

Abstract
Background
The chicken gastrointestinal tract contains a diverse microbiota whose composition and structure play important roles in gut functionality. In this study, microbial shifts resulting from feed supplementation with Bacillus subtilis CSL2 were evaluated in broilers challenged and unchallenged with Salmonella Gallinarum. To analyse bacterial community composition and functionality, 454 GS-FLX pyrosequencing of 16S rRNA gene amplicons was performed.
Results
The Quantitative Insights into Microbial Ecology (QIIME) pipeline was used to analyse changes in the faecal microbiota over a 24-h period. A total of 718,204 sequences from broiler chickens were recorded and analysed. At the phylum level, Firmicutes, Bacteroidetes, and Proteobacteria were the predominant bacterial taxa. In Salmonella-infected chickens (SC), Bacteroidetes were more highly abundant compared to control (NC) and Bacillus-treated (BT) chickens. At the genus level, in the NC and BT groups, Lactobacillus was present at high abundance, and the abundance of Turicibacter, unclassified Enterobacteriaceae, and Bacteroides increased in SC broilers. Furthermore, taxon-independent analysis showed that the SC and BT groups were compositionally distinct at the end of the 24-h period. Further analysis of functional properties showed that B. subtilis CSL2 administration increased gut-associated energy supply mechanisms (i.e. carbohydrate transport and metabolism) to maintain a stable microbiota and protect gut integrity.
Conclusions
This study demonstrated that S. Gallinarum infection and B. subtilis CSL2 supplementation in the diet of broiler chickens influenced the diversity, composition, and functional diversity of the faecal microbiota. Moreover, the findings offer significant insights to understand potential mechanisms of Salmonella infection and the mode of action of probiotics in broiler chickens.
Electronic supplementary material
The online version of this article (doi:10.1186/s40104-016-0130-8) contains supplementary material, which is available to authorized users.
Keywords: Bacillus subtilis, Broiler chicken, Microbiome, Salmonella Gallinarum, 16S rRNA gene
Background
Poultry is one of the most important meat sources for humans [1]. Due to the increasing demand for food, chicken production has increased tremendously in the past few years [1]. Therefore, husbandry and management are vital for preventing infection and maintaining the health of poultry. Animal health is closely associated with the status of the gastrointestinal tract, whose disruption or dysbiosis leads to detrimental effects [2]. Intestinal homeostasis and functionality are influenced by various factors, such as (1) diet and feed additives, (2) farm conditions and practices, and (3) the resident gut microbiota [3]. The microbiota comprises trillions of microorganisms localised primarily at the distal end of the gastrointestinal tract [4]. These microbial communities mediate digestion of feedstuff, control gut homeostasis, and prevent infection.
Salmonella enterica is an important group of gastrointestinal pathogens that causes food-borne diseases, gastroenteritis, and diarrhoea in animals and humans [2, 5, 6]. Several serovars of S. enterica subsp. enterica, specifically S. Enteritidis and S. Gallinarum, are major avian pathogens. These serovars are frequently associated with poultry salmonellosis, which results in severe morbidity and mortality [6]. The pathogenicity and routes of transmission of Salmonella spp. have been investigated extensively; however, few studies have addressed S. Gallinarum infection and its control in poultry [6].
Probiotics are live microorganisms that confer a wide range of benefits on animals, such as stimulation of immune responses, maintenance of gut barrier function, and prevention of pathogen invasion of the gut [1, 7] The gram-positive bacterium Bacillus subtilis has been used as an in-feed probiotic supplement for livestock and poultry [8, 9]. Only recently have studies begun to address the effects of probiotic interventions on the gastrointestinal microbiota. However, previous studies using plating methods, 16S rRNA gene clone libraries, denaturing gradient gel electrophoresis, and so forth have yielded relatively little information [9]. In contrast, high-throughput next-generation sequencing (NGS) methods facilitate rapid quantification and identification of bacterial communities [4, 10]. In addition, simultaneous analysis of multiple samples enables a thorough understanding of microbial communities and therefore prediction of the effect of interventions and infections on microbial functions and community diversity [11]. This study applied 16S rRNA gene sequencing to investigate the composition of the chicken gut microbiota, and the findings suggest a marked effect of S. Gallinarum on the overall composition and metabolic functions of the chicken gut microbiota. In addition, the data suggest that B. subtilis CSL2 exerts protective effects against S. Gallinarum infection by altering the faecal microbiota of broiler chickens.
Methods
Animals and experimental design
A total of 36 Ross-308 broiler chickens were bred and divided into the following three groups: the control group (NC; n = 12), Salmonella-challenged (SC; n = 12) group, and Bacillus-treated (BT; n = 12) group. From day 1, the NC and SC groups were given the standard basal diet (Additional file 1: Table S1), while the BT group was fed a probiotic-supplemented basal diet containing Bacillus subtilis CSL2 (GenBank accession number: KX281166) at 1.0 × 107 colony forming units (CFU)/g feed. Freeze-dried B. subtilis CSL2 (1.0 × 1010 CFU/g) diluted with basal diet by mixing for 2 h with a feed mixer (Daedong Tech, Korea), to obtain final concentration of 1.0 × 107 CFU/g feed. Feeding of the respective diets continued until d 31 (before). On d 32, chickens in the SC and BT groups were orally challenged with S. enterica subsp. Gallinarum KVCC-BA0700722 (S. Gallinarum) at 1.0 × 108 CFU/mL. The Dankook University Animal Care Committee approved all animal protocols.
All 36 chickens (12 birds per treatment) were tagged and placed in cages randomly, which were equipped with a nipple water dispenser for ad libitum access to water, together with a one-sided self-feeder. The room temperature was maintained at 32 °C for the first week, and then reduced 3 °C weekly until the temperature reached 26 °C. The broiler chickens received no antibiotics or other additives during the study period. No additional chickens were introduced during the duration of the experiment. At 31 and 32 d, 12 chickens per treatment were selected for faecal sampling. Fresh faecal samples were collected aseptically from the rectum of the broiler chickens (on d 31: before; on d 33: after). Finally, faecal contents were placed into sterile tubes and kept on ice until used for microbiota analysis on the same day of the sampling.
Sample preparation and DNA isolation
Genomic DNA isolation from freshly collected faeces was carried out using a technique described previously [11]. Briefly, 0.3 g of faecal extract was placed in a bead-beating tube containing garnet beads. Lysis of host and microbial cells was mediated by both mechanical collisions between beads and chemical disruption of cell membranes. DNA was purified using a Power Faecal® DNA Extraction Kit (MO BIO Laboratories, Inc., USA) as per the manufacturer’s instructions. Precipitated DNA was suspended in DNase-free H2O, and its concentration and purity were assessed by UV/vis spectrophotometry and agarose gel electrophoresis, respectively (Mecasys Co., Ltd, Korea).
454-Pyrosequencing analysis
DNA amplicons from individual broiler chicken samples were amplified using primers for the V1-V3 hyper-variable regions of the 16S rRNA gene by polymerase chain reaction (PCR). Forward primers were tagged with 10-bp unique barcode labels at the 5’ end along with the adaptor sequence to allow multiple samples to be included in a single 454 GS FLX Titanium sequencing plate, as described previously [11]. Finally, 16S rRNA amplicons were quantified, pooled, and purified for sequencing.
Data processing
The 16S rRNA sequence data generated by the 454 GS FLX Titanium chemistry (Roche) were processed using the Quantitative Insights into Microbial Ecology (QIIME) pipeline. Briefly, sequences that were less than 200 bp or greater than 600 bp in length, were of low quality, contained incorrect primer sequences, and/or contained more than one ambiguous base were filtered using the split_libraries.py script. After checking for chimeric sequences, sequence data were filtered using the identify_chimeric_seqs.py and filter_fasta.py scripts. Operational taxonomic units (OTU) were picked using the pick_open_reference.py script and the most recent Greengenes reference database (13_8) at a 97% identity threshold. Bacterial composition data from broiler faecal samples were generated using the summarize_taxa_through_plots.py scripts. For alpha diversity measurements, the alpha_diversity.py script was employed to generate Chao1, Shannon, Simpson, and phylogenetic distance (PD) whole-tree values. Rarefaction curves were also generated in the QIIME software. All reads were pooled for each group of broiler chickens. Sequences were rarefied according to sequencing depth to visualise the change in diversity with respect to sampling depth.
Statistical analysis
Statistical and multivariate analyses were performed using the R software (v. 3.1.0; R Core Team, Auckland, New Zealand). The proportions of bacterial taxa (phylum and genus level) were compared between groups before and after the 24 h challenge. To avoid statistical bias and perform valid downstream analysis, the normalisation of OTU table was performed using the base package in R software. For multivariate analysis of bacterial OTU at a 95% identity threshold, the adegenet package in the R software was used to determine the peaks of the bacterial genera that facilitated discrimination according to the defined clustering groups with a user-defined threshold in the canonical loading plot. The NC, SC, and BT groups were labelled accordingly. In discriminant analysis of principal components (DAPC) (dapc {adegenet}), the normalised abundance data of individual samples was employed [12]. The number of principal components was ≥80% of the cumulative variance explained by the eigenvalues of the DAPC plot, and these principal components were subjected to linear discriminant analysis (LDA), resulting in selection of ≥2 linear discriminants for the DAPC plot [12]. The visual outputs of the canonical loading plot and the DAPC plot were then created using (loadingplot {adegenet}) and scatter plot (scatter {ade4}), respectively. Furthermore, functional prediction was carried out using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) based on the Greengenes 16S rRNA database and KEGG Orthologs (KO) [10]. PICRUSt was used to identify differences in the functional potential of bacterial communities among the groups. Using KEGG (level 3) ortholog function predictions, differences among NC, SC, and BT groups were evaluated, and a loading plot was created to identify the most discriminating functions among the groups after 24 h. Tukey’s honestly significant difference (HSD) test was employed to evaluate functional differences among the groups; a P-value <0.05 was considered to indicate significance.
Results
DNA sequence data and quality control
Pyrosequencing analysis generated a total of 718,204 raw sequence reads. The average number of reads per sample was 10,941, and the mean number of sequence reads per group ranged from 7,701 to 14,199 (Table 1). The average number of reads per sample is comparable to previous animal studies that utilised the GS FLX Titanium system [13, 14]. Barcoded primers allowed pooling of samples for individual- and group-based analyses. At a 95% identity cut-off (genus level), 212 unique operational taxonomic units (OTU) were detected in this study using the latest Greengenes database (13_8); these were used for downstream analyses. The recorded mean OTU per group ranged from 421 to 725 (Table 1).
Table 1.
Pyrosequencing data and diversity indices of the faecal microbiota of broiler chickens
| Group* | Diversity indices (Mean ± standard deviation) | |||||
|---|---|---|---|---|---|---|
| No. of readsa | OTU | Chao1 | Shannon | Simpson | PD | |
| NC (Before) | 12,532 ± 6,666 | 725 ± 420 | 1,443 ± 752 | 5.41 ± 1.19 | 0.92 ± 0.05 | 48.8 ± 25.75 |
| NC (After) | 10824 ± 3,322 | 712 ± 369 | 1,555 ± 729 | 5.79 ± 1.20 | 0.94 ± 0.04 | 48.8 ± 21.71 |
| SC (Before) | 12,176 ± 8,221 | 693 ± 447 | 1,418 ± 938 | 5.50 ± 0.85 | 0.93 ± 0.04 | 45.4 ± 28.49 |
| SC (After) | 7701 ± 3,665 | 421 ± 240 | 890 ± 482 | 4.61 ± 1.00 | 0.85 ± 0.07 | 30.7 ± 15.04 |
| BT (Before) | 8212 ± 4,494 | 493 ± 274 | 1,023 ± 574 | 4.98 ± 1.13 | 0.88 ± 0.07 | 36.5 ± 20.07 |
| BT (After) | 14,199 ± 5,013 | 580 ± 254 | 1,047 ± 360 | 5.06 ± 1.03 | 0.90 ± 0.06 | 36.8 ± 14.35 |
*Legend: NC, negative control; SC, Salmonella challenged; BT, Bacillus treated; OTU, operational taxonomic unit; PD, phylogenetic distance (whole tree)
aMean number of raw reads per treatment group. No significant differences (P < 0.05) in alpha diversity were detected between groups using compare_alpha_diversity.py script
Microbial diversity
Alpha diversity was compared among the NC, SC, and BT groups (Table 1; Additional file 2: Fig. S1). α-diversity parameters were calculated based on the OTU using the phylogenetic diversity (PD) whole tree, Chao1, Shannon, and Simpson methods. Diversity (Shannon and Simpson) values were highest in the NC group and lowest in SC broilers after Salmonella challenge (Additional file 2: Fig. S1A). Diversity values are summarised in Table 1. Species richness (Chao1) and bacterial diversity (Shannon and Simpson) exhibited similar trends; i.e. the highest values of both were in the NC group, and lowest in the SC group (Table 1), implying that Salmonella infection negatively affects overall microbial diversity. In addition, the rarefaction curves confirmed that Salmonella infection decreased the level of microbial diversity (Additional file 2: Fig. S1B).
Effect of Salmonella challenge and B. subtilis CLS2 administration on microbiota composition
Irrespective of Salmonella challenge and Bacillus supplementation, the phylum Firmicutes showed the highest abundance (>80%), followed by the phyla Bacteroidetes and Proteobacteria (Fig. 1a). These major bacterial phyla are also major constituents of the gut microbiota of other birds and livestock animals [13]. At the 24-h post-Salmonella challenge, the abundance of Proteobacteria was significantly increased, and that of Firmicutes decreased in the SC group, whereas those in the BT group had recovered to levels similar to the NC group. In previous reports, Proteobacteria was highly abundant in Salmonella-infected animals [15, 16]. Similarly, dietary supplementation with probiotics altered the microbial composition of broiler chickens, resulting in higher abundance of Firmicutes and lower abundance of Bacteroidetes and Proteobacteria [14].
Fig. 1.
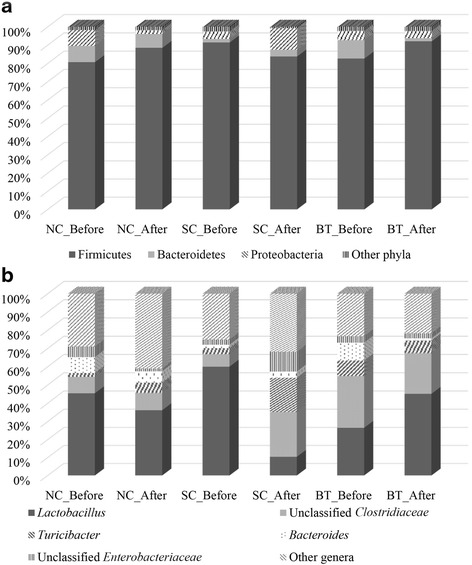
Faecal microbiota composition of broiler chickens at the phylum (a) and genus (b) levels. Broiler chickens were divided into the following three groups before and after Salmonella challenge: NC, control/basal diet; SC, basal diet challenged with S. Gallinarum (SC); and BT, basal diet supplemented with B. subtilis CSL2
At the genus level, a total of 124 bacterial genera were detected in all broiler samples (Additional file 3: Table S2), which is comparable to other avians [17]. Lactobacillus, unclassified Clostridiaceae, Turicibacter, Bacteroides, and unclassified Enterobacteriaceae were the major bacterial genera in broiler faeces (Fig. 1b). Interestingly, the abundance of Lactobacillus, a genus of beneficial bacteria, decreased significantly in the SC group, but increased in the BT group. In addition, the abundance of unclassified Enterobacteriaceae, which comprises several pathogenic species, in the SC group increased from 2.9% to 10.9%. A previous study reported an increase in Lactobacillus abundance after probiotic administration in chicken and pigs [18]. In contrast, SC broilers had a higher abundance of Turicibacter, unclassified Enterobacteriaceae, and Bacteroides than the other groups had. In a previous study, Salmonella infection of mice was also found to result in higher Enterobacteriaceae and lower Lactobacillus abundance [19].
Taxon-independent and functional analyses
Of the 212 bacterial OTU identified in this experiment, the 42 differentially abundant bacterial OTU with >1.0% abundance were used to generate a DAPC plot (Fig. 2a) and canonical loading plot (Fig. 2b). The DAPC plot showed that all groups had similar microbial composition before Salmonella challenge. But separate clusters were formed in response to B. subtilis CSL2 supplementation and/or S. Gallinarum infection, suggestive of distinct microbial communities. These microbial shifts were attributed to subtle changes in the abundance of several bacterial OTU, including unclassified Neisseriaceae, Ruminococcus, and Candidatus Arthromitus (Fig. 2b). Unclassified Neisseriaceae was the strongest indicator of the presence of distinct microbial clusters; however, the other loading peaks might also exert considerable effects [11].
Fig. 2.
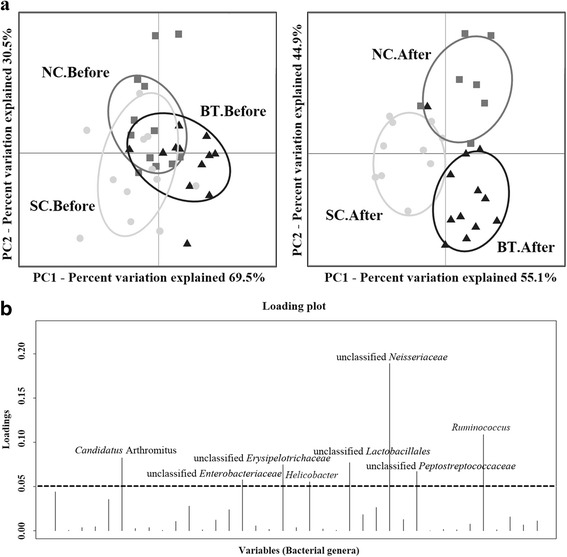
Taxon-independent multivariate analysis and separation of broiler microbiota. a Discriminant analysis of principal components revealed distinct clustering of the control (NC, grey), Salmonella-challenged (SC, white), and Bacillus-treated (BT, black) groups using OTU at the 97% identity level. Significant differences (P < 0.001) were calculated using compare_categories.py using the PERMANOVA test. b Canonical loading plot showing differentially abundant bacterial genera. The individual peaks show the magnitude of the influence of each variable on separation of the NC, SC, and BT groups after challenge of broiler chickens (0.05 threshold level)
Functional analysis identified a total of 137 of 265 KEGG functions as differentially abundant (>0.1% mean relative abundance) (Additional file 4: Table S3). These 137 KEGG functions were analysed using a loading plot to identify vital functions among microbial clusters (Fig. 3a). The three most discriminating KEGG functions in broiler microbiota were the phosphotransferase system (PTS), glycan degradation, and replication/recombination/repair proteins (Fig. 3a). The PTS was significantly decreased by Salmonella challenge, but recovered to almost normal levels following Bacillus administration (NC group) (Fig. 3b). The abundance of genes associated with glycan degradation was significantly decreased in the BT group compared to the NC and ST groups, which might be a unique effect of supplementation of B. subtilis CSL2 (Fig. 3c). In addition, the abundance of genes related to repair and recombination of DNA was highest in the BT group and lowest in the NC group (Fig. 3d).
Fig. 3.
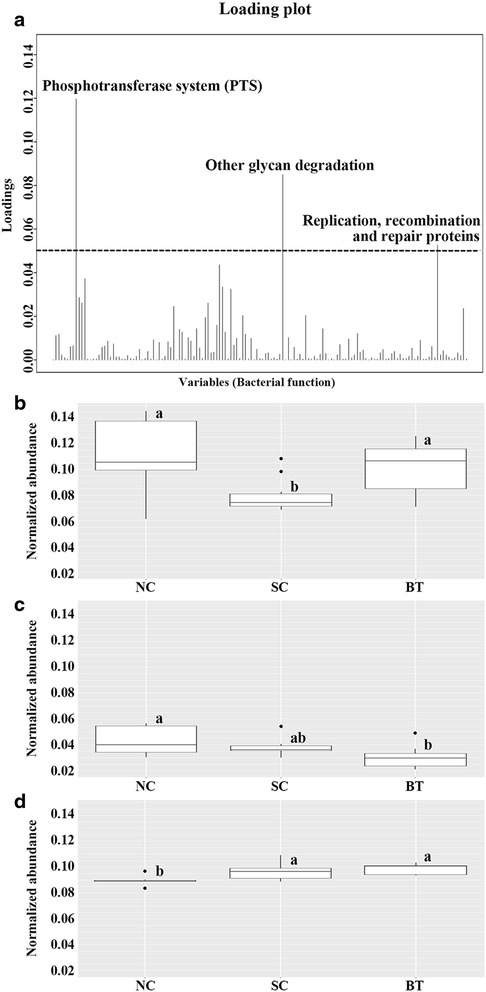
Functional analysis and comparison of microbial communities. a Canonical loading plot showing the magnitude of the influence of each variable on separation of individual peaks in the broiler microbiota (0.05 threshold level). Normalised abundances of functional activities b phosphotransferase system, c glycan degradation, and d recombination and repair proteins after Salmonella challenge. The interquartile ranges are indicated by the outer bounds of the boxes, the median by the midline (black), and the outliers are indicated by black circles (●). Tukey’s honestly significant difference (HSD) test was used to evaluate statistical significance (P < 0.05)
Discussion
In this study, B. subtilis CSL2 administration altered the microbiota of broilers and contributed to protection against Salmonella infection. In previous studies, microbial shifts were evident after probiotic administration and pathogenic invasion [14, 19, 20]. In-feed administration of E. faecium NCIMB 11181 changed the microbial composition in piglets [14]. Furthermore, pathogen infection led to significant alterations in the chicken gut microbiota [5, 15, 16]. Pathogen infection disrupts gut microbial diversity and overwhelms the microbiota by producing toxins and harmful agents [15, 18, 19]. Lower microbial diversity indicates a reduced ability of the microbiota to maintain gut homeostasis and resist invasion. B. subtilis CSL2 might provide protection against pathogen invasion by increasing bacterial diversity and metabolic and cellular functionality. Reduced functional diversity suppresses commensal-microbiota-mediated homeostasis [21].
Firmicutes and Bacteroidetes comprised the majority of the broiler microbiota at the phylum level; these organisms function in energy production and metabolism, particularly starch digestion and microbial fermentation [4, 10, 13]. The increased abundance of Proteobacteria in Salmonella-infected broilers suggests gastrointestinal dysbiosis and imbalance. Indeed, Proteobacteria are closely associated with S. Enteritidis infection in animals [15, 16]. Moreover, significant changes were detected in the abundance of Lactobacillus and Turicibacter after 24 h in the SC and BT groups. Lactobacillus is a dominant and important gut commensal genus present at frequencies of up to 109/g [4]. Increased abundance of Lactobacillus in the gastrointestinal tract of chickens is considered beneficial for their health and performance [22]. Certain strains of Lactobacillus produce antimicrobial substances [23], exopolysaccharides, and short-chain fatty acids as additional energy sources [24]. B. subtilis CSL2 promotes the growth of beneficial lactobacilli, as do other types of in-feed probiotics [9, 14].
Furthermore, Enterobacteriaceae abundance was reduced in the BT group after 24 h of Salmonella challenge, and increased numbers of Enterobacteriaceae in SC broilers might imbalance the microbiota and thus exert harmful effects on the gut [20]. The impact of S. Gallinarum and B. subtilis CSL2 on the overall composition of the gut microbiota can be detected within 24 h post infection [18]. Surprisingly, in this study Salmonella spp. were not detected in the SC group. Previous studies also suggest that Salmonella abundance is lower after 72 h, during which time symptoms are manifested [15, 19]. Furthermore, the abundance of the genus Turicibacter was significantly increased in Salmonella-infected broilers. Turicibacter has been reported in mammalian studies to be relevant to infection [25]. Its immunomodulatory and invasive properties resulted in subclinical infection of the gastrointestinal tracts of livestock and poultry animals [25, 26]. However, its ecological role and pathogenic potential remain unclear due to the dearth of studies.
A taxon-independent analysis was performed to prevent taxonomic bias and calculate variation based on all differentially abundant bacterial OTU. It is plausible that B. subtilis CSL2 maintains microbial community stability, similar to the case in normal broilers. Given that chicken microbial communities are highly dynamic and delicate, abrupt disturbances might cause greater variations in their microbiota than in other animals [4]. In addition, the loading plot of bacterial OTU (i.e. unclassified Neisseriaceae, Ruminococcus, and Candidatus Arthromitus) abundance in the SC group suggested the effects of S. Gallinarum infection in broilers. The identification of bacterial OTU belonging to Neisseriaceae (phylum: Proteobacteria) was highly suggestive of microbial clustering, and these organisms include several diarrhoea-causing pathogens [27]. These results imply that establishment of Salmonella in the gut requires the support of other pathogens or opportunistic bacteria therein.
In this study, functional prediction was performed to evaluate and compare the metabolic activities of the microbial communities among broiler groups [10]. Three metabolic and cellular functions were significantly affected by the administration of B. subtilis CSL2 and S. Gallinarum infection, namely the phosphotransferase system, glycan degradation, and DNA repair mechanisms. The greater abundance of genes associated with the PTS system in the BT group might be beneficial to broiler chickens. The PTS system is the major bacterial transport system for carbohydrates, and is involved in the regulation of bacterial fermentation and feed conversion [13, 21, 28]. This result thus indicates the probability of rapid uptake of available simple sugars rather than the digestion of complex carbohydrates. Furthermore, Pérez-Cobas et al. suggested that the PTS system assists bacterial stabilisation of gut-associated stresses in an unstable environment, as well as increasing energy yield, giving the commensal microbiota a competitive advantage over foreign microorganisms [29]. In addition, the reduced abundance of genes related to glycan degradation in broilers fed B. subtilis CSL2 implies that glycans are not a primary source of nutrients for the microbiota [30]. Higher glycan degradation is significantly correlated with the presence of pathogens (i.e. Salmonella and enterotoxigenic Clostridia) [27, 30]. These results suggest that probiotic supplementation alters the metabolic functions of the microbiota in a way that benefits the host. However, additional studies are required to elucidate the protective effects of B. subtilis CSL2 against S. Gallinarum infection.
Conclusions
In this study, the characterisation of the chicken faecal microbiota community structure and composition by 16S rRNA gene pyrosequencing revealed that the probiotic strain protected against Salmonella infection. The supplementation of B. subtilis CSL2 significantly changed the microbial diversity and composition by increasing the abundance of beneficial microorganisms. Conversely, broiler chickens infected with S. Gallinarum promoted the growth of potentially harmful bacteria such as Turicibacter, Enterobacteriaceae and Neisseriaceae. Furthermore, the potentially probiotic or pathogenic bacteria influenced the microbial functionality, particularly in the energy transport and metabolism capability of the gut. Overall, S. Gallinarum infection and B. subtilis CSL2 supplementation in the diet of broiler chickens influenced the diversity, composition, and functional diversity of the faecal microbiota. These results will facilitate prevention of Salmonella infection before the onset of symptoms using the potentially probiotic strain B. subtilis CSL2. Moreover, the findings offer significant insights to understand potential mechanisms of Salmonella infection and the mode of action of probiotics in broiler chickens.
Acknowledgements
Not applicable.
Funding
This work was supported by a grant from the Next-Generation BioGreen 21 Program (PJ01115903), Rural Development Administration, Republic of Korea.
Availability of data and materials
All standard flowgram format (.sff) files generated by the 454-pyrosequencer containing all raw sequence reads have been deposited at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA), BioProject (accession number: PRJNA327786, http://www.ncbi.nlm.nih.gov/bioproject/327786).
Authors’ contributions
JKO and EABP performed data collection and drafted the manuscript together. JPC carried out data analysis. IH Kim participated in the animal experiment. DSY isolated B. subtilis CSL2 strain. DKK managed the entire experiments and revised the manuscript. All authors read and approved the final manuscript for publication.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
All protocols in this experiment involving animals were approved by the Animal Care and Use Committee of Dankook University.
Abbreviations
- BT
Bacillus subtilis CSL2-treated
- DAPC
Discriminant analysis of principal components
- KEGG
Kyoto encyclopedia of genes and genomes
- NC
Negative control
- NGS
Next generation sequencing
- OTU
Operational taxonomic unit
- PICRUSt
Phylogenetic investigation of communities by reconstruction of unobserved states
- QIIME
Quantitative insights into microbial ecology
- SC
Salmonella-challenged/Salmonella-infected chickens
Additional files
Basal diet composition. (DOCX 19 kb)
Rarefaction curves measuring bacterial diversity among broiler communities. (DOCX 231 kb)
Highly abundant genera in broiler groups. (DOCX 22 kb)
Highly abundant KEGG functions in broiler groups. (DOCX 22 kb)
Contributor Information
Ju Kyoung Oh, Email: supajuko@gmail.com.
Edward Alain B. Pajarillo, Email: eabpajarillo@gmail.com
Jong Pyo Chae, Email: divine1002@naver.com.
In Ho Kim, Email: inhokim@dankook.ac.kr.
Dong Soo Yang, Email: dsyangabson@naver.com.
Dae-Kyung Kang, Phone: +82 41 550 3655, Email: dkkang@dankook.ac.kr.
References
- 1.Leeson S. Future considerations in poultry nutrition. Poult Sci. 2012;91:1281–5. doi: 10.3382/ps.2012-02373. [DOI] [PubMed] [Google Scholar]
- 2.Matulova M, Varmuzova K, Sisak F, Havlickova H, Babak V, Stejskal K, et al. Chicken innate immune response to oral infection with Salmonella enterica serovar Enteritidis. BMC Vet Res. 2013;44:37. doi: 10.1186/1297-9716-44-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanley D, Hughes RJ, Moore RJ. Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. Appl Microbiol Biotechnol. 2014;98:4301–10. doi: 10.1007/s00253-014-5646-2. [DOI] [PubMed] [Google Scholar]
- 4.Oakley BB, Lillehoj HS, Kogut MH, Kim WK, Maurer JJ, Pedroso A, et al. The chicken gastrointestinal microbiome. FEMS Microbiol Lett. 2014;360:100–12. doi: 10.1111/1574-6968.12608. [DOI] [PubMed] [Google Scholar]
- 5.Videnska P, Sisak F, Havlickova H, Faldynova M, Rychlik I. Influence of Salmonella enterica serovar Enteritidis infection on the composition of chicken cecal microbiota. BMC Vet Res. 2013;9:140. doi: 10.1186/1746-6148-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foley SL, Nayak R, Hanning IB, Johnson TJ, Han J, Ricke SC. Population dynamics of Salmonella enterica serotypes in commercial egg and poultry production. Appl Environ Microbiol. 2011;77:4273–9. doi: 10.1128/AEM.00598-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeoman CJ, Chia N, Jeraldo P, Sipos M, Goldenfeld ND, White BA. The microbiome of the chicken gastrointestinal tract. Anim Health Res Rev. 2012;13:89–99. doi: 10.1017/S1466252312000138. [DOI] [PubMed] [Google Scholar]
- 8.Larsen N, Thorsen L, Kpikpi EN, Stuer-Lauridsen B, Cantor MD, Nielsen B, et al. Characterization of Bacillus spp. strains for use as probiotic additives in pig feed. Appl Microbiol Biotechnol. 2013;98:1105–18. doi: 10.1007/s00253-013-5343-6. [DOI] [PubMed] [Google Scholar]
- 9.Park JH, Kim IH. The effects of the supplementation of Bacillus subtilis RX7 and B2A strains on the performance, blood profiles, intestinal Salmonella concentration, noxious gas emission, organ weight and breast meat quality of broiler challenged with Salmonella typhimurium. J Anim Physiol Anim Nutr (Berl) 2015;99:326–34. doi: 10.1111/jpn.12248. [DOI] [PubMed] [Google Scholar]
- 10.Mohd Shaufi MA, Sieo CC, Chong CW, Gan HM, Ho YW. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathog. 2015;7:4. doi: 10.1186/s13099-015-0051-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pajarillo EAB, Chae JP, Kim HB, Kim IH, Kang D-K. Barcoded pyrosequencing-based metagenomic analysis of the faecal microbiome of three purebred pig lines after cohabitation. Appl Microbiol Biotechnol. 2015;99:5647–56. doi: 10.1007/s00253-015-6408-5. [DOI] [PubMed] [Google Scholar]
- 12.Jombart T, Ahmed I. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics. 2011;27:3070–1. doi: 10.1093/bioinformatics/btr521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamendella R, Santo Domingo JW, Ghosh S, Martinson J, Oerther DB. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011;11:103. doi: 10.1186/1471-2180-11-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pajarillo EAB, Chae JP, Balolong MP, Kim HB, Park C-S, Kang D-K. Effects of probiotic Enterococcus faecium NCIMB 11181 administration on swine fecal microbiota diversity and composition using barcoded pyrosequencing. Anim Feed Sci Technol. 2015;201:80–8. doi: 10.1016/j.anifeedsci.2015.01.011. [DOI] [Google Scholar]
- 15.Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, et al. Salmonella enterica serovar Typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5:2177–89. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekirov I, Gill N, Jogova M, Tam N, Robertson M, de Llanos R, et al. Salmonella SPI-1-mediated neutrophil recruitment during enteric colitis is associated with reduction and alteration in intestinal microbiota. Gut Microbes. 2010;1:30–41. doi: 10.4161/gmic.1.1.10950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waite DW, Taylor MW. Exploring the avian gut microbiota: current trends and future directions. Front Microbiol. 2015;6:673. doi: 10.3389/fmicb.2015.00673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mountzouris KC, Dalaka E, Palamidi I, Paraskeuas V, Demey V, Theodoropoulos G, et al. Evaluation of yeast dietary supplementation in broilers challenged or not with Salmonella on growth performance, cecal microbiota composition and Salmonella in ceca, cloacae and carcass skin. Poult Sci. 2015;94:2445–55. doi: 10.3382/ps/pev243. [DOI] [PubMed] [Google Scholar]
- 19.Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, et al. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76:907–15. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Videnska P, Faldynova M, Juricova H, Babak V, Sisak F, Havlickova H, et al. Chicken faecal microbiota and disturbances induced by single or repeated therapy with tetracycline and streptomycin. BMC Vet Res. 2013;9:30. doi: 10.1186/1746-6148-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sergeant MJ, Constantinidou C, Cogan TA, Bedford MR, Penn CW, Pallen MJ. Extensive microbial and functional diversity within the chicken cecal microbiome. PLoS One. 2014;9:e91941. doi: 10.1371/journal.pone.0091941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakphaichit M, Thanomwongwattana S, Phraephaisarn C, Sakamoto N, Keawsompong S, Nakayama J, et al. The effect of including Lactobacillus reuteri KUB-AC5 during post-hatch feeding on the growth and ileum microbiota of broiler chickens. Poult Sci. 2011;90:2753–65. doi: 10.3382/ps.2011-01637. [DOI] [PubMed] [Google Scholar]
- 23.Rybal’chenko OV, Orlova OG, Bondarenko VM. Antimicrobial peptides of lactobacilli. Zh Mikrobiol Epidemiol Immunobiol. 2013;4:89–100. [PubMed] [Google Scholar]
- 24.Pajarillo EAB, Kim SH, Lee JY, Valeriano VDV, Kang, D-K. Quantitative proteogenomics and the reconstruction of the metabolic pathway in Lactobacillus mucosae LM1. Korean J Food Sci Anim Resour. 2015;35:692–2. [DOI] [PMC free article] [PubMed]
- 25.Cuív PÓ, Klaassens ES, Durkin AS, Harkins DM, Foster L, McCorrison J, et al. Draft genome sequence of Turicibacter sanguinis PC909, isolated from human feces. J Bacteriol. 2011;193:1288–9. doi: 10.1128/JB.01328-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosshard PP, Zbinden R, Altwegg M. Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, Gram-positive bacterium. Int J Syst Evol Microbiol. 2002;52:1263–6. doi: 10.1099/00207713-52-4-1263. [DOI] [PubMed] [Google Scholar]
- 27.Deatherage Kaiser BL, Li J, Sanford JA, Kim Y-M, Kronewitter SR, Jones MB, et al. A Multi-omic view of host-pathogen-commensal interplay in Salmonella-mediated intestinal infection. PLoS One. 2013;8:e67155. doi: 10.1371/journal.pone.0067155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JY, Pajarillo EAB, Kim MJ, Chae JP, Kang D-K. Proteomic and transcriptional analysis of Lactobacillus johnsonii PF01 during bile salt exposure by iTRAQ Shotgun proteomics and quantitative RT-PCR. J Proteome Res. 2013;12:432–43. doi: 10.1021/pr300794y. [DOI] [PubMed] [Google Scholar]
- 29.Pérez-Cobas AE, Moya A, Gosalbes MJ, Latorre A. Colonization resistance of the gut microbiota against Clostridium difficile. Antibiotics (Basel) 2015;4:337–57. doi: 10.3390/antibiotics4030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eilam O, Zarecki R, Oberhardt M, Ursell LK, Kupiec M, Knight R, et al. Glycan degradation (GlyDeR) analysis predicts mammalian gut microbiota abundance and host diet-specific adaptations. MBio. 2014;5:e01526–14. doi: 10.1128/mBio.01526-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All standard flowgram format (.sff) files generated by the 454-pyrosequencer containing all raw sequence reads have been deposited at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA), BioProject (accession number: PRJNA327786, http://www.ncbi.nlm.nih.gov/bioproject/327786).