

ABSTRACT
Immunotherapy of advanced melanoma with CTLA-4 or PD-1/PD-L1 checkpoint blockade induces in a proportion of patients long durable responses. In contrast, targeting the MAPK-pathway by selective BRAF and MEK inhibitors induces high response rates, but most patients relapse. Combining targeted therapy with immunotherapy is proposed to improve the long-term outcomes of patients. Preclinical data endorsing this hypothesis are accumulating. Inhibition of the PI3K-Akt-mTOR pathway may be a promising treatment option to overcome resistance to MAPK inhibition and for additional combination with immunotherapy.
We therefore evaluated to which extent dual targeting of the MAPK and PI3K-Akt-mTOR pathways affects tumor immune infiltrates and whether it synergizes with PD-1 checkpoint blockade in a BRAFV600E/PTEN−/−-driven melanoma mouse model. Short-term dual BRAF + MEK inhibition enhanced tumor immune infiltration and improved tumor control when combined with PD-1 blockade in a CD8+ T cell dependent manner. Additional PI3K inhibition did not impair tumor control or immune cell infiltration and functionality. Analysis of on-treatment samples from melanoma patients treated with BRAF or BRAF + MEK inhibitors indicates that inhibitor-mediated T cell infiltration occurred in all patients early after treatment initiation but was less frequent found in on-treatment biopsies beyond day 15.
Our findings provide a rationale for clinical testing of short-term BRAF + MEK inhibition in combination with immune checkpoint blockade, currently implemented at our institutes. Additional PI3K inhibition could be an option for BRAF + MEK inhibitor resistant patients that receive targeted therapy in combination with immune checkpoint blockade.
KEYWORDS: Anti-PD-1, BRAF, checkpoint blockade, immunotherapy, MAPK, MEK, melanoma, mTOR, PI3K, targeted therapy
Introduction
Treatment of advanced melanoma has changed dramatically within the last decade. Immunotherapies that block T cell checkpoint receptors, such as CTLA-4 and/or PD-1/PD-L1, and targeted therapies, such as BRAF and MEK inhibitors that inhibit the activated MAPK pathway in BRAFV600E mutated melanoma, have shown improved response rates and overall survival in randomized trials.1-5 The combination of CTLA-4 and PD-1 antibodies, as well as combined targeting of BRAF and MEK, have further improved patients' outcome.6-8 Although targeted therapy induces a high number of responses, the 3-y progression-free survival is approximately only 20% for patients treated with combined BRAF plus MEK inhibition (BRAFi + MEKi).9 Combined CTLA-4 plus PD-1 immune checkpoint blockade shows a plateau at 2-y, with a progression-free survival of approximately 45–50%.10,11 The characteristics of the high response rates seen with targeted therapy and durable responses often seen with immunotherapy provide a rationale to combine targeted therapy with immunotherapy to further improve patient survival.12,13 Selective BRAFi have been shown to induce an activated CD8+ T cell infiltrate, as well as increase melanoma MHC expression and melanoma antigen presentation early-during treatment both in pre-clinical models and in human melanoma tissue samples.14-20 In melanoma mouse models, BRAFi was shown to be synergistic with adoptive T cell therapy (ACT) in BRAF-driven murine melanoma, while its combination with checkpoint inhibitors has shown limited activity in mice.21-24 Early attempts to combine the selective BRAFi vemurafenib with ipilimumab in melanoma patients failed due to hepatotoxicity.25 Interestingly, Hassel et al. recently reported the feasibility of combining vemurafenib and ipilimumab.26
Addition of MEKi to selective BRAFi improved response rates, progression-free survival and overall survival as compared to single BRAFi in human melanoma.6,9 The addition of MEKi, however, raised the concern that it could dampen immune effector functions.14,27 A recent publication by Hu-Lieskovan et al. has disproven this concern by showing improved antitumor activity of anti-PD-1 immunotherapy when combined with BRAFi + MEKi in a preclinical mouse model of BRAFV600E-driven melanoma.28 In addition, Ebert et al. recently showed that in the CT26 mouse colorectal carcinoma model, MEKi impairs naïve CD8+ T cell priming, antigen-specific CD8+ T cell numbers were increased, and that MEKi worked synergistically with anti-PD-L1 blockade.29 In the same model, Liu et al. showed that MEKi only transiently inhibits T cell functions and was synergistic with anti-CTLA-4, anti-PD-1, and anti-PD-L1 blockade.30 In human melanoma, addition of MEKi to BRAFi did not result in significant reduction in immune infiltration in early treatment biopsies.31 These data together provide a clear rationale for clinical testing of BRAF plus MEK inhibition in combination with PD-1/PD-L1 blockade.
Activation of the PI3K-Akt-mTOR pathway plays an important role in oncogenic transformation and can occur via inactivation of the tumor suppressor protein PTEN.32 Loss of functional PTEN occurs in up to 30% of melanomas, frequently in tumors with the BRAFV600E mutation.33,34 PTEN loss has been associated with reduced response to BRAFi, which can be overcome by co-targeting the PI3K-Akt-mTOR pathway.35,36 Inhibitors of the PI3K-Akt-mTOR pathway that are clinically tested are inhibitors of PI3K (PI3Ki, e.g., BKM120) and mTOR (mTORi, e.g., everolimus).37,38 Combined inhibition of PI3K isoforms α, δ, and γ, but not β, is required to inhibit proliferation of BRAFV600E/PTENNull-driven murine melanoma.39 In addition, inhibition of all isoforms by the pan-PI3Ki BKM120 potentiated the antitumor effect of selective BRAFV600E inhibition.40 Interestingly, a recent study by Peng et al. showed that PTEN loss promotes resistance to T cell-mediated immunotherapy and that selectively targeting PI3Kβ improved the efficacy of immunotherapy in BRAFV600E/PTENNull melanoma.41 Inhibition of mTOR has significant antitumor activity in mouse tumor models and is an approved therapy for several malignancies.38 With respect to immune modulation, mTOR regulates the differentiation of memory CD8+ T cells,42 and leads to improved tumor control when combined with vaccination in the B16 mouse melanoma model.43 However, mTOR inhibition also increases the frequency of intratumoral regulatory T cells (Treg) and thereby limits immune mediated antitumor responses.43-45 These data indicate that single or dual PI3K-Akt-mTOR pathway targeting could be an effective combination partner for immune checkpoint inhibition,41 but it is unclear so far, which combination might be most effective. We therefore evaluated to which extent dual targeting of the MAPK and the PI3K-Akt-mTOR pathways can improve the outcome of PD-1 blockade in BRAFV600E/PTEN−/−-driven mouse melanoma, and analyzed samples from melanoma patients treated with BRAFi +/− MEKi to gain insight into optimal combination therapies.
Results
Addition of MEKi to BRAFi results in improved tumor control and increased tumor lymphocyte infiltration
We have previously shown that synchronous BRAFi + MEKi leads to superior tumor control in an induced BRAFV600E/PTEN−/− mouse melanoma model as compared to either agent alone.46 We confirmed this observation in a syngeneic BRAFV600E/PTEN−/−-driven transplantation model.47 We observed improved tumor control in all combinations of dual MAPK inhibition and PI3K/mTOR targeting as compared to single BRAFi (Fig. 1A). When discontinuing the targeted therapies after 14 d of treatment, we observed that addition of mTORi to BRAFi + MEKi did not improve tumor control. Although not statistical significant, mTORi potentially even antagonized the effect of BRAFi + MEKi +/− PI3Ki (Fig. 1A). Only the BRAFi + MEKi and BRAFi + MEKi + PI3Ki combinations significantly delayed tumor outgrowth as compared to single BRAFi (Fig. 1B). These observed differences in tumor outgrowth for the double, triple, and quadruple therapies could not be explained by a difference in tumor size at treatment withdrawal, as for example, mTORi combinations induced equal or even greater tumor size reductions on treatment (Fig. S1).
Figure 1.
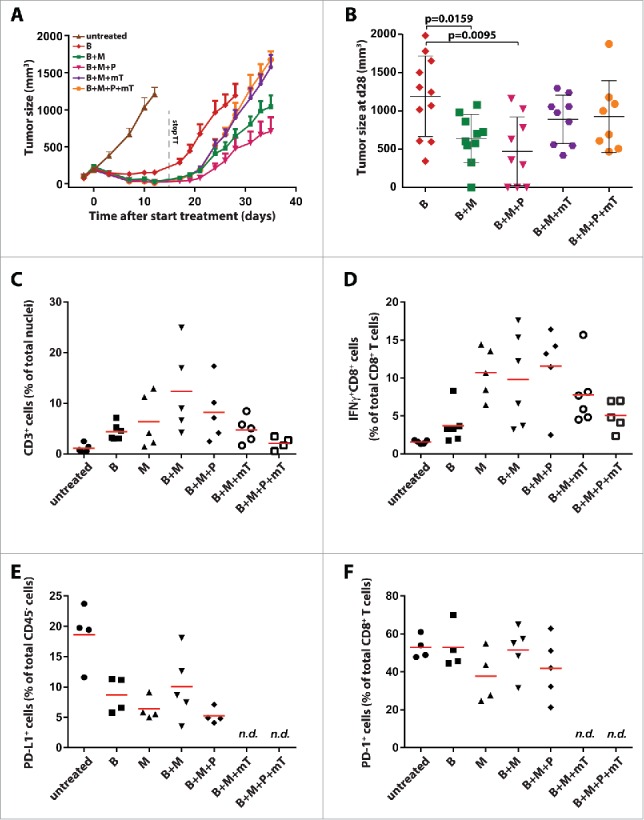
Improved tumor control and lymphocyte infiltration by addition of MEKi and PI3Ki to BRAFi. (A) C57BL/6 mice were injected subcutaneously with 3 × 105 D4M.3A mouse melanoma cells and targeted therapies (14 d) were started after 7 d. BRAFi [B] PLX4720 was provided in chow; MEKi [M] trametinib was dosed by daily oral gavage at 15 µg (on average 0.75 mg/kg); PI3Ki [P] BKM120 daily oral gavage at 400 µg (on average 20 mg/kg); and mTORi [mT] everolimus daily oral gavage at 100 µg (on average 5 mg/kg). Shown are tumor growth curves from mice treated with the indicated combinations (mean ± SEM and n = 9–11). (B) Mean tumor size ± SD at day 28 (no mice removed from experiments) is depicted in a dot plot (Mann–Whitney U-test). (C) D4M.3A tumor-bearing C57BL/6 mice (each group n = 5) were treated for 3 d with MAPK and/or PI3K pathway inhibitors, and tumors were analyzed by immunohistochemistry for CD3+ cell infiltration and analyzed by flow cytometry for (D) proportion of IFNγ producing CD8+ T cells; (E) PD-L1 expression of CD45− cells; and (F) expression of PD-1 on CD8+ T cells.
To evaluate the different MAPKi and/or PI3K/mTORi combinations as possible combination partners for checkpoint inhibition with anti-PD-1/PD-L1, tumor immune cell infiltration was analyzed by immunohistochemistry and flow cytometry on 3 d after the start of targeted therapy (Fig. 1C, Table S1 and Fig. S2). Combined BRAFi + MEKi induced the strongest CD3+ T cell infiltration (Fig. 1C and Table S1). Similar patterns were observed by flow cytometry for the specific CD4+ and CD8+ T cell subsets (Fig. S2). Addition of PI3Ki resulted in less pronounced infiltrates, but still significantly increased CD3+ T cell counts as compared to untreated mice (p = 0.0159). Addition of mTORi to BRAFi + MEKi +/− PI3Ki induced less T cell infiltration as compared to BRAFi + MEKi, but this was only significant for the BRAFi + MEKi + PI3Ki + mTORi combination (Fig. 1C). Qualitative analyses by flow cytometry revealed increased percentages upon targeted therapy for almost all lymphoid populations analyzed, including Tregs and cancer-associated B cells, while the frequency of macrophages with a M2-like phenotype was decreased (Fig. S2).
The proportion of intratumoral IFNγ positive CD8+ T cells was highest in tumors from mice treated with MEKi, BRAFi + MEKi, and BRAFi + MEKi + PI3Ki, while addition of mTORi reduced this amount (Fig. 1D). Combined MAPK and/or PI3K/mTOR targeting had no systemic effect on CD8+ T cells as measured by IFNγ+ CD8+ T cells within the spleen (Fig. S3A). Interestingly, the proportion of PD-L1-expressing cells within the tumor cell compartment (defined as CD45− cells) was decreased upon targeted therapy (Fig. 1E). The infiltrating CD8+ T cells expressed high levels of PD-1 in about 50% of the cells (Fig. 1F), a marker shown to be associated with tumor-antigen-specific T cells,48 and was not altered when targeted agents were applied. Similarly to the systemic absence of IFNγ-producing CD8+ T cells, few PD-1+ CD8+ T cells were found in the spleen and their frequency did not increase upon targeted therapy (Fig. S3B).
Short-term BRAFi + MEKi shows the strongest synergy with anti-PD1
The observation that BRAFi + MEKi led to a strong increase of PD-1+ CD8+ tumor-infiltrating lymphocytes (TILs) raised the question whether additional PD-1 blockade could induce long-term tumor control in our model setting. Tumor-bearing mice were treated with combinations of targeted therapy and anti-PD-1 checkpoint blockade (Fig. 2A). Identical to Fig. 1, targeted therapy was withdrawn after 14 d of treatment, while anti-PD-1 was dosed continuously. Single PD-1 blockade did not affect tumor growth as compared to isotype antibody treated animals (Figs. 2A and C). Short-term BRAFi + MEKi and BRAFi + MEKi + PI3Ki showed the strongest synergy with PD-1 blockade, resulting in significant tumor size reduction at day 32 (Fig. 2B; p < 0.0001 and p = 0.045, respectively). Single BRAFi or MEKi in combination with PD-1 blockade also reduced tumor outgrowth as compared to single BRAFi or MEKi alone, but did not reach statistical significance (Fig. 2B). BRAFi containing combinations combined with PD-1 blockade resulted in complete ongoing responses (CR) in a subset of mice (followed for up to 200 d, data not shown). This was most frequently observed in the BRAFi + MEKi + anti-PD-1 combination (Fig. 2C, 4/9 CRs). Rechallenge of mice that had achieved a complete response with the same tumor cell line did not result in tumor outgrowth in the majority of mice (data not shown).
Figure 2.
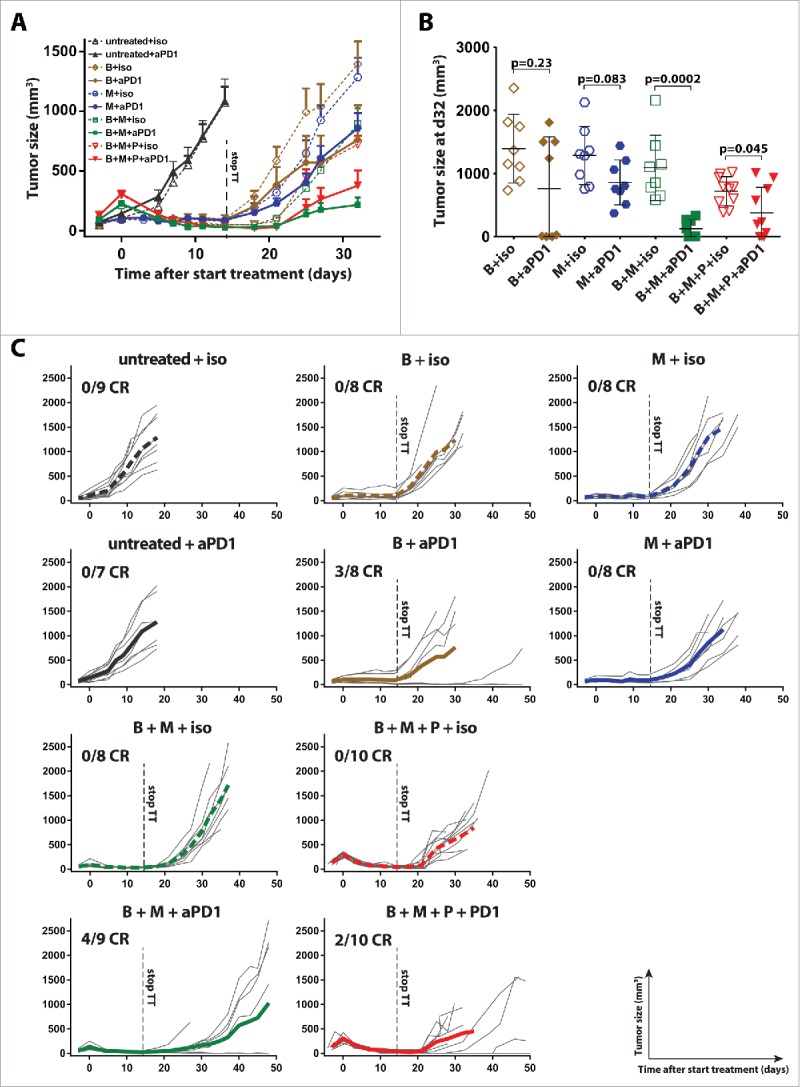
BRAFi + MEKi has the strongest short-term synergy with anti-PD1. (A) Tumor-bearing mice were treated as described in Fig. 1 with the indicated small molecules targeting MAPK and/or PI3K pathway for 14 d and concurrently either with anti-PD-1 or isotype mAb (twice weekly 100 µg intraperitoneal). Anti-PD-1 or control antibody was continued beyond day 14. Shown are the tumor sizes of the different treatment groups (mean ± SEM and n = 8–10). (B) Tumor sizes from 2A at day 32 are depicted in a dot plot (mean ± SD) and statistical significance is analyzed comparing isotype versus anti-PD1 treatment (Mann–Whitney U-test). (C) Individual tumor growth curves of mice are plotted per condition and the average of the group is indicated by a bold line.
Tumor control upon targeted therapy plus PD-1 blockade did not correlate with altered tumor T cell infiltration or changes in T cell function. Specifically, the addition of anti-PD-1 therapy did not increase the number of infiltrating CD3+ T cells as compared to targeted therapy (Figs. 3A and B and Table S2). Addition of anti-PD-1 did not alter the IFNγ+ CD8+ T cell proportion, the granzyme B+ CD8+ T cell proportion, or the proportion of CD44hi CD62Llo CD8+ T cells within the tumors, except for granzyme-positive CD8+ T cells in combination with BRAFi (Figs. 3C–E).
Figure 3.

Addition of anti-PD1 to MAPK and PI3K pathway inhibition does not alter the infiltration and activation/effector status of CTLs. (A) Tumors from mice treated as described in Fig. 2A (n = 4–5 per group) were analyzed by immunohistochemistry for CD3 infiltration on day 3. The black scale bars equal 3 mm and the high magnification inserts are 400 μm squared. Quantification depicted in (B) In the same setup as A and B, tumors were digested and analyzed by flow cytometry for the percentages of (C) IFNγ producing CD8+ T cells (D) granzyme B producing CD8+ T cells; and (E) CD44highCD62Llow expressing CD8+ T cells.
Synergy of targeted therapy with anti-PD-1 is dependent on the presence of CD8+ T cells
The near equal synergy in tumor control with BRAFi + MEKi + PI3Ki + anti-PD-1 as compared to BRAFi + MEKi + anti-PD-1 (Fig. 2) and the absence of changes in tumor T cell infiltrates upon addition of anti-PD-1 (Fig. 3), raised the question whether targeted therapy-induced CD8+ T cell infiltration is the major mechanism for synergy with PD-1 blockade. To address this question, we depleted CD4+ or CD8+ T cells in parallel with BRAFi + MEKi + anti-PD-1 treatment. Depletion of CD8+ cells, but not CD4+ cells, completely abolished the control of tumor outgrowth observed upon BRAFi + MEKi + anti-PD-1 (Figs. 4A and B) and almost completely abolished the induction of complete responders (Fig. S4A and B). Remarkably, depletion of CD8+ cells did not result in a significantly faster outgrowth after BRAFi + MEKi treatment as compared to isotype depletion (Fig. S4B). This might indicate that the initial tumor control is dominated by the targeted therapy and that the observed CD8+ T cell infiltration is required for anti-PD-1-mediated tumor control (Fig. 1A and Fig. 4B). Interestingly, depletion of CD4+ T cells had a minor positive effect on tumor outgrowth control upon BRAFi + MEKi + anti-PD-1 (Fig. 4A), which was preceded by a statistically significant difference in tumor size at the moment targeted therapy was halted (Fig. S4B; p = 0.0002). This might result from interfering with intratumoral CD4+ regulatory T cells (Tregs), which were increased upon BRAFi C MEKi, but not on single agent BRAFi (Fig. S2H).
Figure 4.
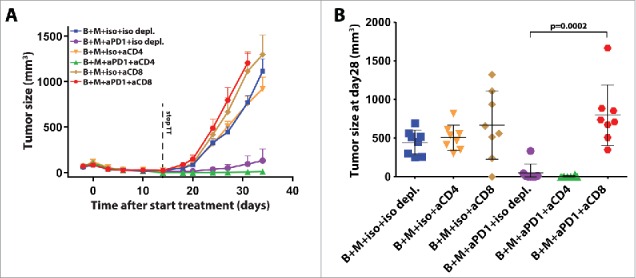
Synergy of targeted therapy with anti-PD-1 is dependent on the presence of CD8+ T cells. (A) Tumor growth curves of D4M.3A tumor-bearing C57BL/6 mice treated for 14 d with BRAFi and MEKi combined with isotype or anti-PD-1 as described in Fig. 2A. In addition, mice were treated with twice weekly intraperitoneal isotype mAb, anti-CD4+ or anti-CD8+ depleting antibodies at 250 µg (mean ± SEM and n = 8–9). (B) Tumor size and mean ± SD at day 28 is depicted in a dot plot (Mann–Whitney U-test).
MAPKi induces transient CD8+ T cell infiltration
Our rationale to combine short-term targeted therapies +/− anti-PD-1 is based on the observations of high T cell infiltration occurring early during treatment, which disappeared at disease progression.17,20,21,49 To investigate the kinetics of immune infiltration upon targeted therapy, we treated tumor-bearing mice with BRAFi or BRAFi + MEKi.6,9 The amount of tumor-infiltrating lymphocytes was analyzed by immunohistochemistry at different time points. An increase of CD3+ T cell infiltrates was found in both treatment groups, starting already on day 3 (Figs. 5A–C). Decreased T cell infiltration was observed upon BRAFi on day 28 (Fig. 5A, upper row, and Fig. 5B p = 0.0635 compared to day 14), which was concurrent with tumor progression (Fig. S5). In contrast, BRAFi + MEKi induced incremental T cell infiltration at all-time points analyzed (Fig. 5A lower row, and Fig. 5C). Moreover, this increase of tumor infiltrates was also accompanied by continuous tumor control by BRAFi + MEKi (Figs. S5A–D). Our effort to identify the optimal treatment duration by simulation in our mouse model was thus hampered by the insufficient tumor control with BRAFi and the incremental tumor control upon BRAFi + MEKi.
Figure 5.
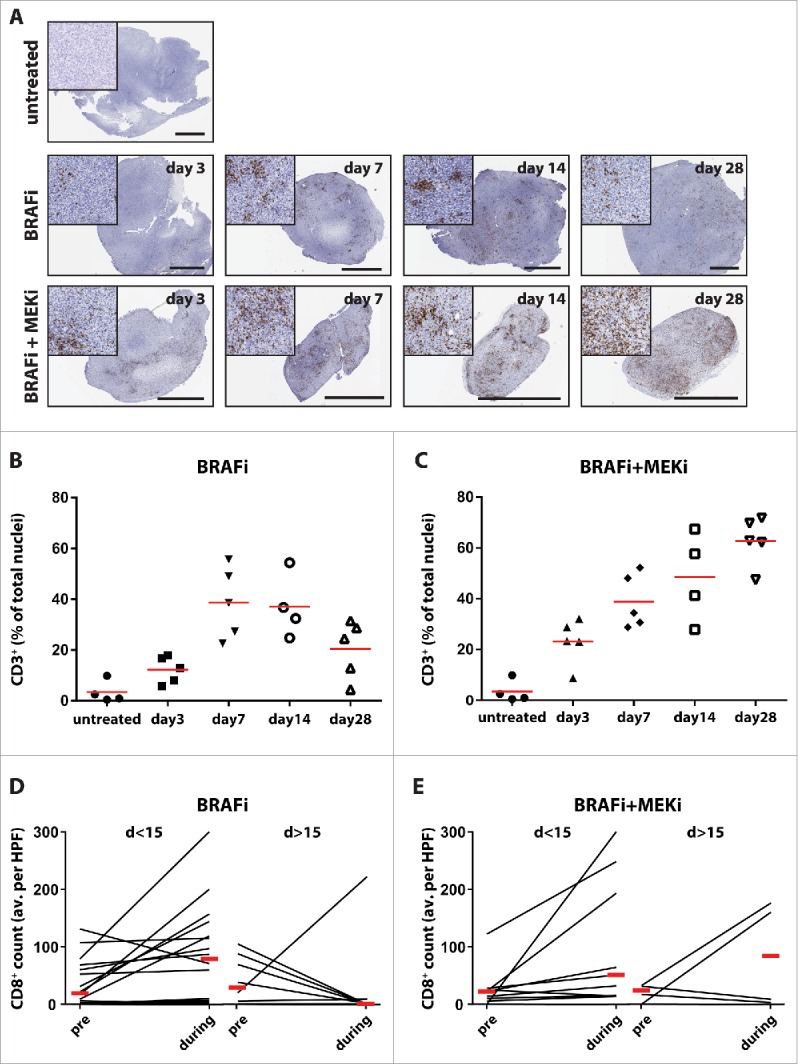
Transient infiltration of T cells upon MAPK pathway inhibition. D4M.3A tumor-bearing C57BL/6 mice were treated with BRAFi and/or MEKi. (A) Tumors treated for the respective time were analyzed by immunohistochemistry for CD3+ cell infiltration. Representative immunohistochemistry stainings are shown. The black scale bars equal 3 mm and the high magnification inserts are 400 μm squared. (B, C) Automated software quantifications for the proportion of infiltrating CD3+ cells are depicted as a fraction of total nuclei (mean indicated by horizontal line). (D, E) Intrapatient pre- and during treatment tumor biopsies from humane melanoma treated with either BRAFi or BRAFi + MEKi. Samples were analyzed by immunohistochemistry for CD8+ cell infiltration in a blinded manner. The average CD8+ cell count per HPF is plotted and median is indicated by horizontal red line. Patient samples were grouped as early on treatment biopsies (< 15 d of treatment) and late on treatment biopsies (> 15 d).
We therefore analyzed CD8+ T cell infiltration in paired pre- and on-treatment human melanoma biopsies from patients responding to BRAFi or BRAFi + MEKi. We selected patients that showed partial responses evaluated by RECIST 1.1 and had undergone on-treatment biopsy during on-going responses, thus not at disease progression. In early biopsies (<15 d of treatment) of BRAFi and BRAFi + MEKi treated patients, we found increased CD8+ T cell infiltrates as compared to the baseline T cell infiltrates (Figs. 5D and E left; shown is fold increase as compared to pre-treatment samples; BRAFi: median 7 d, range 2–13 d; BRAFi + MEKi: median 7 d, range 4–15 d on treatment). However, in the majority BRAFi treated patients (5/7 patients) the amount of CD8+ T cell infiltrates was reduced as compared to pretreatment levels, when the biopsies were taken beyond 15 d (Fig. 5D, right; median 242 d, range 27–365 d on treatment). In contrast, 2/4 treatment biopsies from BRAFi + MEKi treated patients biopsied beyond 15 d maintained an increased level of CD8+ T cell as compared to pre-treatment (Fig. 5E, right; median 227 d, range 34–497 d on treatment).
Discussion
Combining targeted therapy and immunotherapy to potentiate improved long-term patients' outcome was proposed soon after they were established as systemic therapies for advanced melanoma.12,20,50 Unfortunately, toxicities have been observed for the combination of the BRAFi vemurafenib with anti-CTLA-4 immune checkpoint blockade,25,51 leading to suspension of these trials. PD-1 blockade has been shown in large randomized trials to induce less grade 3/4 toxicities, while inducing higher response rates as compared to ipilimumab,7,52 making PD-1/PD-L1 inhibitors possibly a more safe combination partner for targeted therapies. Recent preclinical work has indicated synergistic effects of synchronous BRAFi and ACT.24 The observation that BRAFi + MEKi alleviates some BRAFi-induced toxicities,46,53 provides another argument for combining BRAFi + MEKi with immune checkpoint blockade. However, this raised a concern about possible impairment of T cells functions upon MEKi.27 Recent in vivo studies have reported synergy between MEKi and PD-L1 blockade, and BRAFi + MEKi and PD-1 blockade,28-30 showing that the previously described detrimental effects from MEKi on naïve T cells,27 may not be relevant to intratumoral CD8+ effector T cells. Indeed, in patients receiving BRAFi + MEKi it has been shown that numbers of tumor infiltrating CD8+ T cells were increased after start of treatment.30 Furthermore, increased intratumoral CD8+ T cell numbers have been associated with improved outcome upon PD-1 blockade.54 Based on these data, several trials testing continuous BRAFi + MEKi in combination with PD-L1 or PD-1 blockade have been initiated (NCT02027961 and NCT01656642).
We have aimed to analyze the effect of single and dual MAPK pathway inhibition in combination with additional single or dual PI3K-Akt-mTOR pathway inhibition and its potential synergy with PD-1 blockade. Additional PI3K-Akt-mTOR pathway targeting might be an interesting option for patients unresponsive to BRAFi + MEKi. This urged the preclinical investigation of pan-PI3K and mTOR inhibitors for their assumed T cell inhibitory function.55
We found that short-term MAPK pathway inhibition induced strong tumor control and enhanced tumor immune infiltration as compared to untreated mice or single BRAFi treated mice. This is in line with preclinical and clinical observations showing increased tumor control and T cell infiltration upon dual MAPK-targeted therapies.6,28,53,56,57
Previous data showed that additional blockade of PI3Kα, PI3Kδ, and PI3Kγ slightly improved tumor control and prevented resistance upon MEKi in BRAFV600E/PTENNull melanoma.39 In our experiments testing different targeted therapy combinations, we found no significant additional tumor reduction of short-term pan-PI3K inhibition by BKM120 in addition to BRAFi and BRAFi + MEKi. Furthermore, administration of BKM120 in combination with BRAFi + MEKi did not significantly alter T cell infiltration and effector function. This seems contradictory to the result by Peng et al., who showed that pan-PI3Ki interferes with T cell activation in a vaccination model.41 However, similar to MEKi,29 pan-PI3Ki might predominantly alter early T cell activation, while to a lesser extent the intratumoral effector T cell function.
Additional mTOR targeting by everolimus with BRAFi + MEKi or BRAFi + MEKi + PI3Ki resulted in loss of the significantly improved tumor control upon BRAFi + MEKi +/− PI3Ki. This is in contrast to previous in vitro,35,58 and in vivo studies where mTORi has been shown to improve antitumor immune responses.59,60 Addition of mTORi to BRAFi + MEKi +/− PI3Ki also reduced the tumor infiltration of CD3+ lymphocytes and IFNγ-producing effector CD8+ T cells. Combinations with mTORi were therefore not further tested in combination with PD-1 blockade in our work here.
It was previously shown that increased intratumoral CD8+ T cell infiltration upon BRAFi +/− MEKi is associated with improved outcome with PD-1 blockade in mice.28 In patients, increased CD8+ T cell tumor infiltration was associated with better outcome upon PD-1 blockade.54 Therefore, we have addressed the effect of adding anti-PD-1 treatment to targeted therapy on tumor outgrowth. Indeed, concurrent with increased infiltration of TILs, we observed synergistic effects of the addition of anti-PD-1 to targeted therapy, most strongly in the combination of BRAFi + MEKi. We regularly observed complete responses for all targeted therapies where anti-PD-1 was added, with the exception of the MEKi group. This indicates that early full eradication of the tumors depends mainly on BRAFi + anti-PD-1 combination. However, the addition of MEKi still has an effect on tumor outgrowth in non-complete responders. In that way, addition of MEKi to BRAFi + PD-1 blockade is beneficial for the whole cohort. We found that addition of pan-PI3Ki did not influence the synergy of BRAFi + MEKi with PD-1 blockade. Furthermore, the addition of short-term pan-PI3Ki did not inhibit CD8+ T cell functionality, which seems to be in contrast to Peng et al., who showed that selective PI3Kβ inhibition improved the efficacy of anti–PD-1 checkpoint blockade in an induced BRAFV600E/PTENNull mouse melanoma model.41
Our data suggest that adding pan-PI3Ki in patients that respond to BRAFi + MEKi has no benefit on tumor control. However, our observation that adding pan-PI3Ki to BRAFi + MEKi + anti-PD-1 does not hamper synergistic effects provides a rationale to test pan-PI3K inhibitors in combination with immunotherapy in patients being resistant to BRAFi + MEKi.
Depletion of CD8+ cells completely abolished the synergistic effect of BRAFi + MEKi in combination with anti-PD-1. This confirmed the dogma that the mechanism of anti-PD-1 therapy is at least in part mediated through CD8+ T cells.61,62 Although an increase in IFNγ producing CD8+ T cells was observed upon targeted therapy, we could not find a change in activation markers on bulk CD8+ T cells with the addition of anti-PD-1. Interestingly, we observed rejection of tumor cells when injected in mice that had a previous complete response. This indicates that an immunological memory response is induced upon the combination therapy. Since the nature of this model did not allow us to follow tumor specific T cell clones, we do not know which percentage of effector cells contributed to the response. Kvistborg et al. have shown that tumor-specific T cell populations with a very low frequency can be found in patients treated with anti-CTLA-4.63 This might suggest that only a limited proportion of infiltrating T cells is actually exhibiting antitumor specificity. It is of interest to further study the antitumor specificity of the infiltrating lymphocytes and elucidate the proportion that is responsible for tumor control in the complete responders. Even though the window for improvement is limited, depletion of CD4+ T cells had a beneficial effect on early tumor control. Mice that were treated with BRAFi + MEKi + anti-PD-1 in the absence of CD4+ T cells had significantly smaller tumors at the moment targeted therapy was halted. A likely explanation for this observation could be that Tregs have an important immunosuppressive function on tumor specific T cells. Indeed, we found that about 60% of CD4+ positive T cells in untreated D4M.3A tumors are positive for FoxP3 and this proportion slightly increased upon BRAFi + MEKi, but not upon BRAFi.
In addition to Tregs, other immune regulatory cells, like monocytes, tumor associated B cells, neutrophils, and macrophages were present in this model. No clear correlations were observed between these populations and the synergistic effect of combined targeted therapy and anti-PD-1. However, this does not exclude that depletion of, e.g., Treg, tumor-associated macrophages, neutrophils, or cancer-associated B cells in addition to BRAFi + MEKi +/− PI3Ki might further improve long-term outcome.
In contrast to the work of Hu-Lieskovan et al., we did not apply continuous and concomitant targeted therapy. The rational for this decision was based on our observation of a patient achieving long-term tumor control after receiving short term BRAFi + MEKi subsequent to PD-1 blockade (Blank et al., unpublished), and from another case report of long-term BRAFi being counterproductive for increasing the intratumoral T cell response.21 Indeed, we were able to observe long-term complete responses in the D4M.3A mouse melanoma model, when applying short-term targeted therapy. This has not been described in preclinical work using another subcutaneous mouse melanoma model (harboring BRAFV600E and CDKN2 mutations) testing continuous application of BRAFi + MEKi.24 However, these observed differences could also be due to the different genetic driver mutations present in these cell lines.24,47 Testing continuous and intermittent application of BRAFi + MEKi in combination with PD-1 blockade in the latter model could address this question. On the other hand, we found that infiltration of T cells was already prevalent within the first week after start of treatment, both in mouse and human melanoma. This supports previous ideas that the induction of a favorable antitumor microenvironment occurs early after targeted therapy initiation and might disappear on treatment.20,21,31 In this way, current clinical study designs of long BRAFi + MEKi induction phases before PD-1/PD-L1 blockade might undermine an improved antitumor immune response. This idea is further supported by recent findings, that MAPKi induces an innate anti-PD-1 resistance (IPRES) signature in melanomas, which was found in patients not responding to PD-1 blockade.64
Our results are limited by preclinical mouse modeling and limited availability of human tumor samples. Validation of these results in clinical trials is warranted. Several clinical trials are studying combinations of dual MAPK-pathway inhibition (dabrafenib + trametinib or vemurafenib + cobimetinib) and immune checkpoint blockade (e.g., anti-CTLA-4 or anti-PD-1/PD-L1) in melanoma patients (NCT01940809; NCT01656642; NCT02027961; NCT02130466; NCT02224781). However, none of these trials addresses the question of what the optimal dosing schedule of targeted therapy should be in order to achieve the optimal outcomes for patients. Our data suggest that combining short-term MAPK-pathway +/− PI3K-pathway inhibition with immunotherapies improves antitumor immune responses, and may be superior to continuous combination therapy. Based on our results, we have initiated two phase 2 studies; one study in advanced melanoma comparing the combination of anti-PD-L1 (pembrolizumab) with two different intermittent BRAFi + MEKi (dabrafenib + trametinib) schemes versus continuous BRAFi + MEKi (IMPemBra, NCT02625337) and one neoadjuvant study exploring a short-term induction phase of BRAFi + MEKi followed by anti-PD1 versus continuous triple therapy in resectable stage 3 or 4 melanoma (NCT02858921).
Materials and methods
Syngeneic mouse melanoma model
D4M.3A cells were generated from Tyr::CreER;BrafCA;Ptenlox/lox mice by Jenkins et al.47 and kindly provided to us. The cells were cultured in DMEM/F-12 advanced media (Life Technologies) with 5% FBS, 1× penicillin/streptomycin (Sigma-Aldrich), and 1× glutamine and resuspended in PBS + matrigel (BD Biosciences) before subcutaneous injection (3 × 105 cells) in male C57BL/6 mice (Charles River and Janvier). Tumor outgrowth was followed by two-dimensional digital caliper measurements of the greatest longitudinal diameter (length) and the greatest transverse diameter (width). Tumor volume based on caliper measurements was calculated by the modified ellipsoidal formula. Tumor volume = 1/2 (length × width2). Average tumor sizes are plotted as long as at least 50% of the animals within a group were alive. The last measured tumor size of mice that reached the experimental end point was taken along for the subsequent time points for calculating group averages. All described animal experiments were approved by the Animal Experimentation Committee of the Netherlands Cancer Institute. Mice were treated in accordance with the Dutch law on animal experimentation.
Treatment of tumor-bearing mice
PLX4720, a research analog of the selective BRAF inhibitor vemurafenib (PLX4032) (provided by Plexxikon) was used in chow.65 At the start of the experiments, the mice were switched either to a chow diet containing 417 mg/kg PLX4720 or to control chow containing no compound. On average the food dosing is similar to a daily 50 mg/kg dosing by oral gavage.65 The MEK inhibitor trametinib (Selleckchem) was dissolved in 0.5% hydroxypropyl methylcellulose and 0.2% Tween-80 (Sigma-Aldrich) and was administered daily by oral gavage of 15 µg (on average 0.75 mg/kg), once daily, 5 d per week for the indicated durations. The pan-PI3K inhibitor BKM120 (provided by Novartis) was dissolved in 0.5% methylcellulose and 0.5% Tween-80, and 400 µg (on average 20 mg/kg), once daily, 5 d per week. The mTOR inhibitor everolimus (provided by Novartis), diluted in 5% glucose, was administered by daily oral gavage at 100 µg (on average 5 mg/kg), once daily, 5 d per week for the indicated durations. Anti-PD-1 (clone RMP1-14, BioXcell) was injected intraperitoneal twice weekly 100 µg mAb for the complete duration of the experiments. Control mice, not receiving the anti-PD-1 antibody, were administered with the appropriate isotype control (Rat IgG2a, clone 2A3, BioXcell) at the same dose.
Flow cytometric analysis of immune populations
Tissues were removed from the animals directly after euthanasia. Single-cell suspensions from the tissues were obtained by disruption of the tissue by slicing of the material followed by a 1 h digestion in digestion medium at 37°C. Digestion medium contained 2 mg/mL collagenase A (Sigma-Aldrich) and 1 mg/mL DNAse (Sigma-Aldrich). After digestion the suspension was filtered through a 70 µm filter (BD Biosciences) to get rid of debris. The obtained single-cell suspension was then stained with fluorochrome labeled antibodies and analyzed for expression using a LSR Fortessa (BD Biosciences) and FlowJo software (Tree Star Inc.). Intracellular staining was performed using the Intracellular Fixation & Permeabilization Buffer Set from eBioscience according to the manufacturers' protocol.
Lymphocyte panel; CD45.2-PE-Cy7 (1:200; clone 104, BD Biosciences), CD11b-Alexa Fluor 700 (1:200; clone M1/70, eBioscience), CD19-PE-Cy5.5 (1:400; clone 1D3, eBioscience), NK1.1-APC (1:100; clone PD136, eBioscience), CD3-PE-eFluor610 (1:200; clone 145-2C11, eBioscience), CD4+-APC-H7 (1:200; clone GK 1.5 BD Biosciences), CD8+-V500 (1:200; clone 53–6.7, BD Biosciences), PD-L1-PE (1:200; clone MIH5, eBioscience), PD-1-FITC (1:100; clone J43, eBioscience), DAPI (1:20).
Myeloid panel; CD45.2-PE-Cy7 (1:200; clone 104, BD Biosciences), CD11b-FITC (1:200; clone M1/70, eBioscience), CD11c-PB (1:400; clone N418, Biolegend), F4/80-APC-eFluor780 (1:200; clone BM8, eBioscience), Ly6G-APC (1:400; clone 1A8, BD Bioscience), Ly6C-PE (1:800; clone HK1.4, eBioscience), MHC-II-PerCp-eFluor710 (1:200; clone M5/114.15.2, eBioscience), LIVE/DEAD405 Fixable Aqua Stain, 1:100 in FACS buffer (Invitrogen).
Intracellular panel; CD45.2-PE-Cy7 (1:200; clone 104, BD Biosciences), CD11b-Alexa Fluor 700 (1:200; clone M1/70, eBioscience), CD3-PE-eFluor610 (1:200; clone 145-2C11, eBioscience), CD4+-APC-H7 (1:200; clone GK 1.5 BD Biosciences), CD8+-PerCP-eFluor710 (1:400; clone 53–6.7 eBioscience), FoxP3-APC (1:100; clone FJK-16s, eBioscience), IFNγ-eFluor450 (1:200; clone XMG1.2, eBioscience), CD44-FITC (1:200; clone IM7, eBioscience) CD62L-Alexa Fluor 700 (1:200; clone MEL-14, eBioscience), granzyme B (1:200; clone CLB-GB11, Sanquin), LIVE/DEAD405 Fixable Aqua Stain, 1:100 in FACS buffer (Invitrogen).
Prior to intracellular staining, cells were incubated in IMDM medium containing 8% FCS + Pen/strep + β-mercaptoethanol for 3 h in the presence of 1 µL/mL golgiplug (BD Biosciences). For the analysis of IFNγ, cells were stimulated with PMA (50 ng/mL, Sigma-Aldrich) and Ionomycin (1 µM, Sigma-Aldrich).
Depletion experiments
For depletion of CD4+ and CD8+ T lymphocytes mice were peritoneal injected twice weekly with 250 µg of anti-CD4+ (clone GK1.5, BioXcell) and/or anti-CD8+ (clone 2.43, BioXcell) antibodies, respectively. Control mice were injected with Rat IgG2b isotype control (clone LTF-2, BioXcell). Depletion of the specific populations was confirmed by flow cytometry at day 26 after start of the experiment.
Histological analysis and immunohistochemistry
Tissues were removed from the mice directly after euthanasia. They were fixed in formalin and embedded in paraffin. 4 µm-thick sections were made and stained with anti-CD3 (clone RM-9107, Thermo Scientific) according to standard procedures. CD3 infiltration was scored using SlidePath Tissue Image Analysis software (Leica Microsytems). Human formalin-fixed paraffin-embedded tumor samples were cut into 4 µm-thick sections and stained with anti-CD8+ (clone C8/144B, DAKO) by standard procedures. Slides were scored by counting positive cells in 3–4 random high-power fields (HPF).
Patient material
Melanoma biopsies from patients treated at the Melanoma Institute Australia Sydney and the Netherlands Cancer Institute were taken from patients prior to commencing either vemurafenib (960 mg orally twice daily) or dabrafenib (150 mg orally, twice daily) alone, or dabrafenib combined with trametinib (2 mg orally daily), early during treatment (<15 d, EDT) or late during treatment (> 15 d, LDT). Biopsies were performed under the Treat Excise Analyze for Melanoma (TEAM) protocol,20,49 or the N03LAM protocol (NKI material collection trial). Both protocols were approved by the local human research ethics committees and informed consent from the patients was obtained before inclusion in the analyses.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank David Mullins and Constance Brinckerhoff for providing the D4M.3A cell line. The NKI Experimental Animal Pathology department and the Experimental Animal facility are acknowledged for help with histology and immunohistochemistry and mouse husbandry, respectively. We would like to acknowledge the NKI-AVL Core Facility Molecular Pathology& Biobanking (CFMPB) for supplying NKI-AVL Biobank material and/or lab support. Hojabr Kakavand is thanked for help obtaining the patient material. We would like to thank Karin de Visser and Telma Lança for thoughtful discussions and Chelsea McLean for carefully reading the manuscript. We are grateful to Plexxikon for providing us with PLX4720 and Novartis for providing us with BKM120 and everolimus.
References
- 1.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M et al.. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Eng J Med 2011; 364:2507-16; PMID:21639808; http://dx.doi.org/ 10.1056/NEJMoa1103782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P et al.. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Eng J Med 2012; 367:107-14; PMID:22663011; http://dx.doi.org/ 10.1056/NEJMoa1203421 [DOI] [PubMed] [Google Scholar]
- 3.Mizugaki H, Yamamoto N, Murakami H, Kenmotsu H, Fujiwara Y, Ishida Y, Kawakami T, Takahashi T et al.. Phase I dose-finding study of monotherapy with atezolizumab, an engineered immunoglobulin monoclonal antibody targeting PD-L1, in Japanese patients with advanced solid tumors. Invest New Drugs 2016; 34(5):596-603; PMID:27363843; http://dx.doi.org/ 10.1007/s10637-016-0371-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD et al.. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. The Lancet Oncol 2015; 16:908-18; PMID:26115796; http://dx.doi.org/ 10.1016/S1470-2045(15)00083-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E et al.. Nivolumab in previously untreated melanoma without BRAF mutation. N Eng J Med 2015; 372:320-30; PMID:25399552; http://dx.doi.org/ 10.1056/NEJMoa1412082 [DOI] [PubMed] [Google Scholar]
- 6.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L et al.. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Eng J Med 2014; 371:1867-76; PMID:25265494; http://dx.doi.org/ 10.1056/NEJMoa1408868 [DOI] [PubMed] [Google Scholar]
- 7.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P et al.. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Eng J Med 2015; 373(1):23-34; PMID:26027431; http://dx.doi.org/ 10.1056/NEJMoa1504030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS et al.. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Eng J Med 2015; 372:2006-17; PMID:25891304; http://dx.doi.org/ 10.1056/NEJMoa1414428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long GV, Weber JS, Infante JR, Kim KB, Daud A, Gonzalez R, Sosman JA, Hamid O, Schuchter L, Cebon J et al.. Overall Survival and Durable Responses in Patients With BRAF V600-Mutant Metastatic Melanoma Receiving Dabrafenib Combined With Trametinib. J Clin Oncol 2016; 34:871-8; PMID:26811525; http://dx.doi.org/ 10.1200/JCO.2015.62.9345 [DOI] [PubMed] [Google Scholar]
- 10.Postow MA, Chesney J, Pavlick A, Robert C, Grossmann K, McDermott D et al.. Initial report of overall survival rates from a randomized phase II trial evaluating the combination of nivolumab (NIVO) and ipilimumab (IPI) in patients with advanced melanoma (MEL). AACR meeting abstracts 2016 [Google Scholar]
- 11.Wolchok J, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL et al.. Updated results from a phase III trial of nivolumab (NIVO) combined with ipilimumab (IPI) in treatment-naive patients (pts) with advanced melanoma (MEL) (CheckMate 067). ASCO Meeting Abstracts 2016 [Google Scholar]
- 12.Blank CU, Hooijkaas AI, Haanen JB, Schumacher TN. Combination of targeted therapy and immunotherapy in melanoma. Cancer immunol immunother 2011; 60:1359-71; PMID:21847631; http://dx.doi.org/ 10.1007/s00262-011-1079-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu-Lieskovan S, Robert L, Homet Moreno B, Ribas A. Combining targeted therapy with immunotherapy in BRAF-mutant melanoma: promise and challenges. J Clin Oncol 2014; 32:2248-54; PMID:24958825; http://dx.doi.org/ 10.1200/JCO.2013.52.1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher DE et al.. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res 2010; 70:5213-9; PMID:20551059; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-0118 [DOI] [PubMed] [Google Scholar]
- 15.Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, Escuin-Ordinas H, Chmielowski B, Koya RC, Ribas A. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res 2010; 16:6040-8; PMID:21169256; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donia M, Fagone P, Nicoletti F, Andersen RS, Hogdall E, Straten PT, Andersen MH, Svane IM. BRAF inhibition improves tumor recognition by the immune system: Potential implications for combinatorial therapies against melanoma involving adoptive T-cell transfer. Oncoimmunology 2012; 1:1476-83; PMID:23264894; http://dx.doi.org/ 10.4161/onci.21940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C et al.. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013; 19:1225-31; PMID:23307859; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, Larkin J, Marais R, Sahai E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 2015; 27:574-88; PMID:25873177; http://dx.doi.org/ 10.1016/j.ccell.2015.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAF homozygous melanoma cells. Oncoimmunology 2013; 2:e22890; PMID:3583938; http://dx.doi.org/22156613 10.4161/onci.22890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P, Scolyer RA. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 2012; 18:1386-94; PMID:22156613; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-2479 [DOI] [PubMed] [Google Scholar]
- 21.Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, Lo JA, Hodi FS, Freeman GJ, Bosenberg MW et al.. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res 2014; 2:643-54; PMID:24903021; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hooijkaas A, Gadiot J, Morrow M, Stewart R, Schumacher T, Blank CU. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology 2012; 1:609-17; PMID:22934253; http://dx.doi.org/ 10.4161/onci.20226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC et al.. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest 2013; 123:1371-81; PMID:23454771; http://dx.doi.org/ 10.1172/JCI66236 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T et al.. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res 2012; 72:3928-37; PMID:22693252; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Eng J Med 2013; 368:1365-6; PMID:23550685; http://dx.doi.org/ 10.1056/NEJMc1302338 [DOI] [PubMed] [Google Scholar]
- 26.Hassel JC, Lee SB, Meiss F, Meier F, Dimitrakopoulou-Strauss A, Jager D, Enk AH. Vemurafenib and ipilimumab: A promising combination? Results of a case series. Oncoimmunology 2016; 5:e1101207; PMID:27141385; http://dx.doi.org/ 10.1080/2162402X.2015.1101207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vella LJ, Pasam A, Dimopoulos N, Andrews M, Knights A, Puaux AL, Louahed J, Chen W, Woods K, Cebon JS. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol Res 2014; 2:351-60; PMID:24764582; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0181 [DOI] [PubMed] [Google Scholar]
- 28.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, Pinheiro EM, Koya RC, Graeber TG, Comin-Anduix B et al.. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med 2015; 7:279ra41; PMID:25787767; http://dx.doi.org/ 10.1126/scitranslmed.aaa4691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebert PJ, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, Gould SE, Maecker H, Irving BA, Kim JM et al.. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016; 44:609-21; PMID:26944201; http://dx.doi.org/ 10.1016/j.immuni.2016.01.024 [DOI] [PubMed] [Google Scholar]
- 30.Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, Yang J, Seestaller-Wehr L, Zhang SY, Hopson C et al.. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res 2015; 21:1639-51; PMID:25589619; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-2339 [DOI] [PubMed] [Google Scholar]
- 31.Kakavand H, Wilmott JS, Menzies AM, Vilain R, Haydu LE, Yearley JH, Thompson JF, Kefford RF, Hersey P, Long GV et al.. PD-L1 Expression and Tumor-Infiltrating Lymphocytes Define Different Subsets of MAPK Inhibitor-Treated Melanoma Patients. Clin Cancer Res 2015; 21:3140-8; PMID:25609064; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-2023 [DOI] [PubMed] [Google Scholar]
- 32.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012; 13:283-96; PMID:22473468; http://dx.doi.org/ 10.1038/nrm3330 [DOI] [PubMed] [Google Scholar]
- 33.Aguissa-Toure AH, Li G. Genetic alterations of PTEN in human melanoma. Cell Mol Life Sci 2012; 69:1475-91; PMID:22076652; http://dx.doi.org/ 10.1007/s00018-011-0878-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C et al.. A landscape of driver mutations in melanoma. Cell 2012; 150:251-63; PMID:22817889; http://dx.doi.org/ 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B et al.. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PloS One 2011; 6:e28973; PMID:22194965; http://dx.doi.org/ 10.1371/journal.pone.0028973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR et al.. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 2011; 71:2750-60; PMID:21317224; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Euro J Med Chem 2016; 109:314-41; PMID:26807863; http://dx.doi.org/ 10.1016/j.ejmech.2016.01.012 [DOI] [PubMed] [Google Scholar]
- 38.Saran U, Foti M, Dufour JF. Cellular and molecular effects of the mTOR inhibitor everolimus. Clin Sci 2015; 129:895-914; PMID:26330617; http://dx.doi.org/ 10.1042/CS20150149 [DOI] [PubMed] [Google Scholar]
- 39.Deuker MM, Marsh Durban V, Phillips WA, McMahon M. PI3′-kinase inhibition forestalls the onset of MEK1/2 inhibitor resistance in BRAF-mutated melanoma. Cancer Discov 2015; 5:143-53; PMID:25472943; http://dx.doi.org/ 10.1158/2159-8290.CD-14-0856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marsh Durban V, Deuker MM, Bosenberg MW, Phillips W, McMahon M. Differential AKT dependency displayed by mouse models of BRAFV600E-initiated melanoma. J Clin Invest 2013; 123:5104-18; PMID:24200692; http://dx.doi.org/ 10.1172/JCI69619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X et al.. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov 2016; 6:202-16; PMID:26645196; http://dx.doi.org/ 10.1158/2159-8290.CD-15-0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009; 460:108-12; PMID:19543266; http://dx.doi.org/ 10.1038/nature08155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Wang XY, Subjeck JR, Shrikant PA, Kim HL. Temsirolimus, an mTOR inhibitor, enhances anti-tumour effects of heat shock protein cancer vaccines. Br J Cancer 2011; 104:643-52; PMID:21285988; http://dx.doi.org/ 10.1038/bjc.2011.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scotta C, Esposito M, Fazekasova H, Fanelli G, Edozie FC, Ali N, Xiao F, Peakman M, Afzali B, Sagoo P et al.. Differential effects of rapamycin and retinoic acid on expansion, stability and suppressive qualities of human CD4(+)CD25(+)FOXP3(+) T regulatory cell subpopulations. Haematologica 2013; 98:1291-9; PMID:23242600; http://dx.doi.org/ 10.3324/haematol.2012.074088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Sparwasser T, Figlin R, Kim HL. Foxp3+ T cells inhibit antitumor immune memory modulated by mTOR inhibition. Cancer Res 2014; 74:2217-28; PMID:24574514; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2928 [DOI] [PubMed] [Google Scholar]
- 46.Gadiot J, Hooijkaas AI, Deken MA, Blank CU. Synchronous BRAF(V600E) and MEK inhibition leads to superior control of murine melanoma by limiting MEK inhibitor induced skin toxicity. OncoTargets Ther 2013; 6:1649-58; PMID:24348046; http://dx.doi.org/ 10.2147/OTT.S52552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, Mullins DW, Brinckerhoff CE. Multiple murine BRaf(V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res 2014; 27:495-501; PMID:24460976; http://dx.doi.org/ 10.1111/pcmr.12220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME et al.. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014; 124:2246-59; PMID:24667641; http://dx.doi.org/ 10.1172/JCI73639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Long GV, Wilmott JS, Haydu LE, Tembe V, Sharma R, Rizos H, Thompson JF, Howle J, Scolyer RA, Kefford RF. Effects of BRAF inhibitors on human melanoma tissue before treatment, early during treatment, and on progression. Pigment Cell Melanoma Res 2013; 26:499-508; PMID:23557327; http://dx.doi.org/ 10.1111/pcmr.12098 [DOI] [PubMed] [Google Scholar]
- 50.Ribas A, Wolchok JD. Combining cancer immunotherapy and targeted therapy. Curr Opin Immunol 2013; 25:291-6; PMID:23561594; http://dx.doi.org/ 10.1016/j.coi.2013.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minor DR, Puzanov I, Callahan MK, Hug BA, Hoos A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res 2015; 28:611-2; PMID:25996827; http://dx.doi.org/ 10.1111/pcmr.12383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M et al.. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Eng J Med 2015; 372:2521-32; PMID:25891173; http://dx.doi.org/ 10.1056/NEJMoa1503093 [DOI] [PubMed] [Google Scholar]
- 53.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ et al.. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Eng J Med 2014; 371:1877-88; PMID:25265492; http://dx.doi.org/ 10.1056/NEJMoa1406037 [DOI] [PubMed] [Google Scholar]
- 54.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568-71; PMID:25428505; http://dx.doi.org/ 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol 2009; 9:324-37; PMID:19390566; http://dx.doi.org/ 10.1038/nri2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ribas A, Butler M, Lutzky J, Lawrence DP, Robert C, Miller W et al.. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. ASCO Meeting Abstracts 2015; 33:3003 [Google Scholar]
- 57.Ribas A, Hodi FS, Lawrence D, Atkinson V, Starodub A, Carlino MS et al.. Pembrolizumab (pembro) in combination with dabrafenib (D) and trametinib (T) for BRAF-mutant advanced melanoma: Phase 1 KEYNOTE-022 study. ASCO Meeting Abstracts 2016; 34:3014 [Google Scholar]
- 58.Sweetlove M, Wrightson E, Kolekar S, Rewcastle GW, Baguley BC, Shepherd PR, Jamieson SM. Inhibitors of pan-PI3K Signaling Synergize with BRAF or MEK Inhibitors to Prevent BRAF-Mutant Melanoma Cell Growth. Front Oncol 2015; 5:135; PMID:26137449; http://dx.doi.org/ 10.3389/fonc.2015.00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diken M, Kreiter S, Vascotto F, Selmi A, Attig S, Diekmann J, Huber C, Türeci Ö, Sahin U. mTOR inhibition improves antitumor effects of vaccination with antigen-encoding RNA. Cancer Immunol Res 2013; 1:386-92; PMID:24778131; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0046 [DOI] [PubMed] [Google Scholar]
- 60.Wang X, Hawk N, Yue P, Kauh J, Ramalingam SS, Fu H, Khuri FR, Sun SY. Overcoming mTOR inhibition-induced paradoxical activation of survival signaling pathways enhances mTOR inhibitors' anticancer efficacy. Cancer Biol Ther 2008; 7:1952-8; PMID:18981735; http://dx.doi.org/ 10.4161/cbt.7.12.6944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res 2004; 64:1140-5; PMID:14871849; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-3259 [DOI] [PubMed] [Google Scholar]
- 62.Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol 2005; 17:133-44; PMID:15611321; http://dx.doi.org/ 10.1093/intimm/dxh194 [DOI] [PubMed] [Google Scholar]
- 63.Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, Welters MJ, van der Burg S, Kapiteijn E, Michielin O et al.. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med 2014; 6:254ra128; PMID:25232180; http://dx.doi.org/ 10.1126/scitranslmed.3008918 [DOI] [PubMed] [Google Scholar]
- 64.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G et al.. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016; 165:35-44; PMID:26997480; http://dx.doi.org/ 10.1016/j.cell.2016.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hooijkaas AI, Gadiot J, van der Valk M, Mooi WJ, Blank CU. Targeting BRAFV600E in an inducible murine model of melanoma. Am J Pathol 2012; 181:785-94; PMID:22796458; http://dx.doi.org/ 10.1016/j.ajpath.2012.06.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.