treat‐to‐target trial compared the safety and efficacy of IDeg U200 with IGlar U100 in insulin‐naïve patients with type 2 diabetes and showed that IDeg U200 was non‐inferior to IGlar U100 in terms of HbA1c reduction, and that the rates of both overall and nocturnal hypoglycaemia were similar.75 This was achieved at an 11% lower end‐of‐trial daily insulin dose for patients in the IDeg U200 group versus those in the IGlar U100 group.75
The reason that up‐concentration of IGlar U100 alters its time–action profile and potency whereas IDeg U100 and IDeg U200 are interchangeable has yet to be elucidated, but probably reflects differences in mechanism of protraction. As noted above, for insulin analogues that precipitate, up‐concentration further delays absorption of a given dose by creating larger precipitates and thereby reducing the surface area from which absorption can occur.68, 76 In contrast, the mechanism of protraction of IDeg is such that the release of zinc from multihexamer chains is the rate‐limiting step for IDeg absorption.2, 77 This rate may be inherently less affected by concentration; however, the zinc concentration of IDeg U200 has been optimized to increase its bioequivalence to the U100 formulation.77
An ongoing, randomized, double‐blind, multiple‐dose, 2‐period crossover trial will compare the PK/PD properties of IDeg U200 and IGlar U300 at steady state in people with type 1 diabetes.78
7. OTHER STRATEGIES
7.1. PEGylation
PEGylation is the process of attaching polyethylene glycol (PEG) polymer chains to a molecule to increase its hydrodynamic size. This principle was studied as a potential protraction mechanism for insulin with a PEGylated version of insulin lispro. Trials yielded some promising results before Eli Lilly and Company's decision at the end of 2015 to end the clinical development programme. PEGlispro had a long half‐life of 2 to 3 days in patients with type 2 diabetes, as a result of both prolonged absorption and reduced clearance.79, 80
A randomized crossover study conducted in 8 healthy male subjects revealed a hepato‐preferential effect and relatively decreased peripheral action of PEGlispro on glucose homeostasis that might better recreate the physiological actions of endogenous insulin, which is secreted into the portal vein.81 In the open‐label IMAGINE 1 trial, patients receiving PEGlispro reported a statistically significant higher rate of severe hypoglycaemic events (estimated rate ratio: 2.50); however, in the larger, blinded IMAGINE 3 trial the rate of severe hypoglycaemic events for PEGlispro treatment was numerically lower than for IGlar U100 (19.7 events/100 patient‐years of exposure vs 22.5 events/100 patient‐years of exposure), but not statistically significant.82, 83 In a 52‐week trial comparing safety and efficacy of PEGlispro versus IGlar U100 in patients with type 2 diabetes uncontrolled on basal insulin or ≥3 oral antidiabetic drugs, PEGlispro provided superior HbA1c reductions [least squares mean difference −0.52% (95% CI −0.67; −0.38); P < .001] at a 60% lower rate of nocturnal hypoglycaemia, but with higher mean (standard deviation) levels of triglycerides [174 (4) mg/dL vs 158 (6) mg/dL], alanine aminotransferase [34.3 (0.8) IU/L vs 26.4 (1.1) IU/L], aspartate aminotransferase [27.7 (0.6) IU/L vs 23.5 (0.8) IU/L] and liver fat content [14.9 (0.5)% vs 9.6 (0.8)%] versus IGlar U100 after 52 weeks of treatment.37 PEGlispro was associated with less weight gain versus IGlar U100 in patients with type 2 diabetes not previously using insulin (IMAGINE‐2),84 those using basal insulin with mealtime insulin (IMAGINE‐4),85 and similar weight gain versus IGlar U100 in patients currently using a basal insulin (IMAGINE‐5).37
Future development of PEGylated insulin analogues (or hepato‐preferential basal insulin per se) will have to address possible regulatory concerns about liver fat accumulation.
7.2. Conjugation to the non‐glycosylated Fc portion of human IgG
Hanmi Pharmaceuticals has introduced a novel way of protracting the time–action profile of proteins by conjugating them to the non‐glycosylated Fc portion of human IgG via a short, flexible linker.86 The project is still early in development, but results from animal studies have been promising, indicating that it might be possible to extend the duration of insulin action beyond 7 days and move towards once‐weekly dosing.87, 88 Further data on this novel mechanism of protraction will be of great interest, in particular with regard to safety, but also with regard to PK/PD and clinical efficacy.
8. SUMMARY
Several mechanisms of protraction have been used to improve the PK/PD characteristics of basal insulins and all have resulted in prolonged action to varying extents. The early basal insulin analogues, IGlar U100 (precipitation) and IDet (dimerization and albumin binding), increased duration such that it approached a 24‐hour glucose‐lowering effect, and improved the level of day‐to‐day and intra‐patient variability in insulin action. Newer basal insulin analogues have used up‐concentration in addition to precipitation (IGlar U300), and multihexamer formation in addition to albumin binding (IDeg U100 and U200). The latter mechanism has resulted in a further increase in duration of action and decreased variability versus other available basal insulin therapies. Clinically, the major advantage of these analogues has been a reduction in hypoglycaemia with similar HbA1c control. The recent development of hepato‐preferential or very long‐acting (once‐weekly) insulins promises the potential to achieve further clinical improvements, but the safety of these new preparations and clinical applicability remains to be demonstrated.
ACKNOWLEDGMENTS
Medical writing and submission support was provided by Victoria Atess and Daria Renshaw of Watermeadow Medical, an Ashfield company, part of UDG Healthcare plc. This support was funded by Novo Nordisk. Novo Nordisk reviewed the article for medical accuracy only.
Conflict of interest
T. H. has received research funds from Adocia, Astra Zeneca, BD, Biocon, Boehringer Ingelheim, Dance Pharmaceuticals, Grünenthal, Eli Lily and Company, Medtronic, Novo Nordisk, Novartis, Sanofi and Senseonics, and speaker honoraria and travel grants from Eli Lily and Company, Mylan and Novo Nordisk. C. M. has received advisory panel honoraria from Novo Nordisk, sanofi‐aventis, Merck Sharp and Dohme Ltd., Eli Lilly and Company, Novartis, Bristol‐Myers Squibb, AstraZeneca LP, Pfizer, Jansen Pharmaceuticals and Hanmi, research support from Novo Nordisk, sanofi‐aventis, Merck Sharp and Dohme Ltd., Eli Lilly and Company and Novartis, and speaker honoraria from Novo Nordisk, Sanofi, Merck Sharp and Dohme, Eli Lilly and Company, Novartis and Astra Zeneca.
Author contributions
Both authors (T. H. and C. M.) confirm that they meet the International Committee of Medical Journal Editors uniform requirements for authorship and that they have contributed to the conception of the work, drafting and/or critically revising the article and sharing in the final responsibility for the content of the manuscript and the decision to submit it for publication.
Heise T and Mathieu C. Impact of the mode of protraction of basal insulin therapies on their pharmacokinetic and pharmacodynamic properties and resulting clinical outcomes, 2017;19(1):3–12.
Funding Information Medical writing and submission support was provided by Victoria Atess and Daria Renshaw of Watermeadow Medical, an Ashfield company, part of UDG Healthcare plc. This support was funded by Novo Nordisk.
REFERENCES
- 1. Lindholm A. New insulins in the treatment of diabetes mellitus. Best Pract Res Clin Gastroenterol. 2002;21:492‐505. [DOI] [PubMed] [Google Scholar]
- 2. Jonassen I, Havelund S, Hoeg‐Jensen T, Steensgaard DB, Wahlund PO, Ribel U. Design of the novel protraction mechanism of insulin degludec, an ultra‐long‐acting basal insulin. Pharm Res. 2012;29:2104‐2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hagedorn HC, Norman Jensen B, Krarup NB. Protamine insulin. JAMA. 1936;106:177‐180. [DOI] [PubMed] [Google Scholar]
- 4. Simkin RD, Cole SA, Ozawa H, et al. Precipitation and crystallization of insulin in the presence of lysozyme and salmine. Biochim Biophys Acta. 1970;200:385‐394. [DOI] [PubMed] [Google Scholar]
- 5. Søeborg T, Rasmussen CH, Mosekilde E, Colding‐Jørgensen M. Absorption kinetics of insulin after subcutaneous administration. Eur J Pharm Sci. 2009;36:78‐90. [DOI] [PubMed] [Google Scholar]
- 6. Søeborg T, Rasmussen CH, Mosekilde E, Colding‐Jørgensen M. Bioavailability and variability of biphasic insulin mixtures. Eur J Pharm Sci. 2012;46:198‐208. [DOI] [PubMed] [Google Scholar]
- 7. Jehle PM, Micheler C, Jehle DR, Breitig D, Boehm BO. Inadequate suspension of neutral protamine Hagendorn (NPH) insulin in pens. Lancet. 1999;354:1604‐1607. [DOI] [PubMed] [Google Scholar]
- 8. Lucidi P, Porcellati F, Marinelli Andreoli A, et al. Pharmacokinetics and pharmacodynamics of NPH insulin in type 1 diabetes: the importance of appropriate resuspension before subcutaneous injection. Diabetes Care. 2015;38:2204‐2210. [DOI] [PubMed] [Google Scholar]
- 9. Lepore M, Pampanelli S, Fanelli C, et al. Pharmacokinetics and pharmacodynamics of subcutaneous injection of long‐acting human insulin analog glargine, NPH insulin, and ultralente human insulin and continuous subcutaneous infusion of insulin lispro. Diabetes. 2000;49:2142‐2148. [DOI] [PubMed] [Google Scholar]
- 10. Plank J, Bodenlenz M, Sinner F, et al. A double‐blind, randomized, dose‐response study investigating the pharmacodynamic and pharmacokinetic properties of the long‐acting insulin analog detemir. Diabetes Care. 2005;28:1107‐1112. [DOI] [PubMed] [Google Scholar]
- 11. Heise T, Nosek L, Rønn BB, et al. Lower within‐subject variability of insulin detemir in comparison to NPH insulin and insulin glargine in people with type 1 diabetes. Diabetes. 2004;53:1614‐1620. [DOI] [PubMed] [Google Scholar]
- 12. Yki‐Järvinen H, Dressler A, Ziemen M, HOE 901/300 s Study Group . Less nocturnal hypoglycemia and better post‐dinner glucose control with bedtime insulin glargine compared with bedtime NPH insulin during insulin combination therapy in type 2 diabetes. Diabetes Care. 2000;23:1130‐1136. [DOI] [PubMed] [Google Scholar]
- 13. NICE Guideline (December 2015). Type 2 diabetes in adults: management (NG28). https://www.nice.org.uk/guidance/ng28/resources/type‐2‐diabetes‐in‐adults‐management‐1837338615493. Accessed February 2, 2016.
- 14. Owens DR. Insulin preparations with prolonged effect. Diabetes Technol Ther. 2011;13(suppl 1):S5‐S14. [DOI] [PubMed] [Google Scholar]
- 15. Brange J. Galenics of Insulin: The Physico‐chemical and Pharmaceutical Aspects of Insulin and Insulin Preparations. 1st ed. Berlin, Heidelberg, Germany: Springer‐Verlag; 1987. [Google Scholar]
- 16. Heine RJ, Bilo HJG, Fonk T, van der Veen EA, van der Meer J. Absorption kinetics and action profiles of mixtures of short‐ and intermediate‐acting insulins. Diabetologia. 1984;27:558‐562. [DOI] [PubMed] [Google Scholar]
- 17. Kurtzhals P, Ribel U. Action profile of cobalt(III)‐insulin a novel principle of protraction of potential use for basal insulin delivery. Diabetes. 1995;44:1381‐1385. [DOI] [PubMed] [Google Scholar]
- 18. Kurtzhals P. Engineering predictability and protraction in a basal insulin analogue: the pharmacology of insulin detemir. Int J Obes Relat Metab Disord. 2004;28(suppl 2):S23‐S28. [DOI] [PubMed] [Google Scholar]
- 19. Markussen J, Diers I, Hougaard P, et al. Soluble, prolonged‐acting insulin derivatives. III. Degree of protraction, crystallizability and chemical stability of insulins substituted in positions A21, B13, B23, B27 and B30. Protein Eng. 1988;2:157‐166. [DOI] [PubMed] [Google Scholar]
- 20. Jørgensen S, Vaag A, Langkjaer L, Hougaard P, Markussen J. NovoSol Basal: pharmacokinetics of a novel soluble long acting insulin analogue. BMJ. 1989;299:415‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jorgensen S, Drejer K. Insulin analogs and nasal insulin delivery In: Bailey CJ, Flatt PR, eds. New Antidiabetic Drugs. 1st ed. London, England: Smith‐Gordon; 1990:83‐92. [Google Scholar]
- 22. Vajo Z, Duckworth WC. Genetically engineered insulin analog: diabetes in the new millenium. Pharmacol Rev. 2000;52:1‐9. [PubMed] [Google Scholar]
- 23. Guthrie R. Is there a need for a better basal insulin? Clin Diabetes. 2001;19:66‐70. [Google Scholar]
- 24. Seipke G, Geisen K, Neubauer H‐P, Pittius C, Rosskamp R, Schwabe D. New insulin preparations with prolonged action profiles: A21‐modified arginine insulins. Diabetologia. 1992;35(suppl 1):A4. [Google Scholar]
- 25. Rosenstock J, Park G, Zimmerman J, U.S. Insulin Glargine (HOE 901) Type 1 Diabetes Investigator Group . Basal insulin glargine (HOE 901) versus NPH insulin in patients with type 1 diabetes on multiple daily insulin regimens. Diabetes Care. 2000;23:1137‐1142. [DOI] [PubMed] [Google Scholar]
- 26. Heise T, Pieber TR. Towards peakless, reproducible and long‐acting insulins. An assessment of the basal analogues based on isoglycaemic clamp studies. Diabetes Obes Metab. 2007;9:648‐659. [DOI] [PubMed] [Google Scholar]
- 27. Klein O, Lynge J, Endahl L, Damholt B, Nosek L, Heise T. Albumin‐bound basal insulin analogues (insulin detemir and NN344): comparable time‐action profiles but less variability than insulin glargine in type 2 diabetes. Diabetes Obes Metab. 2007;9:290‐299. [DOI] [PubMed] [Google Scholar]
- 28. Porcellati F, Rossetti P, Ricci NB, et al. Pharmacokinetics and pharmacodynamics of the long‐acting insulin analog glargine after 1 week of use compared with its first administration in subjects with type 1 diabetes. Diabetes Care. 2007;30:1261‐1263. [DOI] [PubMed] [Google Scholar]
- 29. Ashwell SG, Gebbie J, Home PD. Twice‐daily compared with once‐daily insulin glargine in people with type 1 diabetes using meal‐time insulin aspart. Diabet Med. 2006;23:879‐886. [DOI] [PubMed] [Google Scholar]
- 30. Devries JH, Nattrass M, Pieber TR. Refining basal insulin therapy: what have we learned in the age of analogues? Diabetes Metab Res Rev. 2007;23:441‐454. [DOI] [PubMed] [Google Scholar]
- 31. Albright ES, Desmond R, Bell DSH. Efficacy of conversion from bedtime NPH insulin injection to once‐ or twice‐daily injections of insulin glargine in type 1 diabetic patients using basal/bolus therapy. Diabetes Care. 2004;27:632‐633. [DOI] [PubMed] [Google Scholar]
- 32. Ratner RE, Hirsch IB, Neifing JL, Garg SK, Mecca TE, Wilson CA. Less hypoglycemia with insulin glargine in intensive insulin therapy for type 1 diabetes. Diabetes Care. 2000;23:639‐643. [DOI] [PubMed] [Google Scholar]
- 33. Home PD, Fritsche A, Schinzel S, Massi‐Benedetti M. Meta‐analysis of individual patient data to assess the risk of hypoglycaemia in people with type 2 diabetes using NPH insulin or insulin glargine. Diabetes Obes Metab. 2010;12:772‐779. [DOI] [PubMed] [Google Scholar]
- 34. Riddle MC, Rosenstock J, Gerich J, Insulin Glargine 4002 Study Investigators . Randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26:3080‐3086. [DOI] [PubMed] [Google Scholar]
- 35. Rosenstock J, Schwartz SL, Clark CM Jr, Park GD, Donley DW, Edwards MB. Basal insulin therapy in type 2 diabetes 28‐week comparison of insulin glargine (HOE 901) and NPH insulin. Diabetes Care. 2001;24:631‐636. [DOI] [PubMed] [Google Scholar]
- 36. Heise T, Hermanski L, Nosek L, Feldman A, Rasmussen S, Haahr H. Insulin degludec: four times lower pharmacodynamic variability than insulin glargine under steady‐state conditions in type 1 diabetes. Diabetes Obes Metab. 2012;14:859‐864. [DOI] [PubMed] [Google Scholar]
- 37. Buse JB, Rodbard HW, Trescoli Serrano C, et al. Randomized clinical trial comparing basal insulin peglispro and insulin glargine in patients with type 2 diabetes previously treated with basal insulin: IMAGINE 5. Diabetes Care. 2016;39:92‐100. [DOI] [PubMed] [Google Scholar]
- 38. Havelund S, Plum A, Ribel U, et al. The mechanism of protraction of insulin detemir, a long‐acting, acylated analog of human insulin. Pharm Res. 2004;21:1498‐1504. [DOI] [PubMed] [Google Scholar]
- 39. Chaykin L. Insulin detemir and its Unique Mechanism of Action. Internet J Endocrinol. 2006;4:1‐10. [Google Scholar]
- 40. Dornhorst A, Lüddeke HJ, Sreenan S, et al. Safety and efficacy of insulin detemir in clinical practice: 14‐week follow‐up data from type 1 and type 2 diabetes patients in the PREDICTIVE European cohort. Int J Clin Pract. 2007;61:523‐528. [DOI] [PubMed] [Google Scholar]
- 41. Danne T, Datz N, Endahl L, et al. Insulin detemir is characterized by a more reproducible pharmacokinetic profile than insulin glargine in children and adolescents with type 1 diabetes: results from a randomized, double‐blind, controlled trial. Pediatr Diabetes. 2008;9:554‐560. [DOI] [PubMed] [Google Scholar]
- 42. Heller S, Koenen C, Bode B. Comparison of insulin detemir and insulin glargine in a basal‐bolus regimen, with insulin aspart as the mealtime insulin, in patients with type 1 diabetes: a 52‐week, multinational, randomized, open‐label, parallel‐group, treat‐to‐target noninferiority trial. Clin Ther. 2009;31:2086‐2097. [DOI] [PubMed] [Google Scholar]
- 43. Rosenstock J, Davies M, Home PD, Larsen J, Koenen C, Schernthaner G. A randomised, 52‐week, treat‐to‐target trial comparing insulin detemir with insulin glargine when administered as add‐on to glucose‐lowering drugs in insulin‐naive people with type 2 diabetes. Diabetologia. 2008;51:408‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swinnen SG, Dain MP, Aronson R, et al. A 24‐week, randomized, treat‐to‐target trial comparing initiation of insulin glargine once‐daily with insulin detemir twice‐daily in patients with type 2 diabetes inadequately controlled on oral glucose‐lowering drugs. Diabetes Care. 2010;33:1176‐1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Raskin P, Gylvin T, Weng W, Chaykin L. Comparison of insulin detemir and insulin glargine using a basal‐bolus regimen in a randomized, controlled clinical study in patients with type 2 diabetes. Diabetes Metab Res Rev. 2009;25:542‐548. [DOI] [PubMed] [Google Scholar]
- 46. Hollander P, Cooper J, Bregnhøj J, Pedersen CB. A 52‐week, multinational, open‐label, parallel‐group, noninferiority, treat‐to‐target trial comparing insulin detemir with insulin glargine in a basal‐bolus regimen with mealtime insulin aspart in patients with type 2 diabetes. Clin Ther. 2008;30:1976‐1987. [DOI] [PubMed] [Google Scholar]
- 47. Pieber TR, Treichel HC, Hompesch B, et al. Comparison of insulin detemir and insulin glargine in subjects with type 1 diabetes using intensive insulin therapy. Diabet Med. 2007;24:635‐642. [DOI] [PubMed] [Google Scholar]
- 48. Hordern SV, Wright JE, Umpleby AM, Shojaee‐Moradie F, Amiss J, Russell‐Jones DL. Comparison of the effects on glucose and lipid metabolism of equipotent doses of insulin detemir and NPH insulin with a 16‐h euglycaemic clamp. Diabetologia. 2005;48:420‐426. [DOI] [PubMed] [Google Scholar]
- 49. Zachariah S, Sheldon B, Shojaee‐Moradie F, et al. Insulin detemir reduces weight gain as a result of reduced food intake in patients with type 1 diabetes. Diabetes Care. 2011;34:1487‐1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brunner GA, Sendhofer G, Wutte A, et al. Pharmacokinetic and pharmacodynamic properties of long‐acting insulin analogue NN304 in comparison to NPH insulin in humans. Exp Clin Endocrinol Diabetes. 2000;108:100‐105. [DOI] [PubMed] [Google Scholar]
- 51. Derewenda U, Derewenda Z, Dodson EJ, et al. Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer. Nature. 1989;338:594‐596. [DOI] [PubMed] [Google Scholar]
- 52. Krüger P, Gilge G, Cabuk Y, Wollmer A. Co‐operativity and intermediate states in the T R‐structural transformation of insulin. Biol Chem Hoppe Seyler. 1990;371:669‐673. [DOI] [PubMed] [Google Scholar]
- 53. Kaarsholm NC, Ko HC, Dunn MF. Comparison of solution structural flexibility and zinc binding domains for insulin, proinsulin, and miniproinsulin. Biochemistry. 1989;28:4427‐4435. [DOI] [PubMed] [Google Scholar]
- 54. Seested T, Burgess A, Pyke C, Nishimura E. Ultrastructural 3D visualization of insulin degludec multihexamers upon subcutaneous injection in Pig. Diabetes. 2016;65(suppl 1):A236 (Abstract 918‐P). [Google Scholar]
- 55. Steensgaard DB, Schuckebier G, Strauss HM, et al. Ligand‐controlled assembly of hexamers, dihexamers, and linear multihexamer structures by the engineered acylated insulin degludec. Biochemistry. 2013;52:295‐309. [DOI] [PubMed] [Google Scholar]
- 56. Heise T, Hövelmann U, Nosek L, Hermanski L, Bøttcher SG, Haahr H. Comparison of the pharmacokinetic and pharmacodynamic profiles of insulin degludec and insulin glargine. Expert Opin Drug Metab Toxicol. 2015;11:1193‐1201. [DOI] [PubMed] [Google Scholar]
- 57. Heise T, Hövelmann U, Nosek L, et al. Insulin degludec: two‐fold longer half‐life and a more consistent pharmacokinetic profile than insulin glargine. Diabetologia. 2011;54(suppl 1):S425. [Google Scholar]
- 58. Heise T, Meneghini LF. Insulin stacking versus therapeutic accumulation: understanding the differences. Endocr Pract. 2014;20:75‐83. [DOI] [PubMed] [Google Scholar]
- 59. Ratner RE, Gough SCL, Mathieu C, et al. Hypoglycaemia risk with insulin degludec compared with insulin glargine in type 2 and type 1 diabetes: a pre‐planned meta‐analysis of phase 3 trials. Diabetes Obes Metab. 2013;15:175‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lane W, Bailey TS, Gerety G, et al. SWITCH 1: reduced hypoglycemia with insulin degludec (IDeg) vs insulin glargine (IGlar), both U100, in patients with T1D at high risk of hypoglycemia: a randomized, double‐blind, crossover trial [Abstract]. Presented at American Diabetes Association (ADA) 76th Annual Scientific Sessions; June10–14, 2016; New Orleans, LA. Abstract number 87‐LB. [Google Scholar]
- 61. Wysham C, Bhargava A, Chaykin L, et al. SWITCH 2: reduced hypoglycemia with insulin degludec (IDeg) vs. insulin glargine (IGlar), both U100, in patients with T2D at high risk of hypoglycemia: a randomized, double‐blind, crossover trial [Abstract]. Presented at American Diabetes Association (ADA) 76th Annual Scientific Sessions; June 10–14, 2016, New Orleans, LA. Abstract number 90‐LB. [Google Scholar]
- 62. Vora J, Christensen T, Rana A, Bain SC. Insulin degludec versus insulin glargine in type 1 and type 2 diabetes mellitus: a meta‐analysis of endpoints in phase 3a trials. Diabetes Ther. 2014;5:435‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mathieu C, Hollander P, Miranda‐Palma B, et al. Efficacy and safety of insulin degludec in a flexible dosing regimen vs insulin glargine in patients with type 1 diabetes (BEGIN: Flex T1): a 26‐week randomized, treat‐to‐target trial with a 26‐week extension. J Clin Endocrinol Metab. 2013;98:1154‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meneghini L, Atkin SL, Gough SC, et al. The efficacy and safety of insulin degludec given in variable once‐daily dosing intervals compared with insulin glargine and insulin degludec dosed at the same time daily: a 26‐week, randomized, open‐label, parallel‐group, treat‐to‐target trial in individuals with type 2 diabetes. Diabetes Care. 2013;36:858‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Havelund S, Ribel U, Hubálek F, Hoeg‐Jensen T, Wahlund PO, Jonassen I. Investigation of the physico‐chemical properties that enable co‐formulation of basal insulin degludec with fast‐acting insulin aspart. Pharm Res. 2015;32:2250‐2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Greig SL, Scott LJ. Insulin degludec/liraglutide: a review in type 2 diabetes. Drugs. 2015;75:1523‐1534. [DOI] [PubMed] [Google Scholar]
- 67. Christiansen JS, Horne P, Kumar A. IDegAsp (insulin degludec + insulin aspart) for the management of type 2 diabetes: current status. Expert Rev Endocrinol Metabol. 2016;11:103‐111. [DOI] [PubMed] [Google Scholar]
- 68. de la Peña A, Riddle M, Morrow LA, et al. Pharmacokinetics and pharmacodynamics of high‐dose human regular U‐500 insulin versus human regular U‐100 insulin in healthy obese subjects. Diabetes Care. 2011;34:2496‐2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sindelka G, Heinemann L, Berger M, Frenck W, Chantelau E. Effect of insulin concentration, subcutaneous fat thickness and skin temperature on subcutaneous insulin absorption in healthy subjects. Diabetologia. 1994;37:377‐380. [DOI] [PubMed] [Google Scholar]
- 70. Becker RH, Dahmen R, Bergmann K, Lehmann A, Jax T, Heise T. New insulin glargine 300 units.mL‐1 provides a more even activity profile and prolonged glycemic control at steady state compared with insulin glargine 100 units.mL‐1 . Diabetes Care. 2015;38:637‐643. [DOI] [PubMed] [Google Scholar]
- 71. Toujeo SmPC. European Medicines Agency. 2016. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000309/human_med_000955.jsp&mid=WC0b01ac058001d124. Accessed June 28, 2016.
- 72. Ritzel R, Roussel R, Bolli GB, et al. Patient‐level meta‐analysis of the EDITION 1, 2 and 3 studies: glycaemic control and hypoglycaemia with new insulin glargine 300 U/ml versus glargine 100 U/ml in people with type 2 diabetes. Diabetes Obes Metab. 2015;17:859‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ritzel R, Roussel R, Giaccari A, et al. Glycemic control and hypoglycemia with insulin glargine 300 U/mL vs glargine 100 U/mL in T2DM in a patient‐level meta‐analysis of 1‐year phase 3A edition studies. Diabetes Technol Ther. 2016;18(suppl 1):A‐118 (Abstract 294). [Google Scholar]
- 74. Korsatko S, Deller S, Koehler G, et al. A comparison of the steady‐state pharmacokinetic and pharmacodynamic profiles of 100 and 200 U/mL formulations of ultra‐long‐acting insulin degludec. Clin Drug Investig. 2013;33:515‐521. [DOI] [PubMed] [Google Scholar]
- 75. Gough SC, Bhargava A, Jain R, Mersebach H, Rasmussen S, Bergenstal RM. Low‐volume insulin degludec 200 units/mL once daily improves glycemic control similar to insulin glargine with a low risk of hypoglycemia in insulin‐naïve patients with type 2 diabetes: A 26‐week, randomized, controlled, multinational, treat‐to‐target trial: the BEGIN LOW VOLUME trial. Diabetes Care. 2013;36:2536‐2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mader JK, Birngruber T, Korsatto S, et al. Enhanced absorption of insulin aspart as the result of a dispersed injection strategy tested in a randomized trial in type 1 diabetic patients. Diabetes Care. 2013;36:780‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Haahr H, Heise H. A review of the pharmacological properties of insulin degludec and their clinical relevance. Clin Pharmacokinet. 2014;53:787‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Comparing pharmacodynamic and pharmacokinetic properties of insulin degludec and insulin glargine 300 U/mL at steady‐state conditions in subjects with type 1 diabetes mellitus. https://clinicaltrials.gov/ct2/show/NCT02536859?term=NCT02536859&rank=1. Novo Nordisk A/S, 2016. Accessed June 28. 2016.
- 79. Sinha VP, Howey DC, Choi SL, Mace KF, Heise T. Steady‐state pharmacokinetics and glucodynamics of the novel, long‐acting basal insulin LY2605541 dosed once‐daily in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2013;16:344‐350. [DOI] [PubMed] [Google Scholar]
- 80. Caparrotta TM, Evans M. PEGylated insulin Lispro, (LY2605541)–a new basal insulin analogue. Diabetes Obes Metab. 2014;16:388‐395. [DOI] [PubMed] [Google Scholar]
- 81. Henry RR, Mudaliar S, Ciaraldi TP, et al. Basal insulin peglispro demonstrates preferential hepatic versus peripheral action relative to insulin glargine in healthy subjects. Diabetes Care. 2014;37:2609‐2615. [DOI] [PubMed] [Google Scholar]
- 82. Garg S, Jinnouchi H, Dreyer M, et al. Greater HbA1c reduction with Basal Insulin Peglispro (BIL) v Insulin Glargine (GL) in an open‐label, randomized study in T1D patients 9 (pts): IMAGINE 1. Diabetes. 2015;64(suppl 1):A25 (Abstract 95‐OR). [Google Scholar]
- 83. Bergenstal R, Lunt H, Franek E, et al. Superior reduction of HbA1c in a double‐blind, randomized study of Basal Insulin Peglispro (BIL) v Insulin Glargine (GL) in patients (pts) with T1D: IMAGINE 3. Diabetes. 2015;64(suppl 1):A250 (Abstract 986‐P). [Google Scholar]
- 84. Davies MJ, Russell‐Jones D, Selam J‐L, et al. Basal insulin peglispro (BIL) is superior to insulin glargine (GL) in reducing HbA1c at 52 wks in insulin‐naïve T2D patients (pts) treated with oral antihyperglycemicmedications (OAMs): IMAGINE 2. Diabetes. 2015;64(suppl 1):A24 (Abstract 93‐OR). [Google Scholar]
- 85. Blevins T, Pieber TR, Vega GC, Zhang S, Bastyr EJ III, Chang AM. Superior HbA1c reduction with basal insulin peglispro (BIL) vs insulin glargine (GL) and preprandial insulin lispro in a double‐blind study in patients (pts) with type 2 diabetes (T2D): IMAGINE 4. Diabetes. 2015;64(suppl 1):A250. [Google Scholar]
- 86. Hanmi Project . Hanmi Pharmaceuticals. http://www.hanmipharm.com/ehanmi/handler/Rnd‐ProjectBio. Accessed June 28, 2016.
- 87. Kim JK, Park YJ, Lim CK, et al. Once‐weekly combination of GLP‐1R agonist and insulin (HM14220) offers improved glycemic control and reduced weight gain risk. Diabetes. 2015;64(suppl 1):A45 (172‐OR). [Google Scholar]
- 88. Kang J, Kim J‐H, Yi J, et al. The ultra‐long acting LAPSGLP/GCG dual agonist, HM12525A, demonstrated safety and prolonged pharmacokinetics in healthy volunteers: a phase 1 first‐in‐human study. Diabetologia. 2015;58(suppl 1) (Abstract 791). S380–S381. [Google Scholar]