

ABSTRACT
Individuals with robust natural killer (NK) cell function incur lower rates of malignancies. To expand our understanding of genetic factors contributing to this phenomenon, we analyzed NK cells from cancer resistant and susceptible strains of mice. We identified a correlation between NK levels of the X-chromosome-located adaptor protein SLy1 and immunologic susceptibility to cancer. Unlike the case for T or B lymphocytes, where SLy1 shuttles between the cytoplasm and nucleus to facilitate signal transduction, in NK cells SLy1 functions as a ribosomal protein and is located solely in the cytoplasm. In its absence, ribosomal instability results in p53-mediated NK cell senescence and decreased clearance of malignancies. NK defects are reversible under inflammatory conditions and viral clearance is not impacted by SLy1 deficiency. Our work defines a previously unappreciated X-linked ribosomopathy that results in a specific and subtle NK cell dysfunction leading to immunologic susceptibility to cancer.
KEYWORDS: Immunosurveillance, lung cancer, natural killer cells, ribosomopathy
Despite recent advances in immunotherapy, our understanding of individual variability that results in immunologic susceptibility or resistance to cancer is limited. Part of the difficulty in elucidating the tumor immune response stems from the complex and often conflicting role of the adaptive immune system in cancer progression. Natural killer (NK) cells are known for their exclusive cytolytic activity against virally infected and malignantly transformed cells. Population-based studies demonstrate that robust NK cell function correlates with lower lifetime incidence of cancer.1 In mutant mice deficient in the adaptive immune response, NK cells offer significant protection from tumor formation and metastasis.2 For certain malignancies, such as lung cancer, NK cells play a predominant and near-exclusive role in immunologic control of tumor development and progression.3,4 Thus, the study of the genetic basis for differences in NK function between individuals may offer therapeutic insight into cancer biology.
Unlike other lymphocytes, NK cells are required to rapidly produce cytotoxic mediators upon encountering a transformed target cell. In fact, NK cells maintain a pre-existing pool of mRNA for granzyme B and perforin even at rest, relying on translational control of such cytotoxic mediators.5 Here, we show that compared to T and B lymphocytes quiescent NK cells also maintain a relative abundance of ribosomes. The multifunctional adaptor protein SLy1 facilitates ribosomal stability in this cell population. In its absence, ribosomal instability results in the elaboration of free ribosomal proteins that interfere with Mdm2-mediated clearance of p53. Overabundance of p53 leads to NK dysfunction, downregulation of multiple activating receptors and immunologic cancer susceptibility.
Ribosomopathies are traditionally considered as severe or embryonic lethal disorders that result in life threatening diseases such as fragile X syndrome, Diamond-Blackfin anemia or Treacher-Collins syndrome.6 Here, we demonstrate that this group of diseases may also include a previously unrecognized set of subtle immunologic disorders predisposing to cancer susceptibility with few other overt defects.
Results
Expression of the adaptor protein SLy1 correlates with robust NK function
NK cells represent an evolutionary conserved population of granular lymphocytes that recognize stress ligands expressed on virally infected, transformed or malignant cells.7 The interaction of inhibitory receptors such as Ly49 antigens in the NK gene complex (NKC) of mice and killer immunoglobulin-like receptors in man, with self-major histocompatibility complex class I antigens (MHC Class I) plays a role in the licensing or education of NK cells during development.8 Extensive allelic polymorphism in the NKC and MHC Class I is postulated to contribute to individual immunologic susceptibility or resistance to cancer.3,9 We have recently demonstrated that the weak NK function and lung cancer susceptibility of the 129SvEv strain of mice can be improved thorough transgenic expression of the NKC from the C57BL/6 (B6) strain, which has inherently robust NK function.3 The C3H and AJ strains of mice share similar NKC by non-hierarchical genomic clustering.10 Nevertheless, the C3H strain of mice demonstrates a relative resistance to lung cancer development compared to the A/J strain (Figs. S1A and B). C3H mice also have relatively higher numbers of NK cells (Fig. 1A, Fig. S1C) and demonstrate increased NK-mediated lysis of cancer cell lines (Fig. 1B)(Fig. S1D). Thus, despite Ly49 receptor similarity to the AJ strain, NK cells of C3H mice physiologically resemble those of the B6 strain. Taken together, we must conclude that additional factors, other than licensing, must contribute to differences in NK homeostasis in these four strains of mice.
Figure 1.
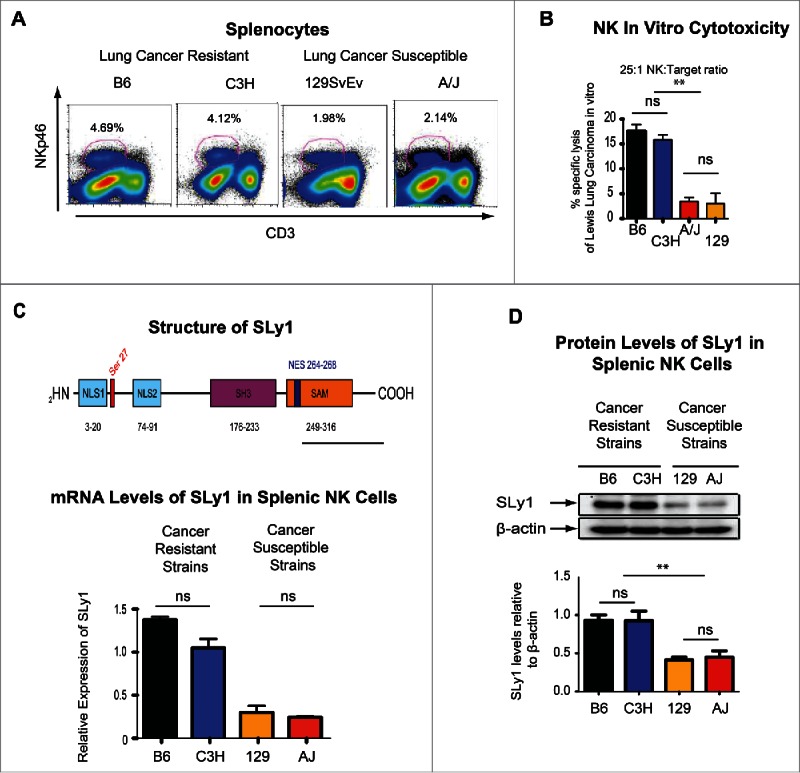
Strain-specific differences in cancer susceptibility and SLy1 levels. (A) Strain-specific differences in the number of splenic NK cells (defined as NKp46+CD3−). Graphic data showing one representative FACS plot from three to six animals. (B) Strain-specific differences in in vitro specific lysis of Lewis Lung Carcinoma (LLC) by splenic NK cells at a 25:1 effector:target ratio by 51Cr-release assay (representative of four separate experiments with comparison performed by unpaired t-test). (C) Structure of SLy1 (top) and relative levels of SLy1 in splenic NK cells of various strains of mice as measured by mRNA gene expression array (bottom). (D) SLy1 levels in freshly isolated splenic NK cells as measured by Western blot analysis (four animals per group). Comparison performed by unpaired t-test. *p < 0.05, **p < 0.01, ns = p > 0.05.
To explore these differences further, we performed genome-wide expression analysis of NK cells from the B6 and C3H cancer-resistant mice, with robust NK function, and compared their mRNA levels to two cancer-susceptible strains with poor NK function (A/J, 129/SvEv). Expression of one candidate gene correlated with NK phenotype between lung cancer resistant and susceptible strains of mice (Figs. 1C and D). This X chromosome-linked gene, known as SLy1 (SH3-domain protein expressed in Lymphocyte 1), has been previously described to play a role in T cell and B cell development and function.11-14 In addition to its expression in T and B lymphocytes,12 it is expressed in mature peripheral NK cells (Fig. 1D) and is a prototypical adaptor protein with a nuclear localization signal as well as protein–protein interacting SH3 and SAM domains. Based on its structure, it has been demonstrated by us to facilitate surface to nuclear signal transduction for rapid T lymphocyte responses.11-14 Nevertheless, its function or biologic significance in NK-mediated immunosurveillance remains elusive. To further explore whether NK licensing or education affects SLy1 expression, we performed Western blot analysis on NK cells from transgenic mice on a B6 background where NK cells are either licensed, or not, based on forced expression of an H2b single chain trimer in otherwise MHC-deficient hosts.15 Licensing status did not alter SLy1 expression (Fig. S1E) suggesting a unique and independent role for this adaptor protein in NK function.
SLy1 deficiency affects NK function
In order to directly evaluate the role of SLy1 on NK function, we took advantage of the knockout mutant mice on a C57BL/6 background created by our group (B6.129Ola-SLy1tm1Sb/P or B6SLy1− from here on).12 Since clearance of RMA-S lymphoma expressing the NKG2D ligand Rae-1 is an NK-mediated process, we injected 2.5 × 106 lymphoma cells intravenously into B6SLy1−or B6wt littermate mice and evaluated pulmonary clearance 18 h later.16 Fewer viable RMA-S cells were evident in B6wt littermates compared to B6SLy1−mice suggestive of poor NK-mediated immunosurveillance in the absence of SLy1 (Fig. 2A). Growth of Lewis Lung Carcinoma (LLC) that, similar to RMA-S lymphoma, is controlled by NK cells3 is accelerated in B6SLy1− mice in an NK-dependent fashion (Fig. 2B). Consistent with this B6SLy1−NK cells lyse LLC less efficiently in vitro (Fig. 2C) and cluster poorly with GFP-expressing LLC in vivo (Fig. 2D) than B6wt NK cells. Decreased in vivo clearance of transgenic splenocytes over-expressing the MCMV antigen m157, which is recognized by the NK activating receptor Ly49H, was also evident in B6SLy1−mice suggesting that SLy1 also plays a role in cytotoxicity toward non-malignant NK cell targets (Fig. 2E). B6SLy1−NK cells also produce less IFNγ and degranulate less efficiently in plate-bound antibody stimulation assays (Fig. 2F and Fig. S2). Taken together, these data demonstrate that SLy1 plays a role in NK effector function.
Figure 2.
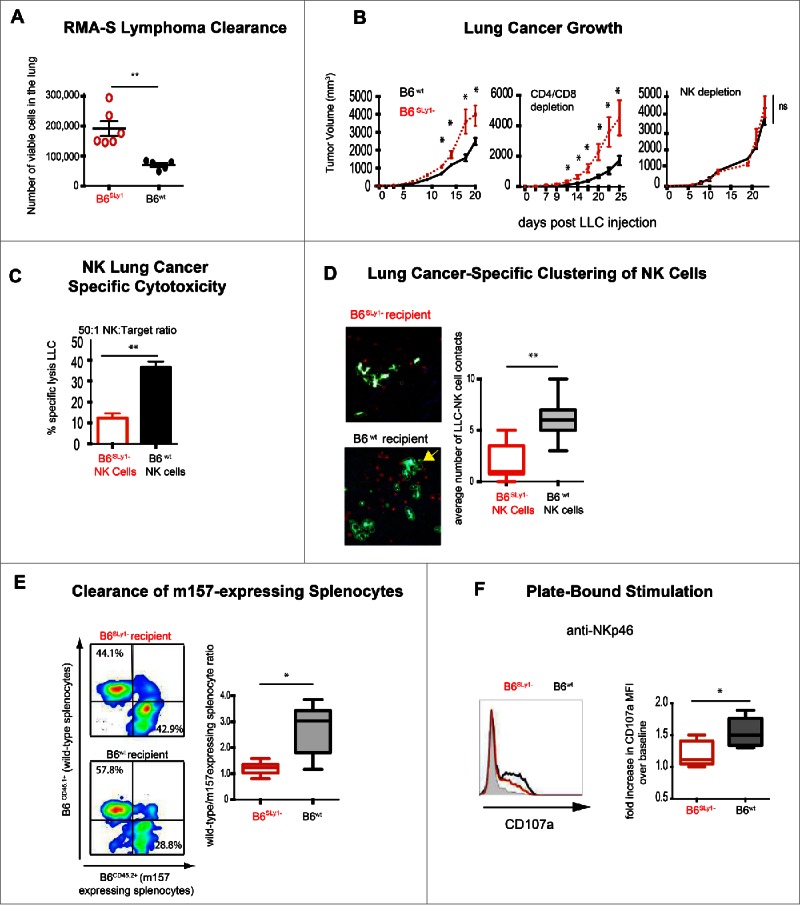
B6SLy1− NK cells demonstrate multiple functional defects. (A) Clearance of RMA lymphoma expressing the NKG2D ligand Rae-1 from lungs of B6wt or B6SLy1−mice 18 h after i.v. injection. Comparison performed by unpaired t-test. (B) Growth of LLC injected into the flank of B6SLy1− (red line) and B6wt mice (black line) in untreated mice (left), those depleted of CD4+ and CD8+ lymphocytes (middle) or NK1.1+ cells (right). Comparison performed by unpaired t-test at each time point. Isotype control-treated animals demonstrated tumor growth identical to unmanipulated mice (data not shown). (C) In vitro lysis of LLC at a 50:1 effector:target ratio. Data representative of four separate experiments with comparison performed by unpaired t-test. (D) Average number of durable contacts between B6SLy1− (top) and B6wt (bottom) NK (crossed to express tomato cherry red on NK1.1 promoter56) cells with GFP-expressing LLC injected into the lung. Yellow arrow points to 4 B6wt NK cells establishing contact with one tumor cell. Data representative of three separate mice per group with comparison performed by unpaired t-test. (E) In vivo clearance of CFSE labeled B6CD45.1+wild-type or B6CD45.2+m157-expressing splenocytes injected into B6SLy1− (top) and B6wt mice (bottom) recipient mice. Data derived from flow cytometric analysis of the spleen, demonstrating one representative experiment on the left and summary of five animals on the right compared by unpaired t-test. (F) Degranulation of B6SLy1−(red) and B6wt (black) NK cells after plate bound stimulation with NKp46 as measured by surface CD107a expression. Left panel representative of one experiment with heavy black line representative of Cd107a in B6wt NK cells after co-culture with plate bound NKp46 and heavy red line representative of B6SLy1− NK cells after co-culture with plate bound NKp46. Shaded black line representative of plate-bound isotype control. Comparison performed by unpaired t-test. Summary of five separate experiments. *p < 0.05, **p < 0.01, ns = p > 0.05.
SLy1 deficiency leads to functional defects of natural killer cells in the periphery but does not affect their development in the bone marrow
B6SLy1−mice had fewer NK cells, defined as NKp46+CD3− or NK1.1+CD3−cells, in spleens (Fig. 3A) or peripheral organs such as lungs (data not shown). Since SLy1 has previously been demonstrated to play a role in T cell development, we next evaluated maturation of NK1.1+CD3− NK cells by stage-specific expression of CD27 and CD11b.17 To our surprise, we were unable to detect differences in the maturation of splenic (Fig. 3B) or lung-resident NK cells (data not shown). We next considered the possibility that SLy1 may affect NK development in the bone marrow. Again, we did not detect differences in the numbers of CD122+ committed NK progenitors, their maturation defined by the acquisition of Ly49 receptors or α-2 integrin CD49b (DX5 antigen), or proliferation as defined by upregulation of Ki67 between B6SLy1−or B6wt mice (Figs. S3A and B). Unlike the case for mature splenic NK cells, SLy1 was undetectable in flow cytometrically sorted CD122+DX5− bone marrow-derived NK progenitors in B6wt mice (Fig. S3C). Such data suggested that unlike the case for T lymphocytes, SLy1 exerts its function only in mature NK cells.
Figure 3.
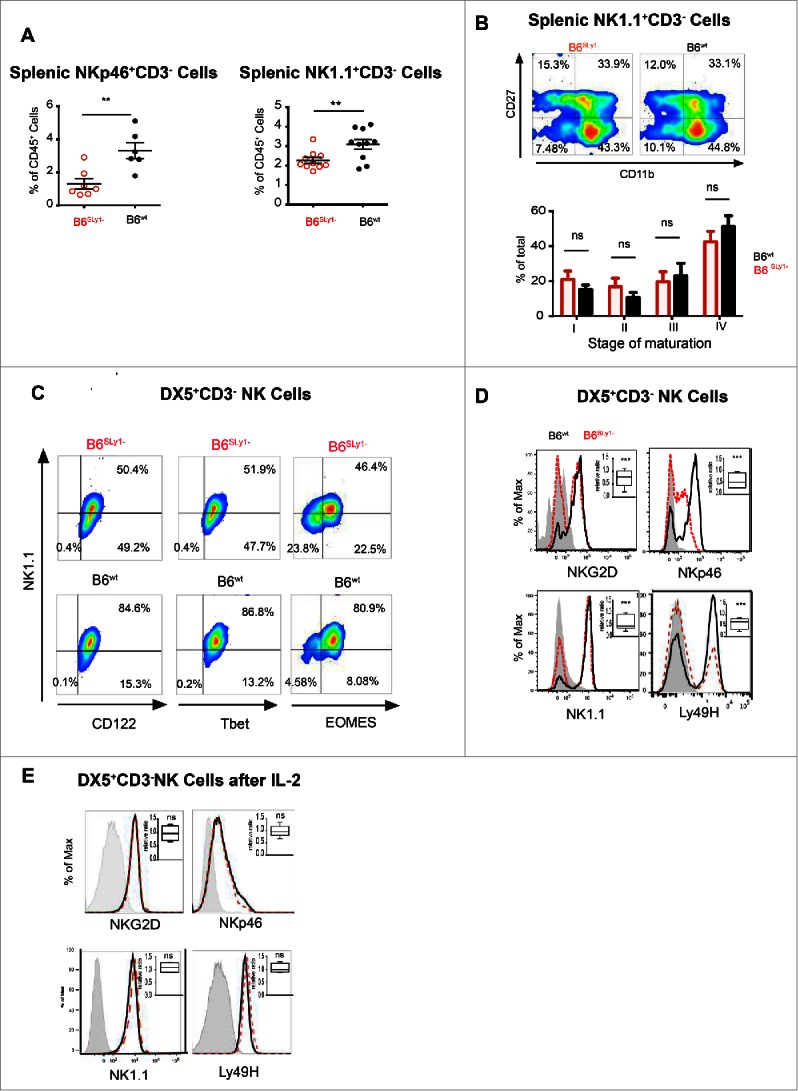
B6SLy1− NK cells demonstrate multiple phenotypic defects. (A) Quantification of splenic NK cells defined as either NK1.1+CD3− or NKp46+CD3−. Comparison performed by unpaired t-test. (B) Comparison of four-stage maturation program of splenic NK1.1+CD3− NK cells in B6SLy1−and B6wt mice as defined by surface expression of CD27 and CD11b. Representative flow cytometry plots at the top and summary data of five separate experiments at the bottom. (C) Expression of NK1.1, CD122, Tbet and EOMES in B6SLy1− and B6wt splenic CD3−DX5+ NK cells. (D) Representative histograms and relative ratios (right upper corner) of NKG2D, NKp46, NK1.1 and Ly49H in DX5+CD3− splenic NK cells in in B6SLy−and B6wt mice. Representative of five separate experiments performed on different animals from different litters. Statistical analysis performed by unpaired t-test to the null hypothesis considering the relative ratio to be 1. (E) Representative histograms and relative ratios (right upper corner) of NKG2D, NKp46, NK1.1 and Ly49H in DX5+CD3− splenic NK cells in in B6SLy1− and B6wt mice after activation in vitro with 1,000 IU/mL of IL-2. Statistical analysis of (D) and (E) performed by unpaired t-test to the null hypothesis which assumes the relative ratio to be 1.
We next considered the possibility that SLy1 may somehow affect activation of peripheral NK cells without altering their development. This raised the concern that the use of activating receptors, such as NKp46 or NK1.1, for identification of this cell population may create a confounding factor if NK activation is altered by SLy1. In order to explore this further, we validated our data using the DX5 antigen, or α-2 integrin CD49b, to identify NK cells. DX5 is expressed on the vast majority of NK cells, is not strain-restricted like other markers such as NK1.1, and is present only on fully mature cells.18,19 Since DX5 can be expressed on other rare lymphocyte subsets such as basophils,20 we next evaluated concomitant surface expression of the activating receptor NK1.1 along with the IL-2 receptor β-chain (CD122), which is constitutively present on NK cells but not basophils.20,21 While a significant portion of DX5+CD3− splenocytes of B6SLy1−mice were NK1.1− they did express surface CD122, confirming their identity as NK cells (Fig. 3C). Furthermore, the transcription factor Tbet was constitutively expressed on both NK1.1+ and NK1.1−DX5+CD3− SLy1− splenocytes, suggestive of NK lineage differentiation. Eomesodermin (EOMES) expression, however, was decreased in NK1.1−DX5+CD3−NK cells compared to NK1.1+DX5+CD3− NK cells (Fig. 3C). Along with NK1.1 expression of Nkp46, Ly49H and NKG2D activating receptors was also decreased on DX5+CD3− NK cells from B6SLy1−mice compared to B6wt littermates(Fig. 3D). This data further suggested altered activation status. Since cytokine stimulation can lead to NK maturation and activation, we next isolated splenic NK cells, based on DX5 expression, and stimulated them in vitro with 1,000 IU/mL of IL-2 for 5 d. Under such conditions, DX5+CD3− cells from B6SLy1−mice uniformly expressed surface activating receptors at similar levels to those from B6wt cell (Fig. 3E). These data confirmed that DX5+CD3− splenocytes of B6SLy1−mice are in fact NK cells with an altered activation state.
In order to explore these differences further, we next isolated NK cells from splenocytes of B6SLy1−or B6wt mice as described above (Fig. S1D) and evaluated mRNA levels by gene expression array. Consistent with flow cytometric data lower mRNA levels for multiple NK cell activating receptors were evident in B6SLy1− mice (Fig. 4A). Lower levels of EOMES were also evident in B6SLy1−mice, confirming flow cytometric observations (Fig. 4A and Fig. 3C, respectively). In addition, we detected alterations affecting multiple other pathways contributing to NK migration, cytotoxicity and metabolism in B6SLy1− NK cells (Figs. S4A–C).
Figure 4.

Gene expression and viability of splenic NK Cells. (A) Relative mRNA expression of NK activating receptors in freshly isolated B6SLy1− and B6wt NK cells. (B) Viability, defined as 7AAD−Annexin−, NK cells in the spleen of B6SLy1− (red) and B6wt mice (black). Representative gated flow cytometry plots on the left and summary of data (middle). Comparison performed by unpaired t-test. Electron microscopy of cytospin preparations of splenic B6wt and B6Sly1−NK cells. Arrows point to apoptotic cells (right).
Decreased NK viability was evident in the spleen of B6SLy1−mice by both flow cytometry and electron microscopy (Fig. 4B). These differences were eliminated by crossing SLy1 deficient mice to those with constitutive Bcl-2 expression in hematopoietic cells22 (Fig. S4D). Furthermore, such differences occurred in a cell-intrinsic manner as mixed bone marrow chimeras of wild-type B6 and B6SLy1− mice failed to rescue deficits associated with SLy1 deficiency (Fig. S4E). A correlation between NK function and SLy1 levels was also apparent in man by increased IFNγ production by NK with the highest expression of SLy1 (Fig. S4F). Taken together, our data suggests that the adaptor protein SLy1 contributes to the viability and activation of mature, peripheral NK cells but is unlikely to control their development in the bone marrow.
SLy1 contributes to ribosomal stability in NK cells
Based on its structure, we previously proposed that in T lymphocytes SLy1 and related family members function as intracellular scaffoldings around which signaling complexes are organized for optimal surface to nuclear signal transduction.11 This assumption is supported by the presence of both a SAM and SH3 domain, which are well-characterized protein–protein interacting motifs.23 Recently, we have shown that in mature T lymphocytes SLy1 regulates Forkhead box protein O1 (Foxo1) shuttling after T cell receptor signaling to facilitate lymphocyte expansion upon Listeria infection.13 It is thus possible that loss of such shuttling function in the absence of SLy1 could affect multiple unrelated pathways of NK activation, due to shuttling of a common signaling intermediate, for example. To study this further, we identified SLy1 binding partners by co-immunoprecipitation and mass spectroscopy. Surprisingly, we were unable to detect differences in signaling proteins or components of the NKG2D, NKp46 or NK1.1 signal transduction pathways. We did detect multiple ribosomal proteins, including several small 40S and large 60S ribosomal subunits as well as accessory ribosomal proteins such as heterogeneous nuclear ribonucleoprotein Q, that co-immunoprecipitated with B6wt but not with B6SLy1−NK cell lysates (Fig. 5A). The compact nature of the eukaryotic ribosome makes it one of the densest structures in the cytoplasm. Thus, ribosomal proteins or RNA can be separated from other cytoplasmic components based on density centrifugation.24 Sucrose density gradients of B6wt NK cells confirmed that SLy1 eluded with the denser ribosomal fraction, specifically the fully formed 80S complex in freshly isolated cells (Fig. 5B). In activated NK cells, SLy1 co-localized with the 80S complex as well as polysomes (Fig. S5A). In direct contrast, SLy1 did not elude with ribosomal proteins in T lymphocytes but localized with other cytoplasmic proteins such as actin in the lightest part of the gradient (Fig. 5B). Taken together, our data demonstrate that the function of SLy1 in NK cells may be unique compared to what we have previously described for T or B lymphocytes11,13 and is linked to ribosomal physiology.
Figure 5.

SLy1 contributes to ribosome stability in NK cells. (A) Mass spectroscopy of anti-SLy1 co-immunoprecipitation of B6wt and B6SLy1−NK cells. Proteins quantitated by spectral counts of peptides. Ribosomal proteins associated with the small 40S subunit are defined as RPS (ribosomal protein small), with the appropriate protein number after the RPS designation. Ribosomal proteins associated with the large 60S subunit are defined as RPL (ribosomal protein large), with the appropriate protein number after the RPL designation. (B) Sucrose density gradient fractionation and Western blot analysis of SLy1 localization of freshly isolated NK cells (left panel) and T lymphocytes (right panel). Representative of four separate experiments. (C) Western blot analysis of cytoplasm and nucleolar SLy1 in the KY1.1 NK cell line utilizing nucleophosmin (NPM) and superoxide dismutase (SOD) for nucleolar and cytoplasmic localization, respectively. (D) Spectral counts of free ribosomal components, not associated with the ribosome, in B6SLy1− in B6wt NK cells by mass spectroscopy. Comparison performed by t-test. (E) Sucrose density gradient fractionation with A254 absorbance evaluation of ribosome formation in freshly-isolated B6wt (black) and B6SLy1−(red) NK cells. Representative of three separate experiments. (F) RT-PCR of 18S rRNA expression in freshly isolated NK cells from B6wt (black) and B6SLy1−(red) mice. (G) RT-PCR analysis of 47S rRNA copy number in freshly isolated NK cells from B6wt (black) and B6SLy1−(red) mice. Comparison performed by unpaired t-test. (H) RT-PCR of 18S rRNA levels in freshly isolated T cells, B cells and NK cells from B6 mice. Expression normalized to β-actin. Comparison performed by One-way ANOVA followed by Bonferroni post-test. *p < 0.05, **p < 0.01, ns = p > 0.05.
It has been reported that the SAM domain of some post-transcription regulator proteins can bind mRNA.25 Although SLy1 SAM shares the hydrophobic conserved residues of a SAM domain, it lacks most of the basic residues essential for RNA binding activity (Fig. S5B). It is thus unlikely that SLy1 is an RNA-binding protein and most likely functions in the ribosome through protein–protein interactions. To directly evaluate its binding of ribosomal proteins, we next cloned SLy1 as well as the SAM and SH3 domains with Glutathione-S-Transferase (GST) fusion tag and evaluated their association with ribosomal proteins by co-immunoprecipitation and Western blot analysis. All domains had the capacity to bind multiple ribosomal proteins (Fig. S5C). Based on this binding pattern and its nuclear to cytoplasmic shuttling capacity described in T lymphocytes,11,14 we next considered the possibility that SLy1 may play a role in ribosome formation or assembly. Such function has been described for other accessory ribosomal proteins such as nucleophosmin, which helps shuttle components of the ribosome from the nucleolus to the cytoplasm during ribosomogenesis.24 Altered ribosomal function in the absence of SLy1 could thus affect protein translation and accumulation of intermediates within multiple divergent signaling pathways. Surprisingly in NK cells SLy1 was located solely in the cytoplasm, with no nuclear or nucleolar localization, even after activation by PMA and ionomycin (Fig. 5C; Fig. S5D). This is unique compared to what we have previously described for T cells.11-13 It is thus highly unlikely that it plays a significant role in ribosomogenesis, which occurs in the nucleolus. We next considered the possibility that SLy1 could contribute to ribosomal stability in the cytoplasm after ribosome formation is complete. Since in healthy lymphocytes cytoplasmic ribosomal proteins should be located solely within the fully assembled ribosome, we decided to evaluate ribosome stability by quantifying free ribosomal components, not associated with the ribosome, in the lightest fractions of an NK cell sucrose density gradient (fractions 1–3 as described in Fig. 5B) by mass spectroscopy. Indeed, a relative increase in multiple free ribosomal proteins was evident in B6SLy1− compared to B6wt NK cells (Fig. S5E). This included components such as small ribosomal protein 3 (RPS3) and large ribosomal protein 5 (RPL5) (Fig. 5D), which have been described to play an important role in detection of cellular stress.26,27 Taken together, based on these data, we can conclude that SLy1 contributes to ribosomal stability and maintenance in resting NK cells.
Loss of ribosomal proteins, such as RPL5 has been shown to impede cell function due to the decrease of ribosomal content and loss of translational capacity.28 Based on our data, we thought it possible that B6SLy1−NK cells may contain lower amount of fully formed functional ribosomes. To investigate this, we fractionated freshly isolated NK cells on a sucrose density gradient and compared ribosomal content by A254 absorbance. Surprisingly, a similar 80S peak was evident in both B6wt and B6SLy1− NK cells suggesting equivalent content of mature assembled ribosomes (Fig. 5E). Since the predominant material within the ribosome is rRNA (rRNA) and 18S rRNA is a critical component of the 40S subunit that facilitates binding to the 5′ untranslated region of mRNA,29 we next verified the results of A254 absorbance by measuring 18S rRNA content by RT-PCR. Similar to A254 absorbance identical amount of 18S rRNA was evident in B6wt and B6SLy1−NK cells (Fig. 5F). Based on these two pieces of data, we must conclude that despite ribosomal instability B6SLy1−NK cells maintain a similar pool of assembled ribosomes. Since structural 18S, 5.8S and 28S rRNAs are produced from the post-transcriptional processing of one 47S transcript, quantification of 47S rRNA has been described as a useful measure of ongoing ribosomogenesis.30 To this end, we quantitated 47S rRNA copy number per cell in resting, freshly isolated B6wt and B6SLy1−NK cells by quantitative PCR. Indeed, B6SLy1−NK cells demonstrated robust ongoing ribosomogenesis, even at rest (Fig. 5G). Interestingly resting, freshly isolated NK cells of wild-type B6 mice have a higher ribosomal content than resting T cells or B cells (Fig. 5H). Taken together, this data suggests that NK cells maintain a larger ribosomal pool at rest compared to other lymphocytes and ribosomal support proteins, such as SLy1, exist to facilitate the stability of this relatively large ribosomal pool. Since the synthesis and assembly of ribosomal components places intense metabolic demands on mammalian cells,31 it is likely that at least some NK dysfunction evident in the absence of SLy1 is the result of metabolic dysregulation associated with ongoing ribosomogenesis.
Ribosomal instability associated with SLy1 deficiency leads to upregulation of p53
Ribosomal stress is often triggered by malignant transformation.32 As a protective mechanism multiple pathways have evolved in mammalian cells to detect such stress and prevent survival and proliferation of transformed cells. Murine double minute 2 (Mdm2) is an E3 ubiquitin ligase that plays a key role in the detection of cellular stress. Under normal conditions, it sequesters p53 and targets it for proteosomal destruction. Ribosomal stress associated with overabundance of ribosomal proteins, such as RPL5, has been demonstrated to interfere with proper function of Mdm2 by specifically displacing p53 binding to the central acidic region of this ubiquitin ligase. This results in decreased ubiquination and intracellular accumulation of active p53.33,34 We thought it possible that overabundance of free ribosomal proteins in B6Sly1−NK cells as detected by mass spectroscopy may similarly interfere with Mdm2 binding of p53. To evaluate this possibility, we quantified levels of Mdm2, RPL5 and p53 in splenic NK cell lysates from B6SLy1−and B6wt mice. A portion of the lysates underwent co-immunoprecipitation with anti-Mdm2 antibody in order to quantify Mdm2 binding partners as well. While similar amounts of Mdm2 and RPL5 were evident in NK cells from B6SLy1−and B6wt mice, higher overall levels of p53 were evident in B6SLy1−NK cells (Fig. 6A). Consistent with this, higher amounts of RPL5 and lower amounts of p53 were bound to Mdm2 in B6SLy1−NK cells (Fig. 6B). In addition to RPL5, increased levels of multiple other ribosomal components were bound to Mdm2 in B6SLy1−NK cells as determined by mass spectroscopy (Fig. S6A). This data confirmed our assumption that in the absence of SLy1 free ribosomal proteins interfere with Mdm2-mediated clearance of p53. Unlike the case for NK cells, little free p53 was evident in B6SLy1− or wild-type T lymphocytes supporting the notion that SLy1 plays different roles in various lymphocyte subsets (Fig. S6B).
Figure 6.

In the absence of SLy1 NK cells upregulate p53. (A) Western blot analysis of freshly isolated NK cells for Mdm2, PRL5 and p53. Left panel representative of one Western blot and right panel demonstrated relative ratios of blot intensities from 5–7 separate experiments. (B) Western blot analysis of Mdm2 co-immunoprecipitates of freshly isolated NK cells for Mdm2, PRL5 and p53. Left panel representative of one Western blot and right panel demonstrated relative ratios of blot intensities from 5–6 separate experiments. Statistical comparison performed by Mann–Whitney U test to the null hypothesis assuming relative ratio to equal 1. 30% of NK cell lysate loaded for Western blot analysis of input lysate as described in panel (A) and 70% used for Mdm2 Co-IP as demonstrated in panel (B). *p < 0.05, **p < 0.01, ***p < 0.001, ns = p > 0.05 (C) Gene set enrichment analysis (left panel) and differences in p53-responsive genes (right panel) as defined by similar analysis in NCI-60 collection of cell lines with or without known p53 mutations.35 (D) Relative mRNA expression of NK activating receptors in expanded EOMES deficient (EOMESf/fX NKp46cre) and B6wt NK cells. (E) Graphic representation of SLy1 function in NK cells.
To evaluate if p53 was functional in B6SLy1−NK cells we compared, by gene set enrichment analysis (GSEA), expression of known p53 target genes.35 Levels of p53 target genes were enriched in B6SLy1−NK cells compared to those of B6wt littermates confirming functional and active p53 (p <0.05 and FDR <0.20) (Fig. 6C). Since p53 is well described to induce apoptosis,36 it is thus not surprising that NK cells from B6SLy1−mice have lower viability. The role of p53 in the downregulation of NK-specific activating receptors still eluded us. Bioinformatic analysis demonstrates that multiple signaling intermediates, cytotoxic mediators, adhesion molecules and transcription factors, such as eomesodermin (EOMES), are known to be transcriptionally regulated by p53.37,38 However, no bioinformatic data exists linking the expression of NK activating receptors to p53.
EOMES has been described as a master regulator of NK development.39 To evaluate whether p53-mediated downregulation of EOMES could affect expression of activating receptors, we performed a genome-wide expression analysis on NK cells expanded from splenocytes of either B6wt or EOMES deficient mice (Eomesf/f X Nkp46icre) with hierarchical clustering of NK specific genes. Similar to differences between NK cells from B6wt and B6SLy1−mice, lower levels of NK cell receptors were evident in EOMES deficient mice (Fig. 6D). Taken together, we can assume that in the absence of SLy1 ribosomal instability affects Mdm-2 mediated homeostasis of p53 which in turn downregulates EOMES, and other target genes, that result in NK cell dysfunction and subsequentially to cancer susceptibility (Fig. 6E).
Functional and phenotypic defects of B6SLy1− NK cells are reversible under inflammatory conditions
In addition to tumors, NK cells have also evolved to eliminate virally infected cells. We thus decided to evaluate the role of SLy1 in an MCMV viral infection model. Interestingly, despite decreased clearance of m157 over-expressing splenocytes in vivo (Fig. 2E) MCMV viral clearance, which relies on Ly49H-mediated recognition of the m157 antigen was identical between B6SLy1−and B6wt littermate mice (Fig. 7A). Even clearance of m157−MCVM, which depends on bystander cytokine production, was identical between B6SLy1−and B6wt mice. Unlike in the tumor microenvironment, viral infection results in significant elaboration of pro-inflammatory cytokines and NK proliferation. Since proliferation is coupled with ribsomogenesis,40 we next considered the possibility that SLy1-related defects in NK cells maybe reversible under inflammatory conditions where NK cells are induced to enter the cell cycle. This notion was already suggested in our in vitro system of NK activating receptor upregulation by high dose IL-2 (Fig. 3E). To evaluate this in vivo, we treated B6SLy1− and B6wt littermate mice with systemic IL-2 prior to challenge with NK cell-specific targets. Differences in m157-expressing splenocyte clearance, LLC cytotoxicity and in vivo tumor growth were eliminated between B6wt and B6SLy1−mice after such treatment (Figs. 7B–D). Treatment with IL-2 also normalized p53 levels (Fig. 7E), mRNA levels of activating receptors (Fig. 7F) and p53 target gene expression (data not shown). Taken together, we can conclude that SLy1 related NK dysfunction is ameliorated by NK proliferation and activation mediated by proinflammatory cytokines.
Figure 7.
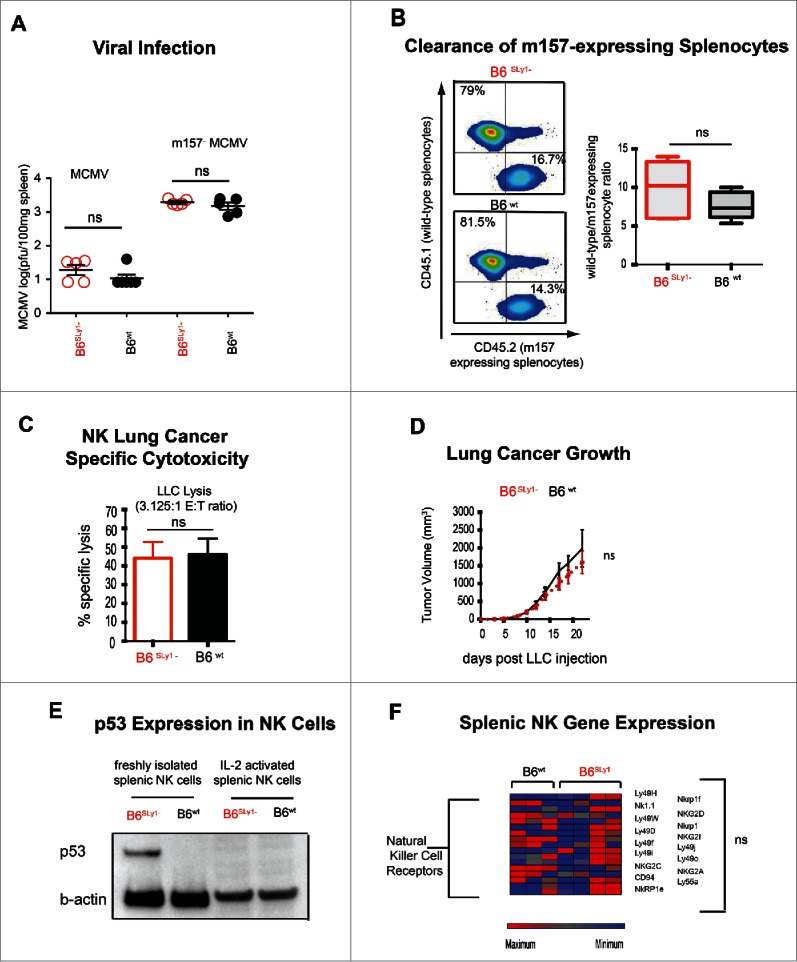
Phenotype of activated B6SLy1−and B6wt NK cells. (A) MCMV and m157−MCMV viral titers in the spleens of B6SLy1− (red) and B6wt (black) mice seventy-two hours after infection. (B) In vivo clearance of B6CD45.1+wild-type or B6CD45.2+m157-expressing splenocytes injected into IL-2 treated B6SLy1− (top) and B6wt mice (bottom) recipient mice. Data derived from flow cytometric analysis of the spleen, demonstrating one representative experiment on the left and summary of five animals on the right. Comparison performed by unpaired t-test between ratios. (C) In vitro lysis of LLC at a 3.125:1 effector:target ratio with NK cells isolated from the spleens of B6SLy1−(red) or B6wt (black) mice and expanded in high dose IL-2 for one week in vitro. (D) Growth of LLC injected into the flank of B6SLy1− (red line) and B6wt mice (black line) treated with 10 doses of 75,000 IU of wild-type IL-2. Representative of two separate experiments with four mice per group per experiment. (E) Comparison of p53 levels in resting and IL-2 activated B6SLy1−and B6wt NK cells (representative of two separate experiments). (F) Relative mRNA expression of various activating receptors, adhesion molecules and signaling intermediates in IL-2 activated NK cells from B6SLy1− and B6wt mice.
Discussion
Multiple etiologic factors can contribute to differences in NK function between individuals or various strains of mice. These include licensing through variability in receptors encoded in the NK gene complex or Ly49 receptors,3,41 expression of the activating receptor encoded by the Chok loci42 as well as individual differences in the density of NK activating receptors.43 Using an established approach of quantitative mRNA analysis, we now identify the SLy1 gene, located on the X chromosome, to also contribute to variability in NK function. SLy1 is a member of a novel family consisting of three adapter proteins with an SH3 and an SAM domain.14,44,45 SLy1 (SH3 lymphocyte protein 1), SLy2 (HACS1) and SLy3 (SASH1) show a similar domain organization and encompass a putative bipartite nuclear localization sequence at the N-terminus of the protein followed by an SH3 and a C-terminally localized SAM domain. The three proteins show different expression patterns. SLy1 is found to be exclusively expressed in lymphoid organs and was identified as a X-chromosomally encoded SH3 protein that is specifically phosphorylated at serine 27 upon B and T cell receptor engagement.14,46 Although SLy1 and SLy2 are mainly expressed in lymphoid organs, SLy3 shows a ubiquitous expression pattern and was postulated as a candidate tumor suppressor gene on chromosome 6q24.3.44 Recently, Klaus-Peter Janssen and coworkers could demonstrate that SLy3 is also significantly downregulated in other malignant tissues, such as liver and colon cancers.47
Previous studies defined SLy1 as an unrecognized target for antigen receptor signal transduction and suggested that it may play a role in adaptive immunity. Here, we demonstrate that SLy1 is repurposed by NK cells to provide ribosomal stability rather than signal transduction. These studies implicate SLy1 as a pleiotropic protein that plays multifaceted roles in different lymphocytes subsets. Such ability of one protein to serve multiple functions, also known as “protein moonlighting,” occurs widely in nature and maybe an evolutionary necessity to conserve the size of the genome.48 Crystallins, for example, function as enzymes in many tissues but in the eye serve a structural role in lens formation.49 It is thus likely that the different roles SLy1 plays in various lymphocytes may represent such a moonlighting function. It is currently unknown, but worthy of future exploration, if other members of the SLy family share in such differences in function based on cell type.
Ribosomal biogenesis is a major metabolic hurdle requiring rRNA transcription, ribosomal protein translation as well as assembly and maintenance of the 40S and 60S subunits to form the 80S translational unit. In most somatic cells, this process is tightly linked to cell growth and division. Lymphocytes of the innate immune system, on the other hand, are required to rapidly synthesize cytotoxic mediators upon encountering a foreign antigen in the absence of proliferation. NK cells, for example, maintain a pool of untranslated perforin and granzyme B mRNA at baseline, prior to activation or antigen encounter.5 It may thus be an evolutional necessity to maintain a large ribosomal pool even in quiescent NK cells to facilitate rapid translation of cytotoxic mediators, a notion that is supported by higher ribosomal content in this cell population (Fig. 5H). It is thus likely that SLy1, and maybe other yet undefined adaptor proteins, have evolved to facilitate ribosomal stability in resting NK cells. In the absence of SLy1, ribosomal instability affects the Mdm2-p53 pathway that has evolved to balance cellular proliferation with ribosomogenesis.50 In other somatic cells or even lymphocytes of the adaptive immune system, such a system may not be as critical given the linkage of proliferation with cytotoxicity.51
Since its initial discovery in 1979 the tumor suppressor protein p53 and related family members have been very well described as master regulators of cellular stress response and “guardians of the genome.”52 The p53 protein accumulates after exposure to ionizing radiation, cytotoxic chemotherapy,53 acquired mutations in ribosomal proteins,54 as well as ribosomal stress of almost any etiology.32 Following its activation p53 regulates multiple cellular processes including proliferation, senescence, apoptosis as well as cellular development.37 This occurs through both direct and indirect transcriptional control of gene expression. To date 44,000 p53 responsive elements have been identified throughout the genome.55 It is thus not surprising that SLy1− NK cells with higher levels of transcriptionally active p53 demonstrate defects in cytotoxicity, viability, activating receptor expression as well as target synapse formation.
As described in Fig. 7 IL-2 treatment restored both cytotoxicity and eliminated differences in surface expression of activating receptors. We speculate that this is the result of cytokine-induced NK cell proliferation. Since NK proliferation is tied to rapid ribosomogenesis stability and maintenance of a large ribosome pool may not be as important as under quiescent conditions in resting NK cells. The ribosome stabilizing function of SLy1 may thus be “dispensable” under inflammatory conditions. Such data is further supported by weakened immune responses of SLy1-deficient NK cells at baseline, or in the context of a quiescent tumor environment, but normalization at sites of inflammation mediated by cytokines or viral infection. While enthusiasm for cytokine therapy in the treatment of cancer has been limited by the inherent morbidity and mortality associated with high-dose IL-2, our data justify the use of moderate or even low dose IL-2 therapy to induce NK cell proliferation and restore NK function in certain susceptible individuals.
Methods
Animals
All animals were housed in a barrier facility in air-filtered cages. As indicated for selected experiments 1 × 106 Lewis Lung Carcinoma cells (ATCC®) in the logarithmic phase of growth were injected subcutaneously into the flank. For specific experiments, mice were depleted of NK cells (clone PK136), CD8+ (clone YTS169.4) and/or CD4+ T cells (clone GK1.5)(BioXcell). C57BL/6 mice expressing cherry tomato-red on an NK1.1 promoter was previously described.56 All animal procedures were approved by the Animal Studies Committee at Washington University School of Medicine, St. Louis, Missouri, USA and University of Tübingen and carried out according to the guidelines of the German Law for the Protection of Animals.
Genome-wide and quantitative gene expression analysis
Genome-wide expression analysis was performed using Agilent Mouse Gene Expression 4 × 44 K microarrays at the Genome Technology Access Center (GTAC), Department of Genetics in Washington University in St. Louis. Quantitative real-time RT-PCR was conducted on an ABI 7900 using TaqMan Gene Expression Assay system (Applied Biosystems) in accordance with the manufacturer's recommendations. Expression was normalized to β-actin and relative expression was calculated in comparison to a calibrator (RNA-probe from a mixture of lymphoid organs from B6wt mice). At least a 2-fold threshold in relative gene expression was used for evaluation. For 47S RT-PCR, total RNA were calculated using a standard curve generated from serial dilutions of a known quantity of cDNA.24
Cell isolation culture and flow cytometry
NK cells were isolated from bulk splenocytes of B6SLy1-and B6wt littermates using either DX5 positive selection (for experiments described in Fig. 3E) or through the use of the NK isolation kit (no-touch NK cell Isolation Kit II) with purity of >85 % confirmed by flow cytometry on multiple occasions (Fig. S1D). Bone marrow-derived CD122+ NK progenitors were obtained by flow cytometric sorting. T lymphocytes were obtained by CD90.2+ selection and B cells by B cell isolation kit (Miltenyi Biotech). For some experiments, Lymphokine Activated Killer (LAK) cells were generated from the spleen by IL-2 activation in vitro. All antibodies for flow cytometry were primarily fluorochrome conjugated and purchased from either BD or eBioscience.
Plate bound and tumor stimulation
For some experiments, six-well tissue culture plates were pre-coated overnight with either PK136, anti-Nkp46 or rat IgG control.57 Bulk splenocytes were cultured for 18 h in such plates and NK activation measured by IFNγ production or degranulation by surface expression of CD107a using flow cytometry. For human lymphocyte experiments, lethally irradiated A549 lung cancer cell line was co-cultured with bulk peripheral blood lymphocytes for 18 h and IFNγ production was measured flow cytometrically. All human peripheral blood lymphocytes were collected from healthy volunteers under an IRB approved protocol at Washington University. Informed consent was obtained from all volunteers prior to blood draw.
Sucrose gradient separation of ribosome components
Sucrose gradients were performed as previously described24 on 2 × 106 B6SLy1−and B6wt littermate NK cells, with a sensitivity setting of 0.2 for freshly isolated cells using a density gradient system (Teledyne ISCO). The sucrose gradients were performed on 4 × 107 B6Sly1−and wild-type littermate NK cells with a sensitivity setting of 2.0 for IL-2 expanded cells. Fractions were collected in 500 μL aliquots.
Western blot analysis
For standard westerns, cellular lysed homogenates in radioimmunoprecipitation assay buffer with 1% protease inhibitors were isolated for protein concentration determination with a bicinchoninic acid assay (ThermoScientific). An equal amount of protein concentration was loaded per well. Samples were prepared with 4X NuPAGE sample buffer containing reducing agent (LifeTechnologies) for 5 min at 95°C. For ribosome sucrose gradient samples, an equal amount of solution was loaded per well. All samples were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to 0.2 μm pore nitrocellulose membranes.
Immunoprecipitation
NK cells isolated from B6SLy1− and B6wt littermates were lysed using radioimmunoprecipitation assay buffer with 1% protease inhibitors and 1% phosphatase inhibitors. Immunoprecipitation was performed on wild-type littermate NK cells using SLy1 Ab (rabbit polyclonal custom-generated by Squarix) or anti-Mdm2 (clone 2A10)(Millipore) cross-linked to magnetic bead immunoprecipitation with Dynabead Protein G Immunoprecipitation kit according to manufacturer instructions (Life Technologies). Lysates were analyzed by Western blot analysis as described above.
Mass spectroscopy
The MS data were acquired on two high-resolution hybrid mass spectrometers, a TripleTOF 5600 Plus (AB SCIEX) and Q-Exactive Plus (ThermoFisher). The TripleTOF mass spectrometer was interfaced to the nano-chromatograph with a Digital Picoview Nanospray source (New Objectives) via a 10 µm Silica PicoTip emitter (New Objectives). The LC system consisted of a 2D Plus (Eksigent, Dublin, CA) pump with a Nanoflex module and AS2 autosampler. The 2D LC system was configured with dual columns to load samples in tandem.
In vitro cytotoxicity
51Chromium release was conducted by incubating the target cells with 100 μCr sodium-51-chromate (PerkinElmer) for 1 h. NK cells were isolated from the spleen and for some experiments were expanded in vitro with high dose IL-2 (1,000 IU/mL) for 7 d. Specific lysis was expressed as (experimental release − spontaneous release)/(maximum release − spontaneous release) ×100% with 0% specific lysis as lowest expressed value.
Statistics
Details of statistical comparison are presented in the figure captions of each graph. For most assays a two-tailed Student's t test was used for two comparisons and ANOVA was used for multiple comparisons, as indicated in the appropriate figure legends. Data in figures are presented as mean ± SEM. A p value of more than 0.05 is assumed to be not statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work is supported by the Rheumatic Diseases Core Center NIH (P30 AR48335), Siteman Flow Cytometry and Biostatistics Core (#P30 CA91842), Institute of Clinical and Translational Sciences (ICTS) of Washington University School of Medicine. ASK supported by VA MERIT grant 1I01BX002299-01, NIH 1R01HL094601, PO1 AI116501, the Barnes Jewish Research Foundation and the Doug and Ann Brown Foundation; SB-H is supported by the Deutsche Forschungsgemeinschaft DFG (BE2813/1-2). Some of the data supported by ICTS/CTSA with funding through NIH Grant Number UL1 TR000448.
Author contributions
SA, DS, SC, RH, AZ, WL and YG performed the experiments; JY analyzed the microarray data; HA performed bioinformatic analysis; AEG, DK WY, SBH and ASK conceptualized the study, designed the research and prepared the manuscript; ARF performed and interpreted the MCMV data; HZ and LM assisted with ribosomal and p53 experimental design and interpretation.
References
- 1.Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet 2000; 356:1795-9; PMID:11117911; http://dx.doi.org/ 10.1016/S0140-6736(00)03231-1 [DOI] [PubMed] [Google Scholar]
- 2.Chen S, Kawashima H, Lowe JB, Lanier LL, Fukuda M. Suppression of tumor formation in lymph nodes by L-selectin-mediated natural killer cell recruitment. J Exp Med 2005; 202:1679-89; PMID:16352740; http://dx.doi.org/ 10.1084/jem.20051473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kreisel D, Gelman AE, Higashikubo R, Lin X, Vikis HG, White JM, Toth KA, Deshpande C, Carreno BM, You M et al.. Strain-specific variation in murine natural killer gene complex contributes to differences in immunosurveillance for urethane-induced lung cancer. Cancer Res 2012; 72:4311-7; PMID:22751136; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-0908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frese-Schaper M, Keil A, Yagita H, Steiner SK, Falk W, Schmid RA, Frese S. Influence of natural killer cells and perforinmediated cytolysis on the development of chemically induced lung cancer in A/J mice. Cancer Immunol Immunother 2014; 63:571-80; PMID:24658838; http://dx.doi.org/ 10.1007/s00262-014-1535-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, Ley TJ. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity 2007; 26:798-811; PMID:17540585; http://dx.doi.org/ 10.1016/j.immuni.2007.04.010 [DOI] [PubMed] [Google Scholar]
- 6.McCann KL, Baserga SJ. Genetics. Mysterious ribosomopathies. Science 2013; 341:849-50; PMID:23970686; http://dx.doi.org/ 10.1126/science.1244156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raulet DH, Held W. Natural killer cell receptors: the offs and ons of NK cell recognition. Cell 1995; 82:697-700; PMID:7671299; http://dx.doi.org/ 10.1016/0092-8674(95)90466-2 [DOI] [PubMed] [Google Scholar]
- 8.Yokoyama WM, Daniels BF, Seaman WE, Hunziker R, Margulies DH, Smith HR. A family of murine NK cell receptors specific for target cell MHC class I molecules. Seminars Immunol 1995; 7:89-101; PMID:7579199; http://dx.doi.org/ 10.1006/smim.1995.0013 [DOI] [PubMed] [Google Scholar]
- 9.Tanimine N, Tanaka Y, Kobayashi T, Tashiro H, Miki D, Imamura M, Aikata H, Tanaka J, Chayama K, Ohdan H. Quantitative effect of natural killer-cell licensing on hepatocellular carcinoma recurrence after curative hepatectomy. Cancer Immunol Res 2014; 2:1142-7; PMID:25135909; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0091 [DOI] [PubMed] [Google Scholar]
- 10.Higuchi DA, Cahan P, Gao J, Ferris ST, Poursine-Laurent J, Graubert TA, Yokoyama WM. Structural variation of the mouse natural killer gene complex. Genes Immunity 2010; 11:637-48; PMID:20861859; http://dx.doi.org/ 10.1038/gene.2010.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beer S, Scheikl T, Reis B, Huser N, Pfeffer K, Holzmann B. Impaired immune responses and prolonged allograft survival in Sly1 mutant mice. Mol Cell Biol 2005; 25:9646-60; PMID:16227612; http://dx.doi.org/ 10.1128/MCB.25.21.9646-9660.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reis B, Pfeffer K, Beer-Hammer S. The orphan adapter protein SLY1 as a novel anti-apoptotic protein required for thymocyte development. BMC Immunol 2009; 10:38; PMID:19604361; http://dx.doi.org/ 10.1186/1471-2172-10-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schall D, Schmitt F, Reis B, Brandt S, Beer-Hammer S. SLy1 regulates T-cell proliferation during Listeria monocytogenes infection in a Foxo1-dependent manner. Eur J Immunol 2015; 45:3087-97; PMID:26306874; http://dx.doi.org/ 10.1002/eji.201545609 [DOI] [PubMed] [Google Scholar]
- 14.Beer S, Simins AB, Schuster A, Holzmann B. Molecular cloning and characterization of a novel SH3 protein (SLY) preferentially expressed in lymphoid cells. Biochim Et Biophys Acta 2001; 1520:89-93; PMID:11470164; http://dx.doi.org/ 10.1016/S0167-4781(01)00242-1 [DOI] [PubMed] [Google Scholar]
- 15.Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, French AR, Sunwoo JB, Lemieux S, Hansen TH et al.. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005; 436:709-13; PMID:16079848; http://dx.doi.org/ 10.1038/nature03847 [DOI] [PubMed] [Google Scholar]
- 16.Ljunggren HG, Ohlen C, Hoglund P, Franksson L, Karre K. The RMA-S lymphoma mutant; consequences of a peptide loading defect on immunological recognition and graft rejection. Int J Cancer Suppl 1991; 6:38-44; PMID:2066183; http://dx.doi.org/ 10.1002/ijc.2910470711 [DOI] [PubMed] [Google Scholar]
- 17.Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood 2009; 113:5488-96; PMID:19234143; http://dx.doi.org/ 10.1182/blood-2008-10-187179 [DOI] [PubMed] [Google Scholar]
- 18.Arase H, Saito T, Phillips JH, Lanier LL. Cutting edge: the mouse NK cell-associated antigen recognized by DX5 monoclonal antibody is CD49b (α 2 integrin, very late antigen-2). J Immunol 2001; 167:1141-4; PMID:11466327; http://dx.doi.org/15032583 10.4049/jimmunol.167.3.1141 [DOI] [PubMed] [Google Scholar]
- 19.Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annual Rev Immunol 2004; 22:405-29; PMID:15032583; http://dx.doi.org/ 10.1146/annurev.immunol.22.012703.104711 [DOI] [PubMed] [Google Scholar]
- 20.Lee JJ, McGarry MP. When is a mouse basophil not a basophil? Blood 2007; 109:859-61; PMID:17003367; http://dx.doi.org/ 10.1182/blood-2006-06-027490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Santo JP. Natural killer cell developmental pathways: a question of balance. Annual Rev Immunol 2006; 24:257-86; PMID:16551250; http://dx.doi.org/ 10.1146/annurev.immunol.24.021605.090700 [DOI] [PubMed] [Google Scholar]
- 22.Ogilvy S, Metcalf D, Print CG, Bath ML, Harris AW, Adams JM. Constitutive Bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc Natl Acad Sci U S A 1999; 96:14943-8; PMID:10611317; http://dx.doi.org/ 10.1073/pnas.96.26.14943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J 2000; 14:231-41; PMID:10657980; http://dx.doi.org/ 10.1096/fj.1530-6860 [DOI] [PubMed] [Google Scholar]
- 24.Maggi LB Jr, Kuchenruether M, Dadey DY, Schwope RM, Grisendi S, Townsend RR, Pandolfi PP, Weber JD. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the Mammalian ribosome. Mol Cell Biol 2008; 28:7050-65; PMID:18809582; http://dx.doi.org/ 10.1128/MCB.01548-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aviv T, Lin Z, Lau S, Rendl LM, Sicheri F, Smibert CA. The RNA-binding SAM domain of Smaug defines a new family of post-transcriptional regulators. Nat Struct Biol 2003; 10:614-21; PMID:12858164; http://dx.doi.org/ 10.1038/nsb956 [DOI] [PubMed] [Google Scholar]
- 26.Yadavilli S, Mayo LD, Higgins M, Lain S, Hegde V, Deutsch WA. Ribosomal protein S3: A multi-functional protein that interacts with both p53 and MDM2 through its KH domain. DNA Repair (Amst) 2009; 8:1215-24; PMID:19656744; http://dx.doi.org/ 10.1016/j.dnarep.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fregoso OI, Das S, Akerman M, Krainer AR. Splicing-factor oncoprotein SRSF1 stabilizes p53 via RPL5 and induces cellular senescence. Mol Cell 2013; 50:56-66; PMID:23478443; http://dx.doi.org/ 10.1016/j.molcel.2013.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teng T, Mercer CA, Hexley P, Thomas G, Fumagalli S. Loss of tumor suppressor RPL5/RPL11 does not induce cell cycle arrest but impedes proliferation due to reduced ribosome content and translation capacity. Mol Cell Biol 2013; 33:4660-71; PMID:24061479; http://dx.doi.org/ 10.1128/MCB.01174-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panek J, Kolar M, Vohradsky J, Shivaya Valasek L. An evolutionary conserved pattern of 18S rRNA sequence complementarity to mRNA 5′ UTRs and its implications for eukaryotic gene translation regulation. Nucleic Acids Res 2013; 41:7625-34; PMID:23804757; http://dx.doi.org/ 10.1093/nar/gkt548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lafontaine DL. Noncoding RNAs in eukaryotic ribosome biogenesis and function. Nat Struct Mol Biol 2015; 22:11-9; PMID:25565028; http://dx.doi.org/ 10.1038/nsmb.2939 [DOI] [PubMed] [Google Scholar]
- 31.Thomson E, Ferreira-Cerca S, Hurt E. Eukaryotic ribosome biogenesis at a glance. J Cell Sci 2013; 126:4815-21; PMID:24172536; http://dx.doi.org/ 10.1242/jcs.111948 [DOI] [PubMed] [Google Scholar]
- 32.Sloan KE, Bohnsack MT, Watkins NJ. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep 2013; 5:237-47; PMID:24120868; http://dx.doi.org/ 10.1016/j.celrep.2013.08.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol 2004; 24:7654-68; PMID:15314173; http://dx.doi.org/ 10.1128/MCB.24.17.7654-7668.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 2003; 3:577-87; PMID:12842086; http://dx.doi.org/ 10.1016/S1535-6108(03)00134-X [DOI] [PubMed] [Google Scholar]
- 35.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545-50; PMID:16199517; http://dx.doi.org/ 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Canman CE, Chen CY, Lee MH, Kastan MB. DNA damage responses: p53 induction, cell cycle perturbations, and apoptosis. Cold Spring Harb Symp Quant Biol 1994; 59:277-86; PMID:7587079; http://dx.doi.org/ 10.1101/SQB.1994.059.01.032 [DOI] [PubMed] [Google Scholar]
- 37.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402-12; PMID:18431400; http://dx.doi.org/ 10.1038/nrm2395 [DOI] [PubMed] [Google Scholar]
- 38.Zhang M. Transcriptional Regulatory Element Database https://cb.utdallas.edu/cgi-bin/TRED/tred.cgi?process=home. 1994 [Google Scholar]
- 39.Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Lindsten T, Reiner SL. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 2012; 36:55-67; PMID:22261438; http://dx.doi.org/ 10.1016/j.immuni.2011.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donati G, Montanaro L, Derenzini M. Ribosome biogenesis and control of cell proliferation: p53 is not alone. Cancer Res 2012; 72:1602-7; PMID:22282659; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3992 [DOI] [PubMed] [Google Scholar]
- 41.Patel R, Belanger S, Tai LH, Troke AD, Makrigiannis AP. Effect of Ly49 haplotype variance on NK cell function and education. J Immunol 2010; 185:4783-92; PMID:20855875; http://dx.doi.org/9858511 10.4049/jimmunol.1001287 [DOI] [PubMed] [Google Scholar]
- 42.Idris AH, Iizuka K, Smith HR, Scalzo AA, Yokoyama WM. Genetic control of natural killing and in vivo tumor elimination by the Chok locus. J Exp Med 1998; 188:2243-56; PMID:9858511; http://dx.doi.org/ 10.1084/jem.188.12.2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marras F, Bozzano F, Ascierto ML, De Maria A. Baseline and dynamic expression of activating NK Cell receptors in the control of chronic viral infections: The Paradigm of HIV-1 and HCV. Front Immunol 2014; 5:305; PMID:25071766; http://dx.doi.org/ 10.3389/fimmu.2014.00305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeller C, Hinzmann B, Seitz S, Prokoph H, Burkhard-Goettges E, Fischer J, Jandrig B, Schwarz LE, Rosenthal A, Scherneck S. SASH1: a candidate tumor suppressor gene on chromosome 6q24.3 is downregulated in breast cancer. Oncogene 2003; 22:2972-83; PMID:12771949; http://dx.doi.org/ 10.1038/sj.onc.1206474 [DOI] [PubMed] [Google Scholar]
- 45.Claudio JO, Zhu YX, Benn SJ, Shukla AH, McGlade CJ, Falcioni N, Stewart AK. HACS1 encodes a novel SH3-SAM adaptor protein differentially expressed in normal and malignant hematopoietic cells. Oncogene 2001; 20:5373-7; PMID:11536050; http://dx.doi.org/ 10.1038/sj.onc.1204698 [DOI] [PubMed] [Google Scholar]
- 46.Astoul E, Laurence AD, Totty N, Beer S, Alexander DR, Cantrell DA. Approaches to define antigen receptor-induced serine kinase signal transduction pathways. J Biol Chem 2003; 278:9267-75; PMID:12515807; http://dx.doi.org/ 10.1074/jbc.M211252200 [DOI] [PubMed] [Google Scholar]
- 47.Rimkus C, Martini M, Friederichs J, Rosenberg R, Doll D, Siewert JR, Holzmann B, Janssen KP. Prognostic significance of downregulated expression of the candidate tumour suppressor gene SASH1 in colon cancer. Br J Cancer 2006; 95:1419-23; PMID:17088907; http://dx.doi.org/ 10.1038/sj.bjc.6603452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huberts DH, van der Klei IJ. Moonlighting proteins: an intriguing mode of multitasking. Biochim Et Biophys Acta 2010; 1803:520-5; PMID:20144902; http://dx.doi.org/ 10.1016/j.bbamcr.2010.01.022 [DOI] [PubMed] [Google Scholar]
- 49.Piatigorsky J, O'Brien WE, Norman BL, Kalumuck K, Wistow GJ, Borras T, Nickerson JM, Wawrousek EF. Gene sharing by delta-crystallin and argininosuccinate lyase. Proc Natl Acad Sci U S A 1988; 85:3479-83; PMID:3368457; http://dx.doi.org/ 10.1073/pnas.85.10.3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teng T, Thomas G, Mercer CA. Growth control and ribosomopathies. Curr Opin Genet Dev 2013; 23:63-71; PMID:23490481; http://dx.doi.org/ 10.1016/j.gde.2013.02.001 [DOI] [PubMed] [Google Scholar]
- 51.Wells AD, Walsh MC, Sankaran D, Turka LA. T cell effector function and anergy avoidance are quantitatively linked to cell division. J Immunol 2000; 165:2432-43; PMID:10946268; http://dx.doi.org/222475 10.4049/jimmunol.165.5.2432 [DOI] [PubMed] [Google Scholar]
- 52.Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979; 17:43-52; PMID:222475; http://dx.doi.org/ 10.1016/0092-8674(79)90293-9 [DOI] [PubMed] [Google Scholar]
- 53.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999; 285:1733-7; PMID:10481009; http://dx.doi.org/ 10.1126/science.285.5434.1733 [DOI] [PubMed] [Google Scholar]
- 54.McGowan KA, Li JZ, Park CY, Beaudry V, Tabor HK, Sabnis AJ, Zhang W, Fuchs H, de Angelis MH, Myers RM et al.. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet 2008; 40:963-70; PMID:18641651; http://dx.doi.org/ 10.1038/ng.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tebaldi T, Zaccara S, Alessandrini F, Bisio A, Ciribilli Y, Inga A. Whole-genome cartography of p53 response elements ranked on transactivation potential. BMC Genomics 2015; 16:464; PMID:26081755; http://dx.doi.org/ 10.1186/s12864-015-1643-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gan Y, Liu Q, Wu W, Yin JX, Bai XF, Shen R, Wang Y, Chen J, La Cava A, Poursine-Laurent J et al.. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci U S A 2014; 111:2704-9; PMID:24550298; http://dx.doi.org/ 10.1073/pnas.1315943111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ho EL, Carayannopoulos LN, Poursine-Laurent J, Kinder J, Plougastel B, Smith HR, Yokoyama WM. Costimulation of multiple NK cell activation receptors by NKG2D. J Immunol 2002; 169:3667-75; PMID:12244159; http://dx.doi.org/ 10.4049/jimmunol.169.7.3667 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.