

ABSTRACT
Purpose: The devastating prognosis of patients with resectable pancreatic ductal adenocarcinoma (PDA) presents an urgent need for the development of therapeutic strategies targeting disseminated tumor cells. Until now, T-cell therapy has been scarcely pursued in PDA, due to the prevailing view that it represents a poorly immunogenic tumor. Experimental design: We systematically analyzed T-cell infiltrates in tumor biopsies from 127 patients with resectable PDA by means of immunohistochemistry, flow cytometry, T-cell receptor (TCR) deep-sequencing and functional analysis of in vitro expanded T-cell cultures. Parallel studies were performed on tumor-infiltrating lymphocytes (TIL) from 44 patients with metastatic melanoma.
Results: Prominent T-cell infiltrates, as well as tertiary lymphoid structures harboring proliferating T-cells, were detected in the vast majority of biopsies from PDA patients. The notion that the tumor is a site of local T-cell expansion was strengthened by TCR deep-sequencing, revealing that the T-cell repertoire in the tumor is dominated by highly frequent CDR3 sequences that can be up to 10,000-fold enriched in tumor as compared to peripheral blood. In fact, TCR repertoire composition in PDA resembled that in melanoma. Moreover, in vitro expansion of TILs was equally efficient for PDA and melanoma, resulting in T-cell cultures displaying HLA class I-restricted reactivity against autologous tumor cells.
Conclusions: The tumor-infiltrating T-cell response in PDA shows striking similarity to that in melanoma, where adoptive T-cell therapy has significant therapeutic impact. Our findings indicate that T-cell-based therapies may be used to counter disease recurrence in patients with resectable PDA.
KEYWORDS: Adoptive T-cell therapy, pancreatic ductal adenocarcinoma, tertiary lymphoid structures, T-cell receptor (TCR) repertoire, tumor-infiltrating lymphocytes
Introduction
Over the past 5 y, cancer immunotherapy has come of age with the FDA approval of checkpoint inhibitor antibodies targeting the T-cell inhibitory receptors CTLA-4 and PD-1, supported by striking clinical results in melanoma and lung cancer.1,2 Nevertheless, unleashing antitumor T-cell immunity by blocking negative signals does not succeed in all cancer types, suggesting that alternative approaches are still needed.
One example in this respect is pancreatic ductal adenocarcinoma (PDA), the most common type of pancreatic cancer. At time of diagnosis, approximately 80% of patients are deemed non-resectable due to local tumor spread and/or the detection of macroscopically visible metastases.3 In spite of recently improved chemotherapeutic regimens, e.g., involving Nab-Paclitaxel or FOLFIRINOX, the median overall survival time of these patients is a mere 9–11 mo.4 Approximately 20–30% of patients diagnosed with PDA have a better prognosis, because their tumor is primary resectable, or secondary resectable after neo-adjuvant chemo/chemo-radiotherapy. Nevertheless, the median survival of these patients is still only 20–24 mo, due to the high recurrence rate, amounting to 70% within 2 y.3,5
Unfortunately, initial trials with checkpoint inhibitors, in particular PD-1-blocking antibodies, did not show a promising outcome in PDA patients, in spite of evidence that these tumors express significant levels of PD-L1.6,7 This has strengthened the longstanding notion that PDA is a poorly immunogenic, “cold” tumor. Approaches to elicit antitumor immunity in PDA by means of vaccination or agonist anti-CD40 immunostimulatory antibodies appear to be more successful.8-11 Even though these clinical trials have not yet shown striking clinical impact, it is evident from these studies that, amidst the hostile microenvironment of pancreatic tumors, T-cells are present that could be mobilized when stimulated properly.
Being confronted with the devastating recurrence rate of PDA patients 12 and based on our experience in charting the T-cell response in “more immunogenic” tumors like melanoma, colorectal cancer and HPV16+ cervical cancer,13-15 we set off to systematically analyze the T-cell infiltrate in biopsies from primary PDA tumors, using tumor-infiltrating lymphocytes (TILs) from metastatic melanoma as our benchmark. Interestingly, we found that most PDA tumors actually contain significant numbers of T-cells and, along with that, tertiary lymphoid structures in which clonal T-cell expansion takes place. Further phenotypic and functional analysis of these TILs revealed striking similarities with T-cells isolated from melanoma. Taken together, our data indicate that adoptive T-cell therapy using T-cells or tumor-reactive T-cell receptors (TCRs) obtained from the primary tumor may be used to combat disease recurrence in patients with primary resectable PDA.
Materials and methods
Patients
127 patients with resected PDA were analyzed. Details on this patient population are summarized in Table S1. 44 patients with metastatic melanoma served as controls for studies on the in vitro expansion of TIL.
Freshly resectable tumor tissue and blood samples from PDA and melanoma patients were obtained via the European Pancreas Center and the Dermatology Department of Heidelberg University Hospital.
While we aim to obtain TILs, xenografts, tumor cell lines, as well as immunohistochemistry and TCR-, exome- and RNA sequencing data for every patient, this is not always feasible, in particular due to limited amounts of primary tumor material and/or failure of xenograft/cell line or TIL outgrowth. For details on sample handling and the generation of xenografts and cell lines see Supplemental Methods. Numbers of samples tested are indicated for all experiments shown.
Informed written consent was obtained from all participants before sample collection. The study was approved by the local ethics committee and conducted in accordance with the declaration of Helsinki.
In vitro expansion of tumor-infiltrating lymphocytes (TILs)
TIL cultures were established following the “young-TIL” protocol16 with minor modifications. Briefly, fresh tumor samples were minced into pieces of approximately 1 mm3 and placed at one piece per well in 24-well culture plates containing X-Vivo 15 medium, supplemented with 2% HSA, 1% Pen-Strep, 20 μg/mL Gentamycine, 2.5 μg/mL Fungizone and 6,000 IU/mL IL-2 (Proleukin, Novartis Pharma, Nürnberg, Germany). After 24 h, half of the medium was replaced with fresh, IL-2-containing medium. Plates were visually monitored every few days and cells were split at approximately 80% confluence. On day 14 of culture all wells containing expanding cells were harvested, pooled, analyzed and a sample of cells was subjected to a rapid expansion protocol: 0.1 × 106 pre-expanded TILs were added to 3 × 107 million feeder cells, consisting of peripheral blood mononuclear cells (PBMC) from three different donors, irradiated at 40 Gy. Cultures were set up in standing T25 flasks in 25 mL of X-Vivo 15 medium supplemented with 2% human AB-serum (Sigma-Aldrich, St. Louis, USA), 1% PenStrep and 30 ng/mL OKT-3 (eBioscience, San Diego, USA). After 24 h, 300 IU/mL IL-2 were added to the cultures. After 5 d, half the medium was exchanged for fresh IL-2-containing medium without OKT-3. After day 5, cultures were split upon visual inspection and harvested after 2 weeks of culture. Expanded TILs were analyzed and cryopreserved (in 90% human AB-Serum + 10% DMSO, using a CoolCell controlled rate freezing device (BioCision, San Rafael, USA)) for further analysis.
Immunohistochemistry (IHC) and whole slide imaging
Immunohistochemistry was performed on cryosections. Details on the general staining procedure and antibody-specific protocols are found in Supplemental Methods and Table S2, respectively. Stained tissue sections were visualized using a computerized image analysis system with a dedicated analysis software (VIS software suite, Visiopharm, Denmark).13,17
Prior to image analysis tumor areas were defined by a pathologist and only samples with ≥50 % of tumor area were analyzed. Full tissue sections were analyzed and all evaluable tumor area on the slide was used for quantification. The number of positively stained cells per mm2 of tumor was counted.
RNA extraction and T-cell receptor (TCR) sequencing
Cryproserved tumor pieces were thawed, homogenized using a pestle and total RNA was extracted using the RNeasy Mini Kit according to the manufacturer's instructions (Qiagen, Hilden, Germany).
Blood samples collected in PaxGene tubes or EDTA-tubes were extracted using Paxgene Blood RNA (Pre AnalytiX GmbH, Hilden, Germany) and RiboPure RNA Purification Kit blood (Thermo Fisher Scientific, Waltham, USA), respectively, according to the manufactures instructions. RNA was quantified using a Nanodrop 8000 (Thermo Fisher Scientific, Waltham, USA) and/or a Qubit (Life Technologies, Carlsbad USA) and RNA quality was measured on a Bioanalyzer 2100 (Agilent, Santa Clara, USA).
For analysis of the TCR repertoire, 250 ng total RNA was reverse transcribed using a template switch oligo (TSO), TRAC- and TRBC- gene-specific primers (RT-TRAC, RT-TRBC), and RevertAid H Minus Reverse Transcriptase (Thermo Scientific, Waltham, USA) in buffer enriched to 6 mM Mg2+. For primer sequences, please refer to Table S3. Incubation time was 90 min at 42°C followed by 15 min at 72°C. One-sixth of cDNA was then amplified using PfuUltra Hotstart DNA Polymerase (Agilent, Santa Clara, USA) with the PCR program: 2 min at 95°C, 20 cycles of 30 s at 94°C, 30 s at 68°C and 45 s at 72°C, followed by 6 min at 72°C (primers: PCR1-TS, PCR1-TRAC and PCR1-TRBC). PCR products were purified with beads (Beckman Coulter, Danvers, USA) and one-third of the purified product was subjected to further amplification: 2 min at 95°C, 25 cycles of 30 s at 94°C, 30 s at 65°C, 45 s at 72°C, followed by 6 min at 72°C (primers: PCR2-TS, PCR2-TRAC, PCR2-TRBC1 and PCR2-TRBC2). Final PCR product was size-selected using gel electrophoresis, purified with QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany), subjected to library preparation using TruSeq Nano DNA LT Sample Preparation Kit (Illumina, San Diego, USA) and sequenced using MiSeq v3 600 cycle Reagent Kit (Illumina, San Diego, USA). The average sequencing depth was ≈106 reads/sample. TCR sequences were identified within raw datasets, translated into amino acids sequences and then divided into V, CDR3 and J regions. V and J genes were then annotated, sequence counts for individual regions as well as for V-CDR3-J patterns were calculated, and sequence frequencies were expressed relative to the number of accepted TCR sequences of the selected chain type (α or β). The range of unique CDR3 sequences detected for TCR-α was 8,112–33,909 for tumor samples and 34,696–126,596 for blood samples and for TCR-β was 10,789–13,307 for tumor and 33,488–94,109 for blood samples.
Based on the clonal abundance (pi) of all clones with unique CDR3-β sequences in each sample, we calculated Shannon's entropy (H) as a measure of diversity:
Shannon's entropy was then normalized by the logarithm of the number of unique CDR3-β sequences (N) for the respective sample (to yield a normalized measure of diversity between zero and one) and subtracted from one to produce our clonality score:
Flow cytometry
Multiparametric flow cytometry was applied to allow characterization of cellular phenotype. Antibody details are provided in Table S4 and staining procedures are detailed in Supplemental Methods. Cells were analyzed using a Fortessa flow cytometer (BD Biosciences, San Jose, CA) and FlowJoX analysis software (Treestar, Ashland, USA).
Enzyme-linked immunospot (Elispot) assays
Antitumor reactivity of expanded TILs against autologous tumor cells, passaged in vivo as xenografts or in vitro as cell lines, was assayed using IFNγ Elispot (Mabtech, Nacka Strand, Sweden). For blocking experiments, tumor targets were pre-incubated with W6/32 antibody (BioLegend, San Diego, USA). For details on the experimental procedure, see Supplemental Methods. A response was considered positive if wells containing TIL + autologous target had significantly higher spot counts than control wells containing TILs alone.
Statistical methods
Statistical analysis was performed using Graph Pad Prism software and SAS software (Release 9.4, SAS-institute, Cary, USA). Differences between groups or experimental conditions were determined using parametric two-tailed t-tests (paired or unpaired, as appropriate). Correlations between TLS-positive and -negative patients and clinical or pathological findings were analyzed using the Fisher's exact-test and the Mann–Whitney-U-test. Survival analysis was performed using the Kaplan–Meyer estimate and the log-rank test was used to compare survival curves statistically. For multivariate survival analysis, the Cox proportional hazards model was used to compute hazard ratios and its 95% confidence interval. Two-sided p-values <0.05 were considered statistically significant and in the figures are indicated as *p < 0.05, **p < 0.01 and ***p < 0.001.
Results
T-cell infiltration and evidence of in situ clonal T-cell expansion in PDA
PDA tumors are generally characterized by a strong desmoplastic reaction and abundant stroma that can constitute up to 70% of the tumor mass.18,19 Our analyses were therefore performed on whole sections of tumor tissue by means of a high-resolution automated microscopy-based procedure that allows accurate enumeration of immune cells in heterogeneous tumors and in immune cell conglomerates.13,17 Representative examples of our findings are shown in Fig. 1A. While approximately 25% of the tumors (18/72 analyzed) are sparsely infiltrated with CD3+ T-cells (mean number of CD3+ cells <100/mm2), the majority of samples showed intermediate (100–300 CD3+ cells/mm2; 32/72 = 44% of samples) to high (>300 CD3+ cells/mm2; 22/72 = 31% of samples) numbers of infiltrating T-cells (Fig. 1C). The mean number of CD3+ cells/mm2 was 276 ± 288. The vast desmoplastic parts of PDA tumors were typically devoid of T-cells (Fig. S1).
Figure 1.
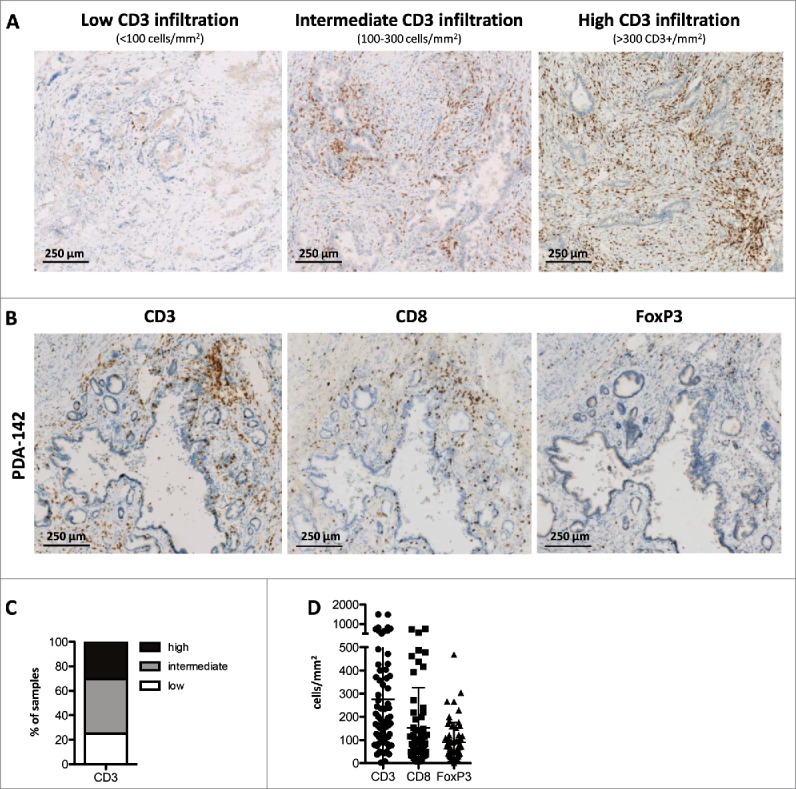
Immune infiltration patterns in primary resectable PDA. (A) Representative examples of PDA tumors showing low (left), intermediate (middle) or high (right) infiltration of CD3+ T-cells; scale bars denote 250 μm; data is representative of 72 patients analyzed. (B) Serial sections of a PDA tumor stained for total CD3+ T-cells (left), CD8+ cytotoxic T-cells (middle) and FoxP3+ regulatory T-cells (right); scale bars denote 250 μm. (C) Proportion of patients (n = 72) displaying low, intermediate or high CD3+ T-cell infiltration (mean CD3 count/mm2 tumor area <100, 100–300 and >300, respectively). (D) Number of CD3+, CD8+ and FoxP3+ T-cells/mm2 tumor area in PDA patients (n≥60 ). Notably, tumor area is defined as the total tumor tissue, including the stromal and fibrotic areas, but excluding surrounding normal pancreatic tissue and chronically inflamed tissue areas that are not directly associated with the tumor.
The infiltration by major T-cell subtypes (CD8+ cytotoxic and FoxP3+ regulatory T-cells) was examined on serial sections as shown in Fig. 1B. Staining for CD4+ was not included in our evaluation, because it is known to also detect myeloid cell subsets. The data summary in Fig. 1D shows that CD8+ T-cells and/or FoxP3+ T-cells dominate the T-cell infiltrate in a subset of tumors, but not in most cases, and thus suggests that CD4+ responder T-cells represent the major fraction of CD3+ T-cells in PDA samples.
During the evaluation of our tumor sections, we noted that the majority of samples (78%) contained multiple dense aggregates of lymphocytes that were dispersed throughout the tissue (exemplified in Fig. 2A). Detailed immunohistochemistry revealed that these conglomerates displayed all hallmarks of ectopic, tertiary lymphoid structures (TLS), in particular T- and B-cell areas with respective co-localization of myeloid and follicular dendritic cells (DC), as well as presence of high-endothelial venules (Figs. S2A and B). Because TLS are known to exert functions similar to secondary lymphoid organs, and contain professional antigen-presenting cells, we hypothesized that the TLS found in PDA tumors might be sites of T-cell activation and proliferation. Concordantly, we observed clusters of Ki-67 positive cells within TLS, indicative of ongoing cell division (Figs. S2C D). In order to substantiate this finding, we also stained with selected antibodies specific for individual TCR-β-chain V-gene families and found that TLS within a single tumor greatly differed with respect to the numbers of T-cells expressing a given Vβ. In the example shown in Figs. 2B and C, Vβ2-positive T-cells were densely packed in only one of the 15 TLS found in this section, while being more diffusely distributed both in other TLS and throughout the tumor. Fig. S3 shows further examples of clustered Vβ2-positive T-cells in TLS that are likely the result of local clonal expansion. Similar results were obtained using antibodies detecting other Vβ families, although the visualization was more challenging due to lower usage of most of these V-genes in the total T-cell repertoire and/or lesser quality of the primary antibody (data not shown).
Figure 2.
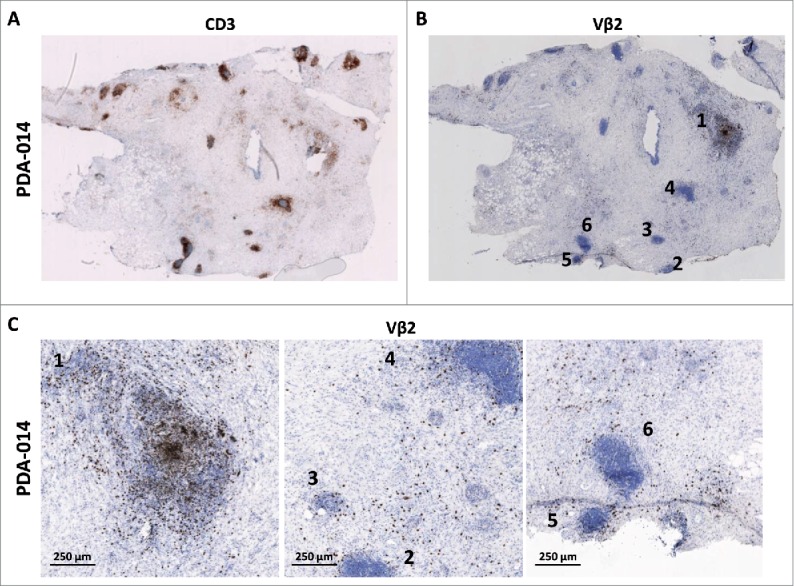
Evidence of clonal expansion in tertiary lymphoid structures (TLS). (A) Example of a tumor biopsy with many TLS as visualized by CD3 staining. (B) Vβ2-specific staining on a serial section of the tumor shown in (A). (C) Magnification of three areas of the aforementioned tumor, showing that Vβ2-positive T-cells are not enriched in TLS other than #1 and are diffusely spread throughout the tumor tissue. Numbers indicate the TLS as denoted in panel (B). Scale bars represent 250 μm. Data shown is representative of 33 tumors analyzed.
Taken together, our immunohistochemistry data suggests that the CD3+ T-cell infiltration found in many of the PDA tumor biopsies is not merely a result of the migration of activated T-cells into the chronically inflamed tumor microenvironment, but also—at least in part—a consequence of local clonal T-cell expansion. Evaluation of the immune infiltrate in the context of post-surgery patient follow-up revealed a significant association between detection of TLS in the resected primary tumors and overall survival (Fig. S4). No correlations of TLS positivity with other clinical or pathological findings were found (Table S1). In line with this, multivariate Cox-regression analysis confirmed the presence of TLS as an independent prognostic factor for prolonged survival (Table S5). Similar analyses on basis of intra-tumoral CD3+, CD8+ and FoxP3+ T-cells, as determined by immunhistochemistry and flow cytometry, did not result in statistically significant correlations with patient survival.
PDA tumors contain highly enriched TCR sequences
We proceeded to systematically compare the TCR repertoire in tumor and blood of patients by means of TCR deep-sequencing. Remarkably, in all cases examined, the 10 most frequent CDR3 sequences in the tumor represent 15–60% of the total number of CDR3-reads, while this percentage is significantly lower in blood samples (Fig. 3A). As shown for two representative PDA patients in Fig. 3B, the largest T-cell clones found in the tumor take up a major part of the infiltrating T-cell repertoire. While abundant CDR3 sequences can also be found in the blood of certain patients (PDA-060 in Fig. 3B; blue bar), their frequency is generally much lower than that of the dominant CDR3 sequences found in the tumor (Fig. 3A and PDA-082 in Fig. 3B). The difference in CDR3 repertoire between tumor and blood is further highlighted in Fig. 3C, in which the detected unique CDR3 sequences are ranked on the horizontal axis on the basis of the number of reads. In essence, these graphs illustrate that the tumor T-cell repertoire is dominated by large clones, represented by frequently detected CDR3 sequences. The blood T-cell repertoire, although also containing larger clones, in addition harbors thousands of small T-cell clones, represented by CDR3 sequences that are detected at very low frequencies. This difference in repertoire diversity is also illustrated by the significantly higher clonality values observed in the TILs as compared to the blood TCR repertoire of PDA patients (Fig. 3D).
Figure 3.
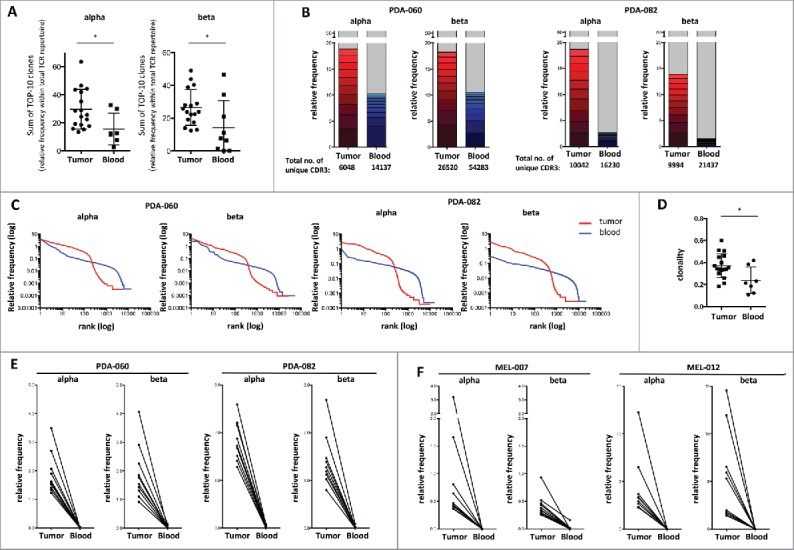
Evidence of highly enriched T-cell clones in PDA and melanoma on basis of TCR deep-sequencing. (A) Sum of the 10 largest clones/unique CDR3 sequences (relative frequency) identified in tumor (n = 17) and blood samples (n = 7) of PDAC patients by deep-sequencing of TCR-α and -β chains. (B) Relative frequency of the 10 largest clones (detected by TCR-α and -β chain sequencing) in tumor (red) and blood (blue) samples of two representative PDA patients versus all remaining clones (gray), where 100% represents the total number of CDR3 reads as given below the bars. (C) All unique CDR3 sequences detected in tumor (red) or blood (blue) of the two aforementioned PDA patients sorted according to their frequency within the sample, showing that larger clones (left) dominate in tumor, whereas smaller clones dominate in the blood (right). (D) Clonality of the TCR-β repertoire in tumor (n = 17) and blood (n = 5) of PDA patients, showing lower TCR diversity in tumor-infiltrating T-cells. Values can range from 0 (maximum diversity) to 1 (minimal diversity). (E) Size of the 10 largest TCR clones (TCR-α and -β sequencing) detected in the tumor of the two aforementioned PDA patients in tumor vs. blood. (E) Size of the 10 largest TCR clones (TCR-α and -β sequencing) detected in the tumor of two representative melanoma patients in tumor versus blood (n = 7 tumor and n = 5 blood samples were analyzed).
The enrichment of selected TCRs in the tumor is vividly illustrated by Fig. 3E, which depicts the relative frequencies in which the top-10 CDR3 sequences from the tumor are found in tumor versus blood. Depending on the TCR sequence, this enrichment can be up to 10,000 fold (mean enrichment ≈3,000 fold). A similar analysis of the top-10 CDR3 sequences in the blood of these patients, in which their relative abundance in blood vs. tumor was compared, confirmed the notion that the TCR repertoires in tumor and blood are clearly distinct (Fig. S5A). Even though large peripheral T-cell clones, most likely representing pathogen-specific T-cell responses, can be detected in the tumor, these do not rank highly in the tumor TCR repertoire. This argues against the possibility that the TIL TCR-repertoire would primarily represent activated T-cells that have been drawn into the inflamed tumor microenvironment and/or would be derived from the blood in the tumor vasculature.
Analysis of the TCR repertoire in tumor and blood samples from patients with metastatic melanoma resulted in data highly similar to those obtained in PDA, both with respect to overall clonal distribution (Fig. S5B) and relative frequencies of top-10 tumor CDR3 sequences in tumor versus blood (Fig. 3F, Fig. S5C). Since the existence of a tumor-reactive TIL repertoire in melanoma has been firmly established 20,21 and a prognostic role of the TIL TCR repertoire has recently been characterized,22,23 this resemblance between melanoma and PDA further supports the existence of an antitumor T-cell response in a majority of patients with primary resectable PDA.
PDA TILs display an antigen experienced phenotype
In parallel to our immunohistochemistry and TCR deep-sequencing efforts, we examined the phenotypes of the TILs derived from freshly dissociated PDA samples by means of flow cytometry (Fig. 4, see Fig. S6A for gating strategies). In accordance with the immunohistochemistry data, flow cytometric analysis revealed a diverse immune infiltrate consisting of lymphocytes and myeloid cells (Figs. S6B and C). Furthermore, discrimination between CD4+ and CD8+ T-cell subsets pointed at an overall dominance of CD4+ over CD8+ cells (Fig. 4A). Importantly, in the majority of cases, this dominance of CD4+ T-cells does not reflect high levels of CD25+CD127-/lowFoxP+ regulatory T-cells (range 2.4–14.4% of CD4+ T-cells; Fig. 4B; see also immunohistochemistry data in Fig. 1D), suggesting that most CD4+ cells are conventional responder/helper T-cells.
Figure 4.

Phenotype of PDA-infiltrating T-cell subsets. Flow cytometric evaluation of (A) the frequency of CD4+ and CD8+ TILs (n = 46) (B) the frequency of regulatory T-cells in CD4+ TILs (n = 14) (C) the distribution of memory cell subsets (naive – CD45RA+/CCR7+; Tcm – CD45RA−CCR7+, Tem – CD45RA−CCR7−, Temra – CD45RA+CCR7−) within tumor-infiltrating CD4+ (left panel) and CD8+ (right panel) T-cells (n = 38). (D–E) Representative flow cytometry data of two primary PDA TIL isolates (totally ≥ 20 PDA patients were analyzed), showing expression of PD1 on CD4+ and CD4− T-cells (left panel), as well as co-expression patterns of activation and exhaustion markers on CD4+ (top) and CD8+ (bottom) TILs.
The majority of CD4+ and CD8+ TILs exhibit a CD45RA-CCR7- effector memory (Tem) phenotype (Fig. 4C) and low expression of the homing receptor CD62L (Figs. 4D and E), which is consistent with their ability to infiltrate tumor tissue. A large fraction of the T-cells are positive for the inhibitory receptor PD-1, while considerably smaller fractions co-express additional inhibitory receptors including CTLA-4, LAG-3 and TIM-3 (Figs. 4D and E), all markers associated with antigen encounter as well as T-cell exhaustion.
The phenotype of PDA TILs is very similar to that observed for melanoma patients (Fig. S7). Of note, melanoma samples show more balanced CD4+ and CD8+ T-cell frequencies (compare Fig. 4A and Fig. S7A) and a more prominent expression of TIM-3 (Fig. S7E).
Expanded PDA TILs react against autologous tumor cells
The striking similarities between PDA and melanoma TILs prompted us to perform in vitro TIL expansions with tumor biopsies from 96 PDA patients using the “young TIL protocol” developed by Rosenberg and colleagues.16 Both the success rate of establishing TIL cultures (≈80%) and the efficiency of in vitro expansion (mean expansion ≈800-fold; Fig. 5A) were very similar for PDA and melanoma, suggesting that expansion of PDA TILs can be scaled up to clinical needs for adoptive TIL therapy as currently performed for melanoma. Monitoring of TIL phenotype during the culture period revealed that biopsies with diverse levels of T-cell infiltration consistently produced cultures with high T-cell content toward the end of the expansion phase (Fig. 5B). In line with their initial phenotype and the activatory culture conditions used, the resulting T-cells predominantly display an effector memory phenotype (Fig. 5C). Importantly, in vitro culture appears to revert the exhausted phenotype observed in freshly isolated TILs, in that PD-1 expression strongly decreased in the course of the culture period (Figs. 5D and E).
Figure 5.
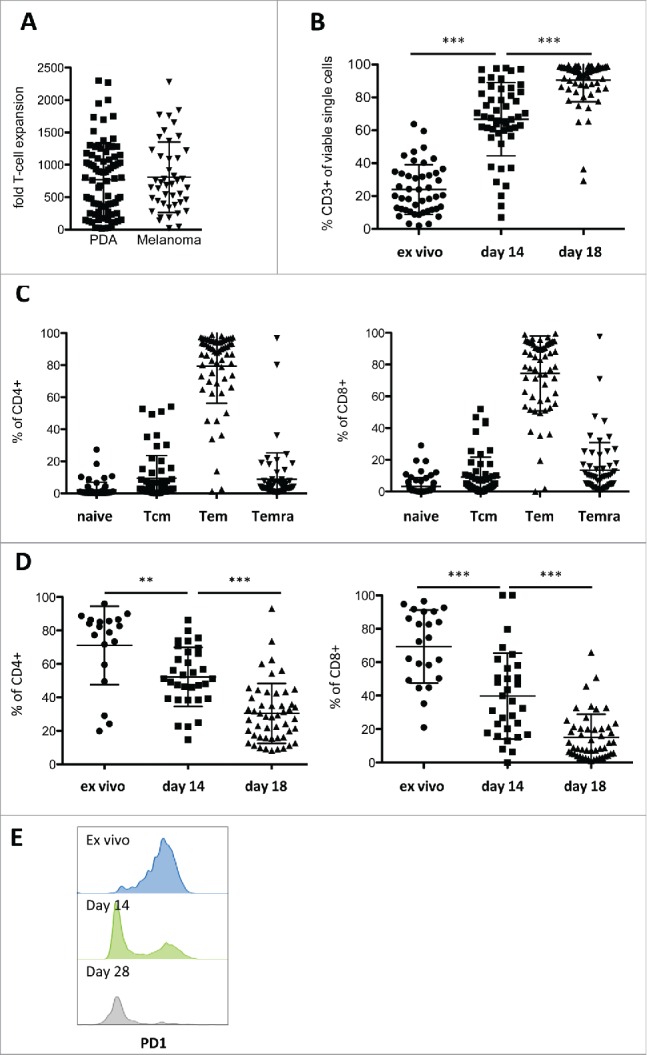
In vitro expansion of PDA-infiltrating lymphocytes. (A) Fold expansion of TIL cultures derived from PDA (n = 86) and melanoma (n = 44) patients during the two-week rapid expansion protocol. (B) Frequency of CD3+ TILs before start of culture (ex vivo, primary isolate), after pre-expansion in 24-well plates (day 14) and after rapid expansion (day 28) (n ≥ 45 ), as measured by flow cytometry. (C) Distribution of memory cell subsets (naive – CD45RA+/CCR7+; Tcm – CD45RA−CCR7+; Tem – CD45RA−CCR7−; Temra – CD45RA+CCR7−) after expansion in CD4+ (left panel) and CD8+ (right panel) T-cells (n = 60), measured by flow cytometry. (D) Expression of the negative regulatory marker PD-1 on CD4+ (left panel) and CD8+ (right panel) T-cells before start of culture (ex vivo), after pre-expansion in 24-well plates (day 14) and after rapid expansion (day 28) shown as percent PD1+ cells (n ≥19 ). (E) PD-1 fluorescence in T-cells of a representative culture ex vivo, on day 14 and day 28, measured by flow cytometry.
Whenever sufficient fresh PDA biopsy material was available, we transplanted histologically validated tumor fragments into NOD.Cg-Prkdcscid Il2rgtm1Wjl (NSG) mice with the aim to establish pairs of TILs and patient-derived xenografts. For the majority (17 of 20) of currently available matched TIL/tumor sets, the expanded TILs were found to react to the autologous tumor cells from freshly dissociated xenografts (Fig. 6). For PDA-161, where in addition to the xenograft an autologous tumor cell line was available, the TIL reactivity against both types of target cells was comparable (Fig. 6E). While autologous non-tumor cells (e.g., B-cells) were not available as control targets and the use of non-autologous tumors as controls would not be meaningful due to the occurrence of allo-reactions, TIL reactivity could be effectively inhibited by the addition of pan-HLA class I antibodies in 15 of 17 of responding cultures (Fig. 6F). This blockade is in line with the expectation that only the CD8+ T-cells should react against the HLA class I-positive, class II-negative tumor cells. The expanded TILs only occasionally displayed reactivity against the NK-cell target K562 (Fig. 6C), but also in this case antitumor reactivity was efficiently blocked by anti-HLA antibodies, arguing that cytotoxic T-cell and NK-like activity co-exist in this TIL culture.
Figure 6.
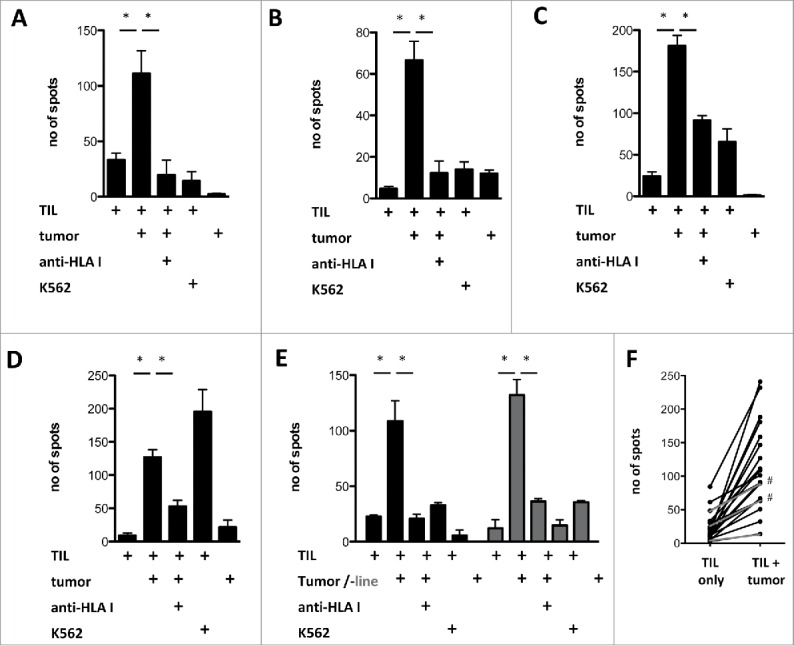
Antitumor reactivity of in vitro expanded PDA TIL. (A–E) Reactivity of in vitro expanded PDA TIL against autologous xenograft tumors (tumor) or a xenograft-derived cell line (“tumor line,” gray bars) measured by IFNγ Elispot, with or without pre-incubation of the tumor cell targets with pan-HLA-class I binding antibody W6/32 (anti-HLA I). (F) Of the 20 expanded TIL cultures for which autologous tumors were available, 17 displayed antitumor reactivity (black lines), whereas 3 did not show a significantly increased spot-count in response to autologous tumor targets (gray lines). In two responding cultures, no significant reduction of IFNγ production was achieved by pre-incubation with the MHC I blocking antibody (marked with #).
Discussion
Our study provides converging evidence for the existence of an antitumor T-cell response in a majority of patients with primary resectable PDA. While the detection of significant numbers of T-cells with activated phenotype does not suffice as evidence in this respect, because activated T-cells are capable of infiltrating inflamed tissues such as tumors in an antigen-independent fashion,24 our data clearly point at local encounter of tumor antigens and clonal T-cell expansion in the tumor. One key finding is the detection of TLS in the vast majority of tumors tested, along with the clustering of T-cells expressing Ki-67 and certain V-β chains within these TLS. Moreover, we are the first to show, by means of TCR deep-sequencing, that the T-cell repertoire in PDA tumors is clearly less diverse and distinct from that in the peripheral blood and greatly enriched for certain T-cell clones. The 10 most frequently detected TCR CDR3 sequences in the tumor take up 15–60% of the total CDR3 reads, while these sequences are detected at much lower frequencies in the patient's blood. Parallel analyses of tumor and blood samples from patients with metastatic melanoma rendered a highly similar picture. The similarity between the T-cell response in PDA and melanoma is further substantiated by the fact that in vitro expansion of TILs worked equally efficient in our hands for both indications. Last but not least, most of the expanded PDA TIL cultures demonstrated in vitro MHC-class I-restricted reactivity against autologous tumor cells. In view of these findings, we are now working toward the development of adoptive T-cell therapy for countering the devastating recurrence rate in patients with primary resectable PDA.
While our data provide strong support for in vitro tumor-reactivity of PDA TILs, we would like to note that the frequency of tumor-reactive T-cells in the expanded cultures is rather low (≤1 %). Accordingly, our pilot in vivo experiments with two selected TIL preparations did not reveal significant efficacy against autologous PDA xenografts, in spite of good in vivo expansion of the TILs (data not shown). Based on longitudinal analysis of the TIL cultures, we have indications that the TCR repertoire changes significantly during ex vivo expansion, including marked decreases in the frequencies of TCRs that are abundant in tumor samples. Our current data suggest that this could be a more prominent problem with PDA than with melanoma TILs (our unpublished observations). Although TIL culture procedures can be optimized to counter the loss of relevant T-cell clones, e.g., by sorting for PD-1 positive cells and/or application of anti-4-1BB co-stimulatory antibodies during culture,25-27 the most effective remedy to this problem likely involves cloning of the dominant TCR-α/β sequences from primary tumor material.20 Our ongoing attempts in this respect are aimed at identifying tumor-dominant TCR-α/β pairs on basis of TCR deep-sequencing data.
A question of definite interest concerns the nature of the T-cell epitopes recognized by tumor-reactive TILs, and in particular whether, in analogy to T-cells from melanoma, lung cancer and colorectal cancer,28,29 a majority of these TILs would be directed against mutanome-encoded neo-epitopes. A counter-indication in this respect is the relatively low number of non-synonymous mutations and indels found in PDA. In line with published reports,30 we find the number of such mutations for primary resectable PDA in the range of 52±15, with an additional 4–6 indels (data not shown). Nevertheless, it was recently demonstrated that T-cell mediated recognition of mutanome-encoded neo-epitopes can also be identified in tumors with lower mutational load, e.g., colorectal, bile duct and esophageal cancer,31 and that mutanome-specific T-cell reactivity can be found in both the CD8+ and CD4+ arms of the T-cell response.32-34 Furthermore, an increasing number of tumor-associated auto-antigens is found to be expressed by the medullary thymic epithelial cells (mTECs), implying that the high affinity T-cell repertoire targeting many of these antigens has been deleted. However, gene expression by mTEC does not cover the full repertoire of auto-antigens,35 leaving the possibility that TILs found in PDA tumors target auto-antigens such as certain cancer testis antigens or other tumor-associated antigens.36,37 In addition over-expression of mutated p53 is known to elicit CD4+ T-helper and IgG antibody responses against non-mutated parts of the antigen, to which the immune system is not tolerant (38 and references therein).
While our study is the first to provide comprehensive, interlocking evidence for the clonal expansion of tumor-reactive T-cells in PDA, several prior reports have pointed toward the existence of T-cell mediated immune surveillance in this disease. Four studies showed that high numbers of CD8+ and low numbers of FoxP3+ regulatory T-cells, as detected by immunohistochemistry in samples of primary resectable PDA, correlate with clinical outcome,39-42 while two other studies primarily highlight the detrimental role of Th2-cells and regulatory T-cells.36,43 In addition, three recent reports refer to the existence of TLS in PDA.10,44,45 Notably, the outcome of one of these studies suggested that TLS could only be observed in tumor samples from patients that underwent whole tumor cell vaccination (GVAX) prior to tumor resection, not in pre-vaccination biopsies or biopsies from control patients, and thereby that the TLS were induced by the vaccination.10 Based on our data and that of Castino45 and Hiraoka,44 in which TLS were found in 75%, 78% and 100% of non-treated patients, respectively, we propose that in the GVAX study 10 the TLS may have been missed in pre-vaccination and control biopsies due to the smaller size of the tumor sections. In fact, our initial estimate of the fraction of TLS-positive tumors based on analysis of a single tumor section was lower (≈50%). However, by sectioning through the negatively scored tumors, we found TLS in an additional ≈30% of samples, illustrating how easily such features may be overlooked. An interesting aspect in the study by Hiraoka et al. is the distinction between peritumoral and intra-tumoral TLS, and the association of intra-tumoral TLS with prolonged overall survival,44 which is in line with Castino et al.45 and similar findings for other cancers.46-48 Evaluation of our own data substantiated the link between presence of TLS and better survival in PDA. The protective effect of TLS is further supported by the higher expression of pro-inflammatory cytokines such as IFNγ, IL-12, IL-6, IL-8 in TLS-positive as compared to TLS-negative tumors.44,45
In conclusion, our data clearly demonstrate that PDA is not an immunologically “cold” tumor, as has been suggested in the context of the failure of checkpoint blockade in this indication.6,7 Nevertheless, the average number of non-synonymous mutations in PDA is ≈10-fold lower than in melanoma.30 In fact, the correlation between the clinical impact of checkpoint blockade and mutational load in melanoma offers a plausible explanation for the failure of this therapeutic approach in PDA.49,50 Furthermore, it is evident that the PDA microenvironment in patients with advanced disease poses additional hurdles for effective T-cell therapy, such as the physical barrier represented by the extensive stroma and the large number of myeloid cells in the infiltrate (Figs. S1 and 6).51,52 In view of these considerations, our strategy for implementing T-cell therapy in PDA focuses on the treatment of early-stage recurrence in patients with primary resectable PDA, by means of adoptive T-cell therapy including use of selected TIL clones or TCRs.20 It is duly noted that the co-application of immunomodulatory antibodies and/or individualized vaccines53,54 is likely to increase the efficacy of this treatment.
Supplementary Material
Disclosure of potential conflicts of interest
U.S. is a shareholder of BioNTech AG. U.S. and M.F. are employed at the BioNTech Corporation (BioNTech AG and its affiliates) and are inventors of the pending patent application published as WO 2015/059277 in the field of TCR deep-sequencing. The other authors have declared that no conflict of interest exists.
Acknowledgments
We thank the biobanks of the National Center for Tumor Diseases (NCT), Heidelberg and of the European Pancreas Center at Heidelberg University Hospital, especially Esther Soyka and Dr Nathalia Giese, for great help with sample collection. For TCR deep-sequencing, we thank Lisa Hebich for excellent technical assistance, Dr Tana Omokoko for valuable comments on data and Dr Michael Blank and Christian Grohmann for software support.
Funding
This work was funded by the K.H. Bauer Foundation (to R.O.), the German National Center for Tumor diseases (to R.O., A.T,. M.S.; project HiPo-015), the FP7 framework program of the European Community (to R.O., project acronym IACT; grant 602262 and I.P. project acronym TIPC_TIL_IP), the German Ministry of education and research (BMBF) (to I.P., M.Fl., O.S. and J.H., project acronyms TIL-REP, 01ZY1403A; to M.S., A.T., T.Ha., O.S., project acronyms PANC-STRAT, 01ZX1305B, 01ZX1305C), the Helmholtz pre-clinical comprehensive cancer center (PCCC) (to M.S. and A.T.) and the Dietmar Hopp Foundation (to M.S. and A.T.). The work of Ma.F. and U.S. has been supported by the Central Innovation Program for SMEs (ZIM) of the Federal Ministry for Economic Affairs and Energy (project EP 131299) and the BioNTech Diagnostics GmbH. Biobanking was supported by the Heidelberger Stiftung Chirurgie, (BMBF grants 01GS08114 and 01ZX1305C) and by the Biomaterial Bank Heidelberg/BMBH (BMBF grant 01EY1101).
References
- 1.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015; 161:205-14; PMID:25860605; http://dx.doi.org/ 10.1016/j.cell.2015.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015; 27:450-61; PMID:25858804; http://dx.doi.org/ 10.1016/j.ccell.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gillen S, Schuster T, Meyer Zum Buschenfelde C, Friess H, Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med 2010; 7:e1000267; PMID:20422030; http://dx.doi.org/ 10.1371/journal.pmed.1000267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C et al.. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011; 364:1817-25; PMID:21561347; http://dx.doi.org/ 10.1056/NEJMoa1011923 [DOI] [PubMed] [Google Scholar]
- 5.Hartwig W, Werner J, Jager D, Debus J, Buchler MW. Improvement of surgical results for pancreatic cancer. Lancet Oncol 2013; 14:e476-85; PMID:24079875; http://dx.doi.org/ 10.1016/S1470-2045(13)70172-4 [DOI] [PubMed] [Google Scholar]
- 6.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al.. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455-65; PMID:22658128; http://dx.doi.org/ 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I et al.. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010; 33:828-33; PMID:20842054; http://dx.doi.org/ 10.1097/CJI.0b013e3181eec14c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL et al.. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011; 331:1612-6; PMID:21436454; http://dx.doi.org/ 10.1126/science.1198443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, Morse M, Zeh H, Cohen D, Fine RL et al.. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol 2015; 33:1325-33; PMID:25584002; http://dx.doi.org/ 10.1200/JCO.2014.57.4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, Solt S, Dorman A, Wamwea A, Yager A et al.. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res 2014; 2:616-31; PMID:24942756; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, Wamwea A, Bigelow E, Lutz E, Liu L et al.. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother 2015; 38:1-11; PMID:25415283; http://dx.doi.org/ 10.1097/CJI.0000000000000062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartwig W, Hackert T, Hinz U, Gluth A, Bergmann F, Strobel O, Büchler MW, Werner J. Pancreatic cancer surgery in the new millennium: better prediction of outcome. Ann Surg 2011; 254:311-9; PMID:21606835; http://dx.doi.org/ 10.1097/SLA.0b013e31821fd334 [DOI] [PubMed] [Google Scholar]
- 13.Halama N, Michel S, Kloor M, Zoernig I, Benner A, Spille A, Pommerencke T, von Knebel DM, Folprecht G, Luber B et al.. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res 2011; 71:5670-7; PMID:21846824; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-0268 [DOI] [PubMed] [Google Scholar]
- 14.Poschke I, Lovgren T, Adamson L, Nystrom M, Andersson E, Hansson J, Tell R, Masucci GV, Kiessling R. A phase I clinical trial combining dendritic cell vaccination with adoptive T-cell transfer in patients with stage IV melanoma. Cancer Immunol Immunother 2014; 63:1061-71; PMID:24993563; http://dx.doi.org/ 10.1007/s00262-014-1575-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Burg SH, Piersma SJ, de Jong A, van der Hulst JM, Kwappenberg KM, van den Hende M, Welters MJ, Van Rood JJ, Fleuren GJ et al.. Association of cervical cancer with the presence of CD4+ regulatory T-cells specific for human papillomavirus antigens. Proc Natl Acad Sci U S A 2007; 104:12087-92; PMID:17615234; http://dx.doi.org/ 10.1073/pnas.0704672104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR, Robbins PF, Rosenberg SA, Dudley ME. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother 2008; 31:742-51; PMID:18779745; http://dx.doi.org/ 10.1097/CJI.0b013e31818403d5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halama N, Zoernig I, Spille A, Westphal K, Schirmacher P, Jaeger D, Grabe N. Estimation of immune cell densities in immune cell conglomerates: an approach for high-throughput quantification. PLoS One 2009; 4:e7847; PMID:19924291; http://dx.doi.org/ 10.1371/journal.pone.0007847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neesse A, Algul H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut 2015; 64:1476-84; PMID:25994217; http://dx.doi.org/ 10.1136/gutjnl-2015-309304 [DOI] [PubMed] [Google Scholar]
- 19.Whatcott CJ, Han H, Von Hoff DD. Orchestrating the tumor microenvironment to improve survival for patients with pancreatic cancer: normalization, not destruction. Cancer J 2015; 21:299-306; PMID:26222082; http://dx.doi.org/ 10.1097/PPO.0000000000000140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348:62-8; PMID:25838374; http://dx.doi.org/ 10.1126/science.aaa4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geukes Foppen MH, Donia M, Svane IM, Haanen JB. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol Oncol 2015; 9(10):1918-35; PMID:26578452; http://dx.doi.org/ 10.1016/j.molonc.2015.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568-71; PMID:25428505; http://dx.doi.org/ 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, Fong L. Improved survival with T-cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med 2014; 6:238ra70; PMID:24871131; http://dx.doi.org/ 10.1126/scitranslmed.3008211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity 2014; 41:694-707; PMID:25517612; http://dx.doi.org/ 10.1016/j.immuni.2014.10.008 [DOI] [PubMed] [Google Scholar]
- 25.Chacon JA, Wu RC, Sukhumalchandra P, Molldrem JJ, Sarnaik A, Pilon-Thomas S, Weber J, Hwu P, Radvanyi L. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One 2013; 8:e60031; PMID:23560068; http://dx.doi.org/ 10.1371/journal.pone.0060031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C, Coukos G, Powell DJ. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T-cells in tumor. Clin Cancer Res 2014; 20:44-55; PMID:24045181; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-0945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME et al.. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014; 124:2246-59; PMID:24667641; http://dx.doi.org/ 10.1172/JCI73639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Zhang C, Pan Y, Xu R, Xu C, Chen Z, Lu Z, Ke Y. T-cell receptor β-chain repertoire analysis reveals intratumour heterogeneity of tumour-infiltrating lymphocytes in oesophageal squamous cell carcinoma. J Pathol 2016; 239(4):450-8; PMID:27171315; http://dx.doi.org/25838375 10.1002/path.4742 [DOI] [PubMed] [Google Scholar]
- 29.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/ 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 30.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L et al.. Signatures of mutational processes in human cancer. Nature 2013; 500:415-21; PMID:23945592; http://dx.doi.org/ 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S et al.. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015; 350:1387-1390; PMID:26516200; http://dx.doi.org/25901682 10.1126/science.aad1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC et al.. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015; 520:692-6; PMID:25901682; http://dx.doi.org/ 10.1038/nature14426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE et al.. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T-cells in human melanoma. Nat Med 2015; 21:81-5; PMID:25531942; http://dx.doi.org/ 10.1038/nm.3773 [DOI] [PubMed] [Google Scholar]
- 34.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS et al.. Cancer immunotherapy based on mutation-specific CD4+ T-cells in a patient with epithelial cancer. Science 2014; 344:641-5; PMID:24812403; http://dx.doi.org/ 10.1126/science.1251102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T-cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol 2014; 14:377-91; PMID:24830344; http://dx.doi.org/ 10.1038/nri3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amedei A, Niccolai E, Benagiano M, Della Bella C, Cianchi F, Bechi P, Taddei A, Bencini L, Farsi M, Cappello P et al.. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol Immunother 2013; 62:1249-60; PMID:23640603; http://dx.doi.org/ 10.1007/s00262-013-1429-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suso EM, Dueland S, Rasmussen AM, Vetrhus T, Aamdal S, Kvalheim G, Gaudernack G. hTERT mRNA dendritic cell vaccination: complete response in a pancreatic cancer patient associated with response against several hTERT epitopes. Cancer Immunol Immunother 2011; 60:809-18; PMID:21365467; http://dx.doi.org/ 10.1007/s00262-011-0991-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lauwen MM, Zwaveling S, de Quartel L, Ferreira Mota SC, Grashorn JA, Melief CJ, van der Burg SH, Offringa R. Self-tolerance does not restrict the CD4+ T-helper response against the p53 tumor antigen. Cancer Res 2008; 68:893-900; PMID:18245492; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-3166 [DOI] [PubMed] [Google Scholar]
- 39.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer 2013; 108:914-23; PMID:23385730; http://dx.doi.org/ 10.1038/bjc.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG et al.. Activated pancreatic stellate cells sequester CD8+ T-cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013; 145:1121-32; PMID:23891972; http://dx.doi.org/ 10.1053/j.gastro.2013.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T, Kato K, Kurokawa T, Suzuoki M, Nakakubo Y et al.. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 2004; 28:e26-31; PMID:14707745; http://dx.doi.org/ 10.1097/00006676-200401000-00023 [DOI] [PubMed] [Google Scholar]
- 42.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T-cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res 2006; 12:5423-34; PMID:17000676; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0369 [DOI] [PubMed] [Google Scholar]
- 43.De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, Braga M, Di Carlo V, Doglioni C, Protti MP. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med 2011; 208:469-78; PMID:21339327; http://dx.doi.org/ 10.1084/jem.20101876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer 2015; 112:1782-90; PMID:25942397; http://dx.doi.org/ 10.1038/bjc.2015.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castino GF, Cortese N, Capretti G, Serio S, Di Caro G, Mineri R, Magrini E, Grizzi F, Cappello P, Novelli F et al.. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology 2016; 5:e1085147; PMID:27141376; http://dx.doi.org/ 10.1080/2162402X.2015.1085147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, Mulé JJ. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am J Pathol 2011; 179:37-45; PMID:21703392; http://dx.doi.org/ 10.1016/j.ajpath.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, Rabbe N, Laurans L, Tartour E, de Chaisemartin L et al.. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol 2008; 26:4410-7; PMID:18802153; http://dx.doi.org/ 10.1200/JCO.2007.15.0284 [DOI] [PubMed] [Google Scholar]
- 48.Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, Rochaix P, Girard JP. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res 2011; 71:5678-87; PMID:21846823; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-0431 [DOI] [PubMed] [Google Scholar]
- 49.Snyder A, Wolchok JD, Chan TA. Genetic basis for clinical response to CTLA-4 blockade. N Engl J Med 2015; 372:783; PMID:25693024; http://dx.doi.org/ 10.1056/NEJMc1415938 [DOI] [PubMed] [Google Scholar]
- 50.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM et al.. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015; 350:207-11; PMID:26359337; http://dx.doi.org/ 10.1126/science.aad0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL et al.. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 2013; 110:20212-7; PMID:24277834; http://dx.doi.org/ 10.1073/pnas.1320318110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hartmann N, Giese NA, Giese T, Poschke I, Offringa R, Werner J, Ryschich E. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin Cancer Res 2014; 20:3422-33; PMID:24763614; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-2972 [DOI] [PubMed] [Google Scholar]
- 53.Offringa R, Glennie MJ. Development of next-generation immunomodulatory antibodies for cancer therapy through optimization of the IgG framework. Cancer Cell 2015; 28:273-5; PMID:26373272; http://dx.doi.org/ 10.1016/j.ccell.2015.08.008 [DOI] [PubMed] [Google Scholar]
- 54.Boisguerin V, Castle JC, Loewer M, Diekmann J, Mueller F, Britten CM, Kreiter S, Türeci Ö, Sahin U. Translation of genomics-guided RNA-based personalised cancer vaccines: towards the bedside. Br J Cancer 2014; 111:1469-75; PMID:25314223; http://dx.doi.org/ 10.1038/bjc.2013.820 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.