

Abstract
Acute renal rejection is a major risk factor for chronic allograft dysfunction and long‐term graft loss. We performed a genome‐wide association study to detect loci associated with biopsy‐proven acute T cell–mediated rejection occurring in the first year after renal transplantation. In a discovery cohort of 4127 European renal allograft recipients transplanted in eight European centers, we used a DNA pooling approach to compare 275 cases and 503 controls. In an independent replication cohort of 2765 patients transplanted in two European countries, we identified 313 cases and 531 controls, in whom we genotyped individually the most significant single nucleotide polymorphisms (SNPs) from the discovery cohort. In the discovery cohort, we found five candidate loci tagged by a number of contiguous SNPs (more than five) that was never reached in iterative in silico permutations of our experimental data. In the replication cohort, two loci remained significantly associated with acute rejection in both univariate and multivariate analysis. One locus encompasses PTPRO, coding for a receptor‐type tyrosine kinase essential for B cell receptor signaling. The other locus involves ciliary gene CCDC67, in line with the emerging concept of a shared building design between the immune synapse and the primary cilium.
Keywords: basic (laboratory) research/science, genetics, immunosuppression/immune modulation, kidney transplantation/nephrology, biomarker, genomics, immunogenetics, microarray/gene array, rejection: T cell mediated (TCMR)
Short abstract
A genome‐wide association study strongly implicates B cell tyrosine kinase PTPRO and ciliary gene CCDC67 in acute renal allograft rejection.
Abbreviations
- BCR
B cell receptor
- CNI
calcineurin inhibitor
- GWAS
genome‐wide association study
- IRB
institutional review board
- LD
linkage disequilibrium
- MAF
minor allelic frequency
- MM
mismatch
- OR
odds ratio
- SNP
single nucleotide polymorphism
- TCMR
T cell–mediated rejection
Introduction
Acute rejection of renal allograft remains a major risk factor for the later development of chronic allograft dysfunction and long‐term graft loss 1. Nonadherence to therapy, HLA mismatches (MM), anti‐HLA immunization, longer period of dialysis before transplantation, younger age, and prolonged cold ischemia time are recognized risk factors of acute renal rejection 2. Besides these classical immunological risk factors, genetic recipient background is likely to modulate the risk of acute rejection. Immune responses involved in the acute rejection process, mediated by T and B lymphocytes, are regulated through a complex, highly regulated network of molecular signals controlled by a large number of encoding genes, among which some could represent a potential candidate that could be associated with an increased alloreactivity.
Numerous association studies of candidate genes have been reported in renal transplantation since the year 2000, and dealt mainly with single nucleotide polymorphism (SNPs) in genes encoding cytokines, chemokines, complement, Toll‐like receptors, and VEGF 3. These studies produced conflicting results, and were prone to false positive, spurious association findings because of inadequate sample size, population stratification, and lack of replication in independent cohorts. To date, no genetic locus has clearly emerged as a risk or as a protection factor for acute rejection of solid organ allograft.
In spite of the coming of age of whole genome sequencing, genome‐wide association studies (GWAS) using arrays of SNPs remain a powerful approach to identify novel genes or loci by analyzing millions of genetic variants, with no a priori hypothesis on gene function, allowing for the discovery of previously unthought‐of pathways. Studying genetic susceptibility of acute rejection is particularly complex. First, acute rejection is not a disease but a complication resulting from alloreactivity that is modulated by factors from the recipient, the donor, and by immunosuppressive therapies. Second, apart from a case–control retrospective study suggesting a trend for familial aggregation in recipients with acute rejection, there is no report from families where multiple members with renal failure received a kidney transplant 4. As transplantation is rarely familial, the existence of some major, Mendelian or near‐Mendelian, genetic factor predisposing to graft rejection would remain practically unnoticed as a hereditary phenotype. In the absence of evidence against such a major gene effect(s), we hypothesized that one or several genetic variants could confer a high relative risk of graft rejection, but no significant risk for disease outside the frame of transplantation, with a relative risk high enough for this gene(s) variant(s) to be amenable to a GWAS with suitable cohorts of transplanted patients. If this hypothesis is true, finding this gene(s) variant(s) would be an important milestone.
Here, we gathered two large European cohorts of kidney transplant recipients, and report the first GWAS of biopsy‐proven acute rejection occurring within the first year after transplant in low‐immunological risk white patients receiving a first renal allograft.
Materials and Methods
Patients
Discovery cohort
We have collected DNA samples and clinical data from a total of 4127 patients transplanted in eight European renal transplant centers (Belgium: ULB‐Hôpital Erasme‐Brussels; France: CHU Tours, CHU Limoges, CHU Brest, CHU St‐Etienne, CHRU Lille, CHU Poitiers, and CHU Bordeaux) with written informed consent and institutional review board (IRB) approval (protocol number: P2007/106), and centralized them at the ULB‐Hôpital Erasme. Among these, we selected white adults (≥18 years) having received a first renal transplantation with induction (anti‐lymphocyte serum or monoclonal IL‐2 receptor antagonist antibody), and calcineurin inhibitor (CNI) therapy at baseline. Exclusion criteria were as follows: the presence of another solid organ transplant, the presence of anti‐HLA antibodies (Luminex®, Austin, TX) or a maximal panel reactive antibody ≥5%, a follow‐up period shorter than 1 year (if the cause was not related to graft loss due to rejection), and lack of DNA or clinical data available. Cases were defined as patients who developed at least one biopsy‐proven acute T cell–mediated rejection (TCMR), defined by BANFF criteria, during the first year after transplantation 5. Patients with episodes of pure antibody‐mediated rejection, untreated borderline or unexpected rejection (discovered in a protocol biopsy) were not eligible. Controls were defined as patients with neither acute nor chronic rejection—defined on the basis of a stable graft function (rise in serum creatinine between 6 and 12 months < 20%) and absence of significant proteinuria (<0.5 g/day or negative urinary dipstick at 12 months)—during the same period. Most participating centers did not perform systematic protocol biopsies; hence most controls were not biopsied. Among those, we selected for each case two center‐matched hypercontrols (graft recipients who did not present acute rejection in spite of an initially less favorable HLA match) with the highest possible number of HLA mismatches in the order: 2×DR > 1×DR, 2×B > 1×B, 2×A > 1×B, 1×A mismatches. Patients older than 55 years receiving antilymphocyte serum at baseline (n = 72) were not considered as hypercontrols, as they were felt to be at lower risk of developing acute rejection. A total of 328 cases and 588 hypercontrols were eligible in the database. After exclusion of patients with DNA of poor quality, 275 cases and 503 hypercontrols transplanted between 1986 and 2010 were genotyped (Figure 1).
Figure 1.

Discovery cohort: flowchart of patients included. *Death, lost to follow‐up, graft loss for another reason than rejection. **Not biopsy‐proven, protocol biopsy, borderline not treated, or pure humoral rejection. CNI, calcineurin inhibitor; MM, mismatch; PRA, panel reactive antibody; Tx, transplantation.
Replication cohort
DNA samples and clinical data were collected with written informed consent and IRB approval (protocol number: P2007/106), from 2765 patients transplanted in two renal transplantation centers (Belgium: KUL Leuven, n = 1068; Czech Republic: IKEM Prague, n = 1697). Inclusion criteria for cases and controls were the same as for the discovery cohort, except for the requirement of induction therapy. We did not select hypercontrols for replication. This resulted in the selection of 333 cases and 593 controls in the database. A total of 313 cases and 531 controls transplanted between 1984 and 2011, with a genotyping rate >90% were included in the association analyses (Figure 2).
Figure 2.
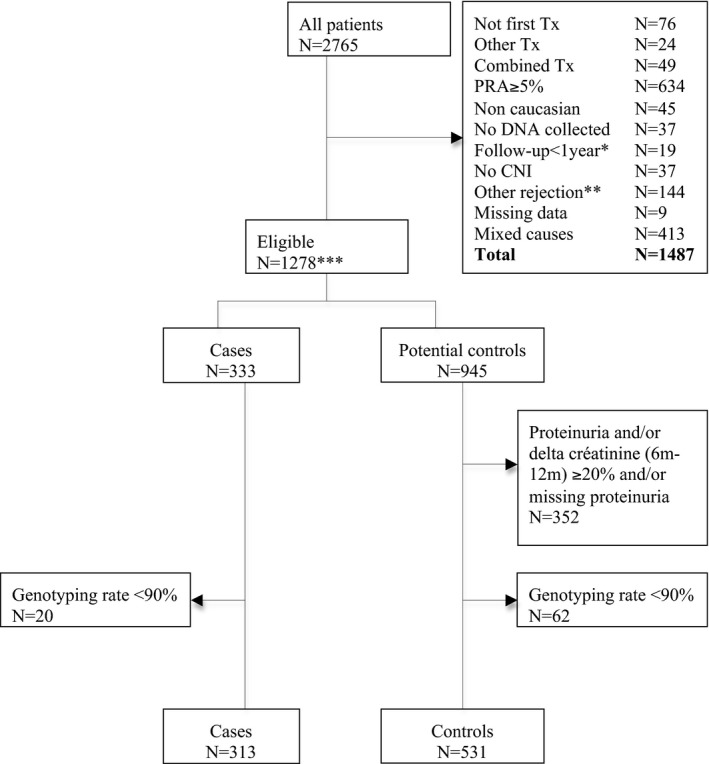
Replication cohort: flowchart of patients included. *Death, lost to follow‐up, graft loss for another reason than rejection. **Not biopsy‐proven, protocol biopsy, borderline not treated, or pure humoral rejection. ***Including 951 patients without induction. CNI, calcineurin inhibitor; PRA, panel reactive antibody; Tx, transplantation.
Genotyping
Discovery cohort
Genomic DNA was extracted using standard procedures. DNA quality was assessed for all samples by agarose gel electrophoresis, and samples with degraded DNA were excluded. DNA concentrations were estimated by fluorometry using Picogreen® (Invitrogen, Carlsbad, CA), as the average of three independent measurements with a coefficient of variation <0.10. Equimolar case and hypercontrol pools were generated by mixing 60 ng of DNA from each of the 275 cases and 503 hypercontrols, respectively. Pools were generated in triplicate, yielding three case (CA, CB, CC) and three hypercontrol (HA, HB, HC) pools. DNA (250 ng) from each pool was hybridized on Human Omni 2.5‐4 v1 DNA analysis BeadChip arrays® (Illumina, San Diego, CA). A (CA and HA) and B (CB and HB) pools were hybridized in duplicate, yielding five measurements for both cases and hypercontrols. Allelic frequencies in the pools were estimated from the B‐allele frequencies computed with Genome Studio® (Illumina). We genotyped the 778 DNA samples individually for nine unlinked SNPs (rs11543947, rs2279804, rs17421009, rs2476601, rs3087243, rs3087456, rs7528684, rs4839469, and rs10804682) using Taqman SNP assays® as recommended by the manufacturer (Applied Biosystems, Foster City, CA) to evaluate the accuracy of the B‐allele frequency estimates over a range of allelic frequencies.
Replication cohort
Genomic DNA was extracted and quantified using standard procedures. A total of 313 cases and 531 controls were genotyped (genotyping rate > 90%) individually for 18 SNPs selected for highest difference of B allelic frequency between cases and hypercontrols at loci identified as significant in the discovery cohort, using a Sequenom Mass Array iPLEX assay® (San Diego, CA). We genotyped at least three SNPs per locus. In addition, we genotyped SNP rs10846175 because the difference in B allele frequency was very high (0.21) in the discovery cohort, despite the fact that the variance of the allele frequency estimates was >0.001 for cases and hypercontrols. Three SNPs with a call rate <90% were excluded from the analysis, leaving the other 15 SNPs eligible for the analyses.
Association analyses
Power calculation
Considering a rejection prevalence of 15% during the first year, the sample size of this two‐stage GWAS (cases, n = 588 and controls, n = 1034) has a theoretical power of ≥80% to identify TCMR alleles with relative risks of 2.4, 1.63, 1.59, and 1.62 for minor allele frequencies (MAFs) of 0.05, 0.2, 0.3, and 0.5, respectively, under an additive genetic model (CATS calculator) 6, 7.
Significance of associated SNP clustering
Categorical data were analyzed using Pearson's χ2 or Fisher's exact tests as appropriate. t‐test or Mann–Whitney test were used to compare normally or non‐normally continuous data. A bilateral p‐value smaller than 0.05 was used to reject the null hypothesis except for SNPs variables. First, we performed a univariate analysis, evaluating the association between the selected SNPs and acute rejection in the replication cohort using PLINK v1.07 (http://pngu.mgh.harvard.edu/purcell/plink/ 8). We estimated the statistical significance of the association from permutations performed within cohorts (respectively Leuven and Prague) to account for potential stratification. We applied a one‐sided test by imposing that the difference in allelic frequency between cases and controls in the replication cohort would have the same sign as in the discovery control. Second, the association of SNPs with acute rejection after adjustment for other risk factors was assessed by multivariate logistic regression modeling. The model was constructed by progressively adding independent variables starting with those that had the strongest univariate association with the outcome of interest. In case of strong linkage disequilibrium (LD) between significant SNPs, only one SNP was included in the logistic regression to avoid co‐linearity problems. The Wald test was used to test the null hypothesis of a log odds ratio (OR) (coefficient) equal to zero. The Hosmer and Lemeshow test was used to check the goodness‐of‐fit of the model. A likelihood ratio test was used to assess whether adding a new variable to the model increased the overall log‐likelihood. To test for a potential interaction between two risk factors, we calculated stratum‐specific ORs and tested the null hypothesis of no difference between stratum‐specific ORs by a χ2 test of homogeneity.
Results
A pool‐based GWAS reveals five candidate risk loci for acute renal graft rejection
From an initial cohort of 4127 patients having undergone a first renal transplantation, we selected 275 cases with acute TCMR within 1 year, and 503 hypercontrols without TCMR, despite being at higher risk of rejection using the specific criteria outlined above (Figure 1). Baseline characteristics of the ensuing case–control cohort are reported in Table 1. As expected from our study design, hypercontrols had a significantly higher number of HLA mismatches than cases, in particular HLA‐DR mismatches (p < 0.0001). The proportion of patients under steroids at 6 months was higher in cases, as a consequence of acute rejection occurrence (p < 0.0001). Donors were significantly older in cases (p = 0.035). The other characteristics were well balanced between the two groups.
Table 1.
Discovery cohort: baseline characteristics of patients (n = 778)
| Characteristics | Cases (N = 275) | Hypercontrols (N = 503) | p |
|---|---|---|---|
| Recipient age: mean ± SD (years) | 48.3 ± 14.1 | 48.6 ± 13.4 | 0.77 |
| Recipient sex (male): n (%) | 179 (65.1) | 331 (65.8) | 0.84 |
| Type of donor (deceased): n (%) | 263 (95.6) | 481 (95.8) | 0.91 |
| Donor age: mean ± SD (years) | 47.2 ± 15.9 | 44.7 ± 15.9 | 0.035 |
| Donor sex (male): n (%) | 148 (54) | 294 (60) | 0.11 |
| Cold ischemia time: mean ± SD (h) | 19.4 ± 7.7 | 18.3 ± 7.9 | 0.07 |
| Dialysis duration: median (P25–P75) (m) | 18 (8.9–36) | 18 (9.5–30.8) | 0.97 |
| Primary nephropathy: n | |||
| Glomerulopathy | 80 | 143 | 0.45 |
| Nephroangiosclerosis/hypertension | 25 | 24 | |
| Polycystic kidney disease | 52 | 107 | |
| Diabetic | 13 | 22 | |
| Chronic interstitial nephropathy | 30 | 54 | |
| Uncertain | 33 | 73 | |
| Other | 18 | 35 | |
| Congenital/hereditary | 22 | 44 | |
| Steroids at 6 months (yes): n (%) | 249 (93.3) | 328 (65.7) | <0.0001 |
| Tacrolimus/cyclosporin: n | 92/183 | 204/299 | 0.05 |
| Induction (thymoglobulin/IL2R antagonist): n | 75/200 | 154/349 | 0.33 |
| HLA‐A MM (0/1/2): n | 38/149/86 | 47/270/186 | 0.09 |
| HLA‐B MM (0/1/2): n | 25/129/119 | 21/215/267 | 0.003 |
| HLA‐DR MM (0/1/2): n | 30/154/88 | 10/272/221 | 0.0003 |
| HLA B+DR MM: mean ± SD | 2.56 ± 0.93 | 2.91 ± 0.71 | <0.0001 |
| HLA A+B+DR MM: mean ± SD | 3.73 ± 1.24 | 4.20 ± 0.95 | <0.0001 |
MM, mismatch; SD, standard deviation.
After very stringent evaluation of DNA quantity and quality, we generated equimolar DNA pools of the 275 cases and 503 hypercontrols in triplicates. The DNA pools were hybridized to arrays interrogating 2.5 million SNPs covering the entire genome, and allele frequencies were computed using Genome Studio® (Illumina). We genotyped the 778 DNA samples individually for nine unlinked SNPs showing a large range of allelic frequency using Taqman SNP assay, to evaluate the accuracy of the B‐allele frequency estimates by Genome Studio software. The global coefficient of correlation (r2) exceeded 0.98, demonstrating the accuracy of our pooling method (Table S1).
We first excluded 42 526 SNPs for which the variance of the allele frequency estimates exceeded 0.001 (i.e. cases or hypercontrols). We then selected 1109 SNPs for which the average allele frequency between cases and hypercontrols differed by ≥0.10. We reasoned that true positive association would tend to involve multiple contiguous SNPs as a result of LD, and used a 50‐kb sliding window to scan the genome for clusters of positive SNPs. We identified five loci encompassing at least six such SNPs in a 50‐kb window. Iterative in silico permutations of our experimental data showed that more than 5 contiguous SNPs were never observed by chance alone in a 50‐kb window (after 100 in silico permutations, Table S2). The corresponding loci were assumed to be highly enriched in true risk loci for acute renal graft rejection.
Two risk loci are replicated by individual SNP genotyping in an independent cohort
From two independent cohorts totaling 2765 patients transplanted in Leuven or Prague, we selected 333 cases with biopsy‐proven acute TCMR and 593 matched controls. A total of 313 cases (Belgian cohort, n = 116; Czech cohort, n = 197) and 531 controls (Belgian cohort, n = 212; Czech cohort, n = 319) with a genotyping rate >90% were eligible for association analyses (Figure 2).
Baseline characteristics of patients are reported in Table 2. As observed in the discovery cohort, donors were older in patients with acute rejection (p = 0.0006). Cases had significantly higher numbers of HLA mismatches, in particular HLA‐DR mismatches (p < 0.0001). The proportion of patients under tacrolimus was higher in cases (p = 0.03). Other characteristics were well balanced between groups.
Table 2.
Replication cohort: baseline characteristics of patients (n = 844)
| Characteristics | Cases (N = 313) | Controls (N = 531) | p |
|---|---|---|---|
| Recipient age: mean ± SD (years) | 51.3 ± 13.2 | 52 ± 12.9 | 0.44 |
| Recipient sex (male): n (%) | 205 (65.5) | 350 (65.9) | 0.90 |
| Type of donor (deceased): n (%) | 268 (85.6) | 457 (86.1) | 0.86 |
| Donor age: mean ± SD (years) | 50 ± 14.3 | 46.4 ± 14.8 | 0.0006 |
| Donor sex (male): n (%) | 166 (53.2) | 299 (56.5) | 0.35 |
| Cold ischemia time: mean ± SD (h) | 14.3 ± 6.8 | 14.4 ± 6.8 | 0.90 |
| Dialysis duration: median (P25–P75) (m) | 24 (12–41.8) | 23 (11.5–38) | 0.24 |
| Primary nephropathy: n | |||
| Glomerulopathy | 95 | 177 | 0.34 |
| Nephroangiosclerosis/hypertension | 25 | 29 | |
| Polycystic kidney disease | 49 | 107 | |
| Diabetic | 28 | 34 | |
| Chronic interstitial nephropathy | 30 | 50 | |
| Congenital/hereditary | 48 | 84 | |
| Uncertain | 22 | 27 | |
| Other | 16 | 23 | |
| Steroids at 6 m (yes): n (%) | 276 (92.3) | 488 (92.1) | 0.91 |
| Tacrolimus/cyclosporin: n | 251/62 | 390/141 | 0.03 |
| Induction therapy: n (%) | 95 (30.4) | 130 (24.5) | 0.06 |
| HLA‐A MM (0/1/2): n | 37/175/98 | 94/285/146 | 0.06 |
| HLA‐B MM (0/1/2): n | 38/162/110 | 67/286/172 | 0.72 |
| HLA‐DR MM (0/1/2): n | 83/166/61 | 203/273/48 | <0.0001 |
| HLA B+DR MM: mean ± SD | 2.2 ± 1.0 | 1.9 ± 0.9 | 0.0002 |
| HLA A+B+DR MM: mean ± SD | 3.4 ± 1.3 | 3.00 ± 1.3 | 0.0001 |
MM, mismatch; SD, standard deviation.
We individually genotyped all samples from the replication cohort using a Sequenom Mass ARRAY iPLEX assay interrogating 18 SNPs including at least 3 SNPs for each of the 5 selected loci. SNPs with a call rate <90% (n = 3) and individuals with a genotyping rate <90% were excluded (n = 63). From 15 SNPs with a genotype rate ≥90%, 14 did not deviate significantly from Hardy–Weinberg equilibrium (p ≥ 0.05), and were retained for further analysis. We first performed an association analysis under an additive model using Plink. We estimated the statistical significance of the observed association by permutations. Permutations were performed within cohorts to account for possible stratification that might differentiate the Belgian and Czech cohorts. Two SNPs replicating with a nominal p‐value <0.05 were excluded because the difference in allelic frequency (between cases and controls) in the replication cohort did not have the same sign as in the discovery cohort (chr5: rs2416500 and chr5: rs6859254). Three SNPs in two from the five loci replicated with nominal p‐value ≤0.05: rs10765602 (p = 0.007) on chr11:93048165, rs10846175 (p = 0.007) and rs7976329 (p = 0.004) on chr12:15584624, and chr12:15602639, respectively. They remained significantly associated with TCMR after Sidak correction (Table 3). Genotype distribution of rs10765602 and rs7976329 in cases and controls is reported in Table S3.
Table 3.
Replicated SNPs: corresponding MAF in the discovery cohort and univariate analysis in the replication cohort
| SNP | Discovery cohort | Replication cohort | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr | SNP | Position | Minor allele | MAF in cases | MAF in CTRLS | Delta MAF | MAF in cases | MAF in CTRLS | Delta MAF | OR | 95% CI | pa | pb |
| 5 | rs182190 | 70840233 | A | 0.41 | 0.56 | −0.15 | 0.43 | 0.44 | −0.02 | 0.94 | 0.77–1.14 | 0.333 | 0.868 |
| 5 | rs277978 | 70926559 | G | 0.45 | 0.54 | −0.09 | 0.42 | 0.44 | −0.02 | 0.92 | 0.76–1.13 | 0.153 | 0.564 |
| 5 | rs2416500 | 117376303 | G | 0.36 | 0.2 | 0.16 | 0.19 | 0.25 | −0.05 | 0.73 | 0.57–0.93 | 0.007 | 0.033 |
| 5 | rs10079827 | 117424611 | C | 0.41 | 0.27 | 0.14 | 0.23 | 0.26 | −0.02 | 0.88 | 0.69–1.11 | 0.163 | 0.589 |
| 5 | rs6859254 | 117438003 | G | 0.34 | 0.22 | 0.12 | 0.19 | 0.24 | −0.04 | 0.77 | 0.60–0.98 | 0.017 | 0.081 |
| 11 | rs10765602 | 93048165 | G | 0.36 | 0.26 | 0.1 | 0.35 | 0.29 | 0.06 | 1.32 | 1.07–1.63 | 0.007 | 0.036 |
| 11 | rs200848508 | 93082760 | G | 0.49 | 0.6 | −0.11 | 0.47 | 0.49 | −0.03 | 0.90 | 0.74–1.10 | 0.144 | 0.541 |
| 11 | rs3020071 | 93105965 | G | 0.42 | 0.52 | −0.1 | 0.45 | 0.47 | −0.02 | 0.91 | 0.75–1.12 | 0.262 | 0.781 |
| 12 | rs1461039 | 15577935 | C | 0.44 | 0.55 | −0.11 | 0.43 | 0.47 | −0.04 | 0.90 | 0.74–1.10 | 0.073 | 0.316 |
| 12 | rs10846175 | 15584624 | G | 0.51 | 0.3 | 0.21 | 0.36 | 0.30 | 0.06 | 0.85 | 0.69–1.03 | 0.007 | 0.037 |
| 12 | rs7976329 | 15602639 | C | 0.49 | 0.31 | 0.18 | 0.37 | 0.30 | 0.06 | 1.30 | 1.06–1.61 | 0.004 | 0.020 |
| 14 | rs1952836 | 28576698 | A | 0.28 | 0.16 | 0.12 | 0.27 | 0.28 | 0.00 | 1.33 | 1.08–1.63 | 0.500 | 0.969 |
| 14 | rs1191395 | 28693834 | G | 0.63 | 0.47 | 0.16 | 0.46 | 0.47 | −0.02 | 0.99 | 0.80–1.25 | 0.184 | 0.639 |
| 14 | rs942630 | 28702660 | A | 0.56 | 0.43 | 0.13 | 0.46 | 0.47 | −0.02 | 0.93 | 0.77–1.15 | 0.333 | 0.868 |
Chr, chromosome; CI, confidence interval; CTRLS, controls; MAF, minor allelic frequency; OR, odds ratio; SNP, single nucleotide polymorphism.
p‐value after permutation (to control for potential stratification for the two subcohorts Leuven and Prague).
p‐value after Sidak correction (for five loci).
We then performed a genotype‐based association test of the two corresponding regions jointly using a multivariate logistic regression analysis including donor age, type of CNI, administration of induction therapy or not, and number of HLA‐DR mismatches as covariates. We only included one SNP per locus in these analyses because of the high LD between the SNP pair mapping to the same locus. Both rs10765602 (p = 0.02) and rs7976329 (p = 0.01) remained significant independent risk factors of TCMR. Genotype‐specific OR suggested a recessive effect of the risk allele for the chr11 locus, and a dominant effect of the risk allele for the chr12 locus (Table 4).
Table 4.
Multivariate logistic regression analysis (n = 829/cases = 309)
| Variable | OR | 95% CI | p |
|---|---|---|---|
| Donor age (increase per year) | 1.02 | 1.01–1.03 | 0.002 |
| Calcineurin inhibitor | |||
| Tacrolimus | 1 | ||
| Cyclosporin | 0.70 | 0.5–1.00 | 0.05 |
| Induction | |||
| No induction | 1 | ||
| Induction | 1.37 | 0.99–1.91 | 0.06 |
| HLA‐DR MM (n) | |||
| 0 | 1 | ||
| 1 | 1.38 | 0.99–1.92 | |
| 2 | 2.88 | 1.80–4.60 | 0.0001 |
| rs10765602 (genotype) | |||
| TT | 1 | ||
| GT | 1.07 | 0.79–1.47 | |
| GG | 1.98 | 1.21–3.25 | 0.02 |
| rs7976329 (genotype) | |||
| TT | 1 | ||
| CT | 1.59 | 1.16–2.17 | |
| CC | 1.61 | 0.96–2.70 | 0.01 |
CI, confidence interval; MM, mismatch; OR, odds ratio.
SNP rs10765602 is located upstream CCDC67 (coiled‐coil domain containing 67) alias DEUP1, a gene involved in centriole biogenesis in multiciliated cells 9.
Variants rs10846175 and rs7976329 are in strong LD (r2 = 0.93) and lie in the first intron of the PTPRO gene‐encoding protein tyrosine phosphatase receptor type O. PTPRO alias Glomerular Epithelial Protein‐1 (GLEPP1) has two major isoforms. The PTPRO‐FL (full‐length form) is a receptor‐type protein tyrosine phosphatase expressed at the apical membrane of the podocyte foot processes. Rare, highly penetrant mutations cause a Mendelian glomerulopathy characterized by a steroid‐resistant childhood‐onset nephrotic syndrome 10. PTPRO‐T (truncated isoform) is encoded by an alternatively spliced form of PTPRO initially found to be expressed in naïve quiescent B cells and memory B cells 11. PTPRO regulates both the amplitude and timing of tyrosine phosphorylation‐based B cell receptor (BCR) signaling events and modulates protein tyrosine kinase‐mediated cellular response. Both Lyn kinase and ZAP‐70 tyrosine kinases are substrates of PTPRO‐T 12, 13.
Discussion
We here report what is to our knowledge the first GWAS of acute rejection in vast numbers of kidney transplant recipients.
In a discovery cohort comparing TCMR and non‐TCMR graft recipients, we identified five candidate loci tagged by a number (more than five) of contiguous SNPs that was never observed by iterative in silico permutations of our experimental data, indicating strong enrichment for true positive signals. In an independent replication cohort, we confirmed the association with two loci. These loci remained independent risk factors in a multivariate analysis integrating significant clinical risk factors. The OR associated with these SNPs was modest, except for rs10765602 where the GG genotype increased the risk of acute rejection by nearly twofold.
The number of renal graft recipient patients is limited and recruiting large cohorts is notoriously difficult. We were hence impelled to include in our replication cohort patients who did not receive induction therapy. This choice may have caused loss of association power in our replication study.
Variants rs10846175 and rs7976329 lie in the first intron of the PTPRO gene. PTPRO would have been an excellent a priori candidate gene for acute renal graft rejection as this gene might modulate alloreactivity through regulation of TCR and BCR signaling and regulation of cytokine production. PTPRO plays several roles at the immune synapse. PTPRO substrate ZAP‐70 is directly involved in TCR signaling and promotes TCR degradation by inducing receptor ubiquitination and targeting to lysosomes 13, 14. PTPRO or ZAP‐70 defects cause immune deficiency. ZAP‐70‐deficient patients have no functional T cells in their peripheral blood and suffer a severe combined immunodeficiency 15. Induced fulminant hepatitis in PTPRO‐knockout (KO) mice showed that PTPRO deficiency led to inflammation attenuation and to a significant decrease in cytokine secretion by both T and natural killer cells leading to a marked impairment of NF‐κB activation 16. The association between PTPRO SNPs and acute rejection was not tighter in the subgroup of patients with a glomerulopathy, excluding the potential association with acute rejection due to stratification only.
Unexpectedly, we found an association of acute TCMR with ciliary gene CCDC67 alias DEUP1 9. Although the GWAS methodology does not demonstrate that the genes at, or near, the associated SNPs are the cause of the association, it allows for a reasonable hypothesis. Lines of evidence indicate tight similarities between the primary cilium and the immune synapse. Indeed, there are important architectural similarities, shared signaling platforms, and common pathways for the two structures, supporting the idea that the immune synapse is derived from the primary cilium 14, 17. The CCDC67 locus association is unlikely to result from stratification of our cohorts for patients whose primary nephropathy was a known or even an unrecognized ciliopathy as there were no imbalances between groups regarding the proportion of glomerulopathies, tubulopathies, or naturally, recognized primary cilium‐related nephropathies (polycystic kidney disease, nephronophthisis, and Bardet‐Biedl syndrome) 18.
We have to acknowledge several limitations of our study. The DNA pooling approach is not as accurate as individual genotyping. However, genotyping using pooled DNA samples allows measurement of allele frequencies at affordable costs and we applied a stringent methodology that minimizes errors 19, 20, 21. First, we pooled high‐quality DNAs with strict quantification in order to ensure that each individual DNA was represented in the same equimolar amount. Second, we obtained a good correlation between B allele frequency estimates by arrays and true B allele frequency calculated by Taqman individual genotyping (r2 > 0.98), similar to previous reports. Third, we minimized pipetting variability by constructing triplicates and batch (array) variability by constructing duplicates. Fourth, in order to reduce the chance of false positive results, we ranked SNPs based on B allele frequency differences and we excluded SNPs with a variance above 0.001. Finally, we considered for replication only loci with 6 or 7 contiguous SNPs, a significant number that was never observed by chance alone after 100 in silico permutations of our experimental data, minimizing the risks of false positive results.
We studied TCMR instead of a more fixed phenotype such as long‐term graft failure. Indeed, TCMR, which is associated with poorer long‐term graft outcome, is closely related to immune causes, whereas graft failure is related to immune as well as nonimmune processes. Likewise, we did not include pure antibody‐mediated acute rejection, which is associated with heterogeneous immunological risk factors and involves different pathways, likely to be associated with different genetic risk factors 22. The timing (TCMR within the first year) is well justified by the fact that most TCMRs occur during the first 3 months, while late acute rejection episodes (after 1 year) are often the consequences of nonadherence.
Some heterogeneity in immunosuppression must be acknowledged, due to the differences in immunosuppression protocols in the centers. First, regarding tacrolimus, the proportion of patients is higher in the replication cohort. This difference might be related to a lower use of induction therapy (100% in the discovery cohort vs. 27% in the replication cohort). The use of tacrolimus (instead of cyclosporine) is likely due to an effort to balance the absence of induction, in order to minimize the risk of acute rejection. Second, regarding steroids, part of the difference is also likely to be due to centers’ practices, with more centers in the discovery cohorts discontinuing steroids at 6 months if no rejection had occurred since transplantation. Along the same line, we must also acknowledge the slight differences with regard to cold ischemia time and dialysis duration. The higher number of HLA MM in controls (discovery cohort) is intentional and related to the selection of hypercontrols. Hypercontrols are control individuals from the lower extremity of the relevant trait distribution and a study design using hypercontrols is a powerful approach in GWAS focusing on one disease 23. Among controls, we have selected recipients with highest number of HLA MM, at theoretically higher risk of acute rejection, in order to maximize the chance to find at‐risk variants. Conversely, in the replication cohort, there was a higher number of HLA MM in cases, because we did not select for hypercontrols.
These differences are unlikely to bias our results. The fact that rs10765602 and rs7976329 were significant in both cohorts and remained independent risk factors in the multivariate analysis (Table 4) strongly supports a causal risk independently of other factors.
Studies in renal transplantation are notoriously limited in the number of available patients with sufficient homogeneity, as opposed to frequent complex traits such as diabetes or hypertension. The present GWAS was therefore not powered to detect frequent alleles conferring a low risk of acute rejection, or low frequency or rare alleles (MAF<0.005). In addition, true associations might have been missed by the exclusion of SNPs, using the stringent quality filters set in our discovery cohort.
In conclusion, the present GWAS addressed the important scientific issue of the genomic basis for immune rejection of the allograft and provides strong evidence for PTPRO, a lymphocyte receptor‐type tyrosine kinase gene and CCDC67, a ciliary gene, being involved in the acute rejection of renal transplants. These novel genes may help in understanding the molecular pathways involved in acute rejection, which may in turn lead to the development of novel antirejection approaches. Furthermore, novel genetic biomarkers that reflect individual susceptibilities to graft rejection could provide the rationale for customized immunosuppression by allowing the pre‐graft identification of low‐ and high‐risk patients.
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting information
Table S1: MAF correlation between individual genotyping and pools (array).
Table S2: Clusters of SNPs: real array data and in silico permutation (100X) data in three sliding windows.
Table S3: Genotype distribution of rs10765602 and rs7976329.
Acknowledgments
Ondřej Viklický received Grants from Czech Ministry of Health and Frame Programs of the European Union: 15‐26865A, NT14102‐3/2013, NT11227‐5/2010, and Bio‐DrIM (EU FP7 program). Lidia Ghisdal received the Astellas European Foundation Transplantation Grant in 2009 and research Grants from Erasme Fund (Université Libre de Bruxelles) for years 2009–2010 and 2010–2011, for this work. Marc Abramowicz was supported by the Transdisciplinary grant of the Erasme Fund and by the Fund for Scientific Medical Research, Belgium (Fonds de la Recherche Scientifique Médicale, FRSM) of the National Fund for Scientific Research FRS‐FNRS, grant T.0174.15, and by the Fund for Scientific Medical Research, Belgium (Fonds de la Recherche Scientifique Médicale, FRSM). Elisa Docampo is supported by a Marie Curie Intraeuropean Fellowship (IEF). We thank Brigitte Borre and Nicole Lietard for data collection. We thank Michaela Prokopova for her invaluable secretarial assistance.
Ghisdal L, Baron C, Lebranchu Y, Viklický O, Konarikova A, Naesens M, Kuypers D, Dinic M, Alamartine E, Touchard G, Antoine T, Essig M, Rerolle JP, Merville P, Taupin JL, Le Meur Y, Grall‐Jezequel A, Glowacki F, Noël C, Legendre C, Anglicheau D, Broeders N, Coppieters W, Docampo E, Georges M, Ajarchouh Z, Massart A, Racapé J, Abramowicz D & Abramowicz M. Genome‐Wide Association Study of Acute Renal Graft Rejection. Am J Transplant 2017; 17: 201–209
References
- 1. Wissing KM, Fomegne G, Broeders N, et al. HLA mismatches remain risk factors for acute kidney allograft rejection in patients receiving quadruple immunosuppression with anti‐interleukin‐2 receptor antibodies. Transplantation 2008; 85: 411–416. [DOI] [PubMed] [Google Scholar]
- 2. Ekberg H, Tedesco‐Silva H, Demirbas A, et al. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med 2007; 357: 2562–2575. [DOI] [PubMed] [Google Scholar]
- 3. Goldfarb‐Rumyantzev AS, Naiman N. Genetic predictors of acute renal transplant rejection. Nephrol Dial Transplant 2010; 25: 1039–1047. [DOI] [PubMed] [Google Scholar]
- 4. Goldfarb‐Rumyantzev AS, Shihab F, Emerson L, et al. A population‐based assessment of the familial component of acute kidney allograft rejection. Nephrol Dial Transplant 2009; 24: 2575–2583. [DOI] [PubMed] [Google Scholar]
- 5. Solez K, Colvin RB, Racusen LC, et al. Banff 07 classification of renal allograft pathology: Updates and future directions. Am J Transplant 2008; 8: 753–760. [DOI] [PubMed] [Google Scholar]
- 6. Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication‐based analysis for two‐stage genome‐wide association studies. Nat Genet 2006; 38: 209–213. [DOI] [PubMed] [Google Scholar]
- 7. Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science 1996; 273: 1516–1517. [DOI] [PubMed] [Google Scholar]
- 8. Purcell S, Neale B, Todd‐Brown K, et al. PLINK: A tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 2007; 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao H, Zhu L, Zhu Y, et al. The Cep63 paralogue Deup1 enables massive de novo centriole biogenesis for vertebrate multiciliogenesis. Nat Cell Biol 2013; 15: 1434–1444. [DOI] [PubMed] [Google Scholar]
- 10. Ozaltin F, Ibsirlioglu T, Taskiran EZ, et al. Disruption of PTPRO causes childhood‐onset nephrotic syndrome. Am J Hum Genet 2011; 89: 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aguiar RC, Yakushijin Y, Kharbanda S, Tiwari S, Freeman GJ, Shipp MA. PTPROt: An alternatively spliced and developmentally regulated B‐lymphoid phosphatase that promotes G0/G1 arrest. Blood 1999; 94: 2403–2413. [PubMed] [Google Scholar]
- 12. Chen L, Juszczynski P, Takeyama K, Aguiar RC, Shipp MA. Protein tyrosine phosphatase receptor‐type O truncated (PTPROt) regulates SYK phosphorylation, proximal B‐cell‐receptor signaling, and cellular proliferation. Blood 2006; 108: 3428–3433. [DOI] [PubMed] [Google Scholar]
- 13. Motiwala T, Datta J, Kutay H, Roy S, Jacob ST. Lyn kinase and ZAP70 are substrates of PTPROt in B‐cells: Lyn inactivation by PTPROt sensitizes leukemia cells to VEGF‐R inhibitor pazopanib. J Cell Biochem 2010; 110: 846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Finetti F, Paccani SR, Rosenbaum J, Baldari CT. Intraflagellar transport: A new player at the immune synapse. Trends Immunol 2011; 32: 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan AC, Kadlecek TA, Elder ME, et al. ZAP‐70 deficiency in an autosomal recessive form of severe combined immunodeficiency. Science 1994; 264: 1599–1601. [DOI] [PubMed] [Google Scholar]
- 16. Jiang R, Chen D, Hou J, et al. Survival and inflammation promotion effect of PTPRO in fulminant hepatitis is associated with NF‐kappaB activation. J Immunol 2014; 193: 5161–5170. [DOI] [PubMed] [Google Scholar]
- 17. de la Roche M, Ritter AT, Angus KL, et al. Hedgehog signaling controls T cell killing at the immunological synapse. Science 2013; 342: 1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tran PV. Dysfunction of intraflagellar transport proteins beyond the primary cilium. J Am Soc Nephrol 2014; 25: 2385–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Macgregor S, Zhao ZZ, Henders A, Nicholas MG, Montgomery GW, Visscher PM. Highly cost‐efficient genome‐wide association studies using DNA pools and dense SNP arrays. Nucleic Acids Res 2008; 36: e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pearson JV, Huentelman MJ, Halperin RF, et al. Identification of the genetic basis for complex disorders by use of pooling‐based genomewide single‐nucleotide‐polymorphism association studies. Am J Hum Genet 2007; 80: 126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bosse Y, Bacot F, Montpetit A, et al. Identification of susceptibility genes for complex diseases using pooling‐based genome‐wide association scans. Hum Genet 2009; 125: 305–318. [DOI] [PubMed] [Google Scholar]
- 22. Noel C, Abramowicz D, Durand D, et al. Daclizumab versus antithymocyte globulin in high‐immunological‐risk renal transplant recipients. J Am Soc Nephrol 2009; 20: 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wellcome Trust Case Control Consortium . Genome‐wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 2007; 447: 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: MAF correlation between individual genotyping and pools (array).
Table S2: Clusters of SNPs: real array data and in silico permutation (100X) data in three sliding windows.
Table S3: Genotype distribution of rs10765602 and rs7976329.