

ABSTRACT
Middle T antigen (MT), the principal oncoprotein of murine polyomavirus, transforms by association with cellular proteins. Protein phosphatase 2A (PP2A), YAP, Src family tyrosine kinases, Shc, phosphatidylinositol 3-kinase (PI3K), and phospholipase C-γ1 (PLCγ1) have all been implicated in MT transformation. Mutant dl1015, with deletion of residues 338 to 347 in the C-terminal region, has been an enigma, because the basis for its transformation defect has not been apparent. This work probes the dl1015 region of MT. Because the region is proline rich, the hypothesis that it targets Src homology domain 3 (SH3) domains was tested, but mutation of the putative SH3 binding motif did not affect transformation. During this work, two point mutants, W348R and E349K, were identified as transformation defective. Extensive analysis of the E349K mutant is described here. Similar to wild-type MT, the E349K mutant associates with PP2A, YAP, tyrosine kinases, Shc, PI3 kinase, and PLCγ1. The E349K mutant was examined to determine the mechanism for its transformation defect. Assays of cell localization and membrane targeting showed no obvious difference in localization. Src association was normal as assayed by in vitro kinase and MT phosphopeptide mapping. Shc activation was confirmed by its tyrosine phosphorylation. Association of type 1 PI3K with MT was demonstrated by coimmunoprecipitation, showing both PI3K subunits and in vitro activity. Nonetheless, expression of the mutants failed to lead to the activation of two known downstream targets of PI3K, Akt and Rac-1. Strikingly, despite normal association of the E349K mutant with PI3K, cells expressing the mutant failed to elevate phosphatidylinositol (3,4,5)-trisphosphate (PIP3) in mutant-expressing cells. These results indicate a novel unsuspected aspect to PI3K control.
IMPORTANCE The gene coding for middle T antigen (MT) is the murine polyomavirus oncogene most responsible for tumor formation. Its study has a history of uncovering novel aspects of mammalian cell regulation. The importance of PI3K activity and tyrosine phosphorylation are two examples of insights coming from MT. This study describes new mutants unable to transform like the wild type that point to novel regulation of PI3K signaling. Previous mutants were defective in PI3K because they failed to bind the enzyme and bring the activity to the membrane. These mutants recruit PI3K activity like the wild type, but fail to elevate the cellular level of PIP3, the product used to signal downstream of PI3K. As a result, they fail to activate either Akt or Rac1, explaining the transformation defect.
KEYWORDS: polyomavirus, transformation, middle T antigen, PI3 kinase, Akt, Rac
INTRODUCTION
Murine polyomavirus causes a wide variety of tumors (1–3). The gene coding for middle T antigen (MT) is the major viral oncogene. It is always necessary for viral transformation and often sufficient to transform cells (see references 4 and 5) for reviews). The MT gene expressed as a transgene causes tumors in a broad range of tissues. Important insights into regulation of both normal and cancer cells have come from its study. Tyrosine phosphorylation (6) and phosphatidylinositol 3-kinase (PI3K) (7) are just two important areas of investigation opened by analysis of MT. Recent work showing the importance of the Hippo effector YAP (8) and protein phosphatase 2A (PP2A) Aβ isoforms (9) to polyomavirus phenotypes demonstrates the continuing value of the system. Studies of the PI3K isoform dependence in a genetically engineered MT model of breast cancer aided the design of clinical trials of p110α isoform-specific PI3K inhibitors (10).
MT has never been shown to possess intrinsic catalytic activity. Instead, it functions by targeting cellular proteins involved in growth regulation (Fig. 1A). Binding of PP2A (11–13) allows recruitment of protein tyrosine kinases (PTKs) of the Src family (Src, Yes, and Fyn) (14–17). In this role, PP2A seems to function as a scaffold, since it does not have to be catalytically active for recruitment of Src (13). Src associated with MT is activated (18) and phosphorylates MT on three major tyrosine sites: residues 315, 322, and 250 (19–22). Each of these sites allows recruitment of an important signaling system: residue 250 to SHC (23, 24) and then to Grb2 and SOS, 315 to PI3K (25, 26), and 322 to phospholipase C-γ1 (PLCγ1) (27). These connections all play a role in transformation. PI3 kinase binding (22) and SHC binding (23, 24) are important for transformation in vitro. An effect of the PLCγ1 binding site at residue 322 is seen under some conditions (27), but not others (28). Minor tyrosine phosphorylations seem also to contribute (29). YAP and TAZ, the effectors of the Hippo signaling pathway, have recently been added to the list of important associations (8, 30, 31). YAP is quite important for MT transformation, since knocking it down or preventing its binding to MT affects transformation. Examination of tumorigenesis in animals shows that different signal transduction pathways may be more or less important, depending on the tissue background (reviewed in references 4 and 5). PI3 kinase is widely important (32). While apparently less important than PI3K, SHC binding (33, 34) is significantly involved in MT tumorigenesis. Binding of 14-3-3 proteins appears to be important only in few tissues (35). A key aspect of MT's transforming ability is its dependence on association with membranes (36). Because of this, it has been compared to an activated growth factor receptor tyrosine kinase (37), with Src family members substituting for the intrinsic kinase activity of the receptors.
FIG 1.

(A) Murine polyomavirus MT showing associated proteins. (B) dl1015 is defective in transformation. Rat-1 fibroblasts were transfected with control vector DNA or DNA expressing either wild-type or dl1015 MT. After 2 weeks, cells were fixed and stained with crystal violet. (C) Wild-type (WT) and mutant MT sequences from residues 336 to 350. (D) MT interactions with the N-terminal SH3 of GRB2. Anti-MT immunoprecipitates from control cells or cells expressing wild-type, E349K, or 2XP MT were labeled in vitro with 32P. MT was then reisolated and incubated with either GST-agarose or Grb2 N-SH3-GST-agarose. Washed precipitates were analyzed by PAGE and autoradiography. (E) SH3 association does not correlate with transformation. Transformation experiments performed as described for panel B compared wild-type MT to dl1015 or 2XP (P338A P341A). Expression is shown by MT blotting of equal amounts of cell protein from each cell type.
The mutant dl1015 was one of the very early mutants of MT defective in transformation (38, 39). This mutant has been an enigma, because it has been unclear whether it is defective in associations of MT known to be important for transformation. The deletion removes 10 residues (residues 338 to 347) (Fig. 1A and C) from a region rich in proline residues. Deletion of just three of these prolines causes defects in transformation and tumorigenesis (40). One hypothesis is that this region of MT is responsible for binding a host target protein that contains a Src homology domain 3 (SH3) domain. SH3 domains have long been recognized to interact with proline-rich regions. Specifically, interactions require the core binding motif of PxxP, where “x” is any amino acid (reviewed in reference 41). The work described here set out to test the SH3 hypothesis by site-directed mutagenesis of MT. As shown below, it seems unlikely that the effect of the dl1015 mutations is related to a defect in SH3 binding. The work uncovered two single mutants, W348R and E349K, which were highly defective in transformation. Here we provide a detailed analysis of the E349K mutant. This mutant associates with all of the proteins known to be important for transformation. This specifically includes PI3 kinase. However, despite PI3K binding and its associated activity, E349K MT fails to elevate phosphatidylinositol (3,4,5)-trisphosphate (PIP3) levels in cells and is unable to activate PI3K-mediated pathways activating the downstream targets Akt and Rac1. The importance of PI3K signaling in cancer can hardly be overstated (see reference 42 for one review). The p110α catalytic subunit is frequently mutated in different cancers, such as colon, breast, prostate, liver, and brain tumors (43). The mutated p110α found in human tumors has been shown to be activating (44–47). Knockout of p110α is sufficient to block MT transformation in mouse embryo fibroblasts (48). The defects we observed for E349K MT are sufficient to explain the transformation defect, but raise new questions about how PI3K signaling is regulated.
RESULTS
MT genetics argues against a role for SH3 interactions and uncovers new point mutants defective in transformation.
dl1015 is known to be transformation defective (38, 39). This is confirmed in a focus formation assay as shown in Fig. 1B and by additional quantitation in Fig. 1E. The dl1015 mutant has always been an enigma, because there has been no consensus on the basis for its transformation defect. The dl1015 deletion impinges on a region of MT that is proline rich (Fig. 1C). SH3 domains are known to bind proline-rich regions. Specifically, interactions utilize the core-binding motif PxxP (41, 49–51). In principle MT residues 338 to 341 could provide such a sequence to associate with one or more SH3 domains. Figure 1D shows that wild-type MT reisolated from immunoprecipitates could associate with the N-terminal SH3 domain of the adaptor Grb2. This is almost certainly an artifact since Grb2 binding is associated with the binding of Shc at pY250 (52). However, it might support the idea that MT associates with some SH3-containing target. Nonetheless, we created a P338A P341A double mutant (2XP) to test the association and its possible function. As shown in Fig. 1D, this 2XP double mutant binds weakly, if at all, to the Grb2 SH3, confirming the role of the prolines in the interaction. However, 2XP seemed to be fully capable of transformation in focus formation assays (Fig. 1E). This argues that function of this region of MT is unlikely to involve canonical SH3 interactions.
Extensive mutation of MT by site-directed mutagenesis did yield two point mutants, W348R and E349K, which were strikingly defective in transformation (Fig. 2A). Because we were concerned about possible structural alterations caused by mutation of W348 to R, we concentrated on the E349K mutant. As shown in Fig. 2B, this mutant is defective in soft agar transformation as well as in focus formation.
FIG 2.
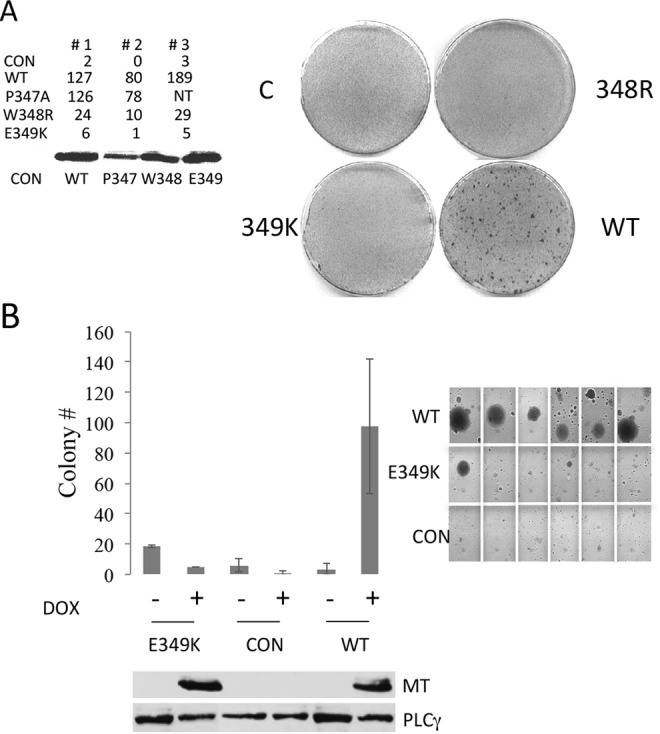
E349K mutant is defective in transformation. (A) Focus formation assays in Rat1 cells show an E349K defect in transformation. Cells were transfected with the empty vector control (CON) or vectors expressing wild-type, P347A, W348R, or E349K MT. MT expression was assessed by Western blotting. Focus formation assays were carried out as described in the legend to Fig. 1B. (B) E349K MT is defective in soft agar assays of transformation. NIH/3T3 cells were exposed to doxycycline for 24 h to induce expression of EGFP, WT MT, or E349K MT, after which the cells were seeded into soft agar in the continued presence of doxycycline. Colonies were counted after 4 weeks. Results are shown on the left. Representative images of individual NIH/3T3 colonies are shown on the right. Control Western blotting of NIH/3T3 cell extract was conducted to detect MT. PLCγ1 served as a loading control.
E349K MT is unaffected in binding known MT targets important for transformation.
The story of MT transformation has been the story of binding to cellular targets that regulate growth. Cells transiently transfected and cell lines expressing E349K have been tested for MT binding to its targets. The left panel of Fig. 3 shows that PP2A, Src, Shc, the PI3K p85 subunit, and YAP all bind to E349K MT in a similar fashion as to wild-type MT in NIH/3T3 cells. For reasons of antibody sensitivity, we used human cells to look for the catalytic subunits of PI3K and PLCγ1 (Fig. 3, right panel). Again E349K MT associated with these targets like wild-type MT.
FIG 3.
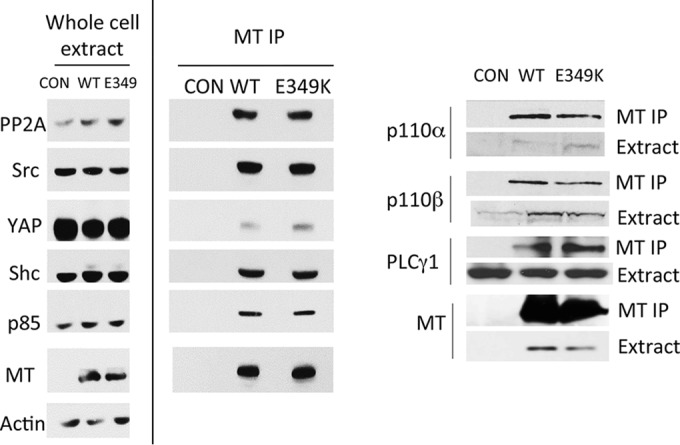
E349K MT associations with cellular proteins. (Left panel) Inducible NIH/3T3 cells were induced to express EGFP (CON), WT MT, or E349K MT by exposure to doxycycline for 24 h. Western blotting of MT immunoprecipitates (IP) shows WT binding of PP2A, Src, YAP, Shc, and PI3K (p85) to E349K MT. Actin was used as a loading control. (Right panel) 293 cells were induced to express EGFP, WT MT, or E349K MT by exposure to doxycycline for 6 h. Cell extracts were made and MT immunoprecipitated. After SDS-PAGE, blots were probed for p110 subunits of PI3K, PLCγ1, and MT.
Given the key role of MT-associated tyrosine kinase activity, it was important to determine whether the association with Src was functional. MT immunoprecipitates from controls or cell lines expressing either wild-type, dl1015, W348R, or E349K MT were tested in in vitro kinase assays. Washed immunoprecipitates were then incubated with [γ-32P]ATP. Figure 4A demonstrates both E349K and W348R mutants were capable of associating with a PTK activity at a level close to or the same as that of wild-type MT.
FIG 4.
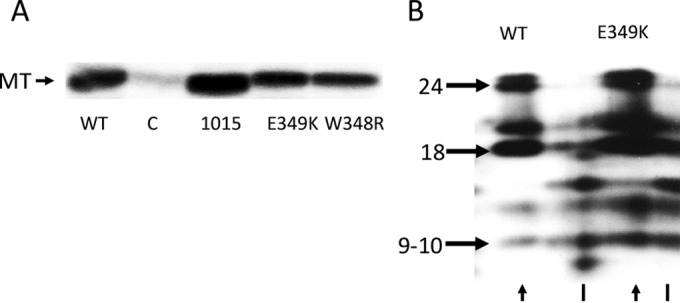
E349K MT associates normally with Src family PTKs. (A) Equal protein amounts of extracts from control cells or stable cell lines expressing either wild-type or mutant MT were immunoprecipitated with an MT-specific antibody. An in vitro kinase assay was performed on washed immunoprecipitates. 32P-labeled proteins were resolved by SDS-PAGE. The gel was treated with alkaline to remove serine and threonine phosphate backgrounds. The image was developed using a Molecular Dynamics PhosphorImager screen. (B) Partial proteolytic digests of E349K MT resulted in the expected phosphopeptides. WT and E349K MT labeled in vitro with 32P were resolved in the first dimension in 7.5% acrylamide cylinders. After the appropriate regions of the cylinders were cut out, digestion was carried out with S. aureus V8 on a second-dimension 12.5% acrylamide gel. After alkaline treatment, that gel was dried and exposed to a Molecular Dynamics PhosphorImager screen. Full-length wild-type and E349K MTs are indicated by the arrowheads. A known shorter MT fragment is indicated by solid lines. The phosphopeptides are indicated by molecular weight.
Association with members of the Src family of PTKs results in phosphorylation of middle T antigen on critical residues, including tyrosines 250, 315, and 322. To determine if 348R and 349K MT association with PTK activity resulted in the tyrosine phosphorylation of the critical middle T antigen residues, the mutant E349K MT in vitro phosphorylations were compared to those of the wild type by partial proteolytic digestion with the Staphylococcus aureus V8 protease (Fig. 4B). After resolution on first-dimension cylindrical gels, the appropriate region was cut out and digested in the second dimension with Staphylococcus aureus V8 protease. The familiar peptide pattern with fragments of molecular weights 24,000, 18,000, and 9,000 to 10,000 was the same as that for the wild type. Digestion with chymotrypsin (not shown) also gave identical fragments for the wild type and E349K mutant. Therefore, the association with Src family kinases appeared to be normal.
Next we turned our attention to PI3 kinase. Since both regulatory 85-kDa and catalytic 110-kDa subunits were associated with E349K MT, enzymatic activity was expected. Figure 5A shows that this was the case. Phosphatidylinositol 3-phosphate (PI3P) product was detected after incubations of either wild-type or E349K MT immunoprecipitates with phosphatidylinositol and [γ-32P]ATP. The initial observation of PI3K association with MT was seen by phosphorylation of the p85 regulatory subunit in in vitro tyrosine kinase reactions (53, 54). Phosphopeptide mapping with chymotrypsin was used to confirm that p85 was modified in the same way in wild-type and E349K immunoprecipitates (Fig. 5B). The overall conclusion from these experiments is that association of E349K MT with PI3 kinase appeared to be normal.
FIG 5.
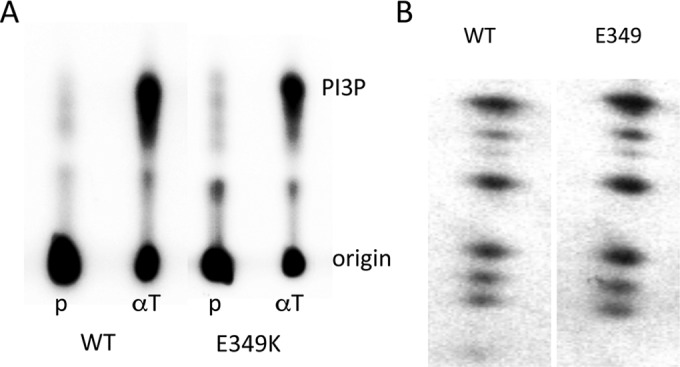
E349K MT association with PI3 kinase. (A) E349K MT has associated PI3 kinase activity. Washed MT immunoprecipitates from cell lines expressing wild-type or E349K MT were analyzed in an in vitro lipid kinase assay. As described in Materials and Methods, phosphatidylinositol was used as the substrate. Phosphorylated lipids were extracted in CHCl3-methanol (1:1) and spotted onto TLC plates. The plates were developed by chromatography in n-propanol–2 N acetic acid (65:35), dried, and exposed to a Molecular Dynamics PhosphorImager screen. (B) PI3 kinase p85 associated with E349K MT is phosphorylated normally. Washed wild-type or E349K MT immunoprecipitates were labeled in an in vitro tyrosine kinase reaction. Complexes were disrupted with SDS and p85 isolated by immunoprecipitation. p85 was analyzed by two-dimensional chymotryptic mapping as described in the legend to Fig. 4.
E349K MT activates Shc signaling normally, but is defective for PI3 kinase signaling.
Activation of Shc in response to signals leads to phosphorylation of Shc on tyrosine residues (55–57). In turn this leads to the recruitment of Grb2 and SOS, leading to Ras activation. Figure 6 shows two experiments confirming that E349K MT is able to activate Shc. In Fig. 6A, antiphosphotyrosine (anti-ptyr) immunoprecipitations were carried out. Comparison of anti-ptyr-precipitated Shc to total Shc showed that both wild-type and mutant MTs lead to ptyr-associated Shc. In Fig. 6B, blotting with phosphospecific antibody directed against Shc pY317 confirmed that E349K MT could signal causing Shc phosphorylation. Figure 6B also shows that induction of E349K MT in NIH/3T3 cells by treatment with doxycycline also increases the level of Erk phosphorylation. This is consistent with the activation of Shc by the mutant MT. The results with Shc also support the conclusion that the assembly and activity of Src in the MT complex are normal.
FIG 6.
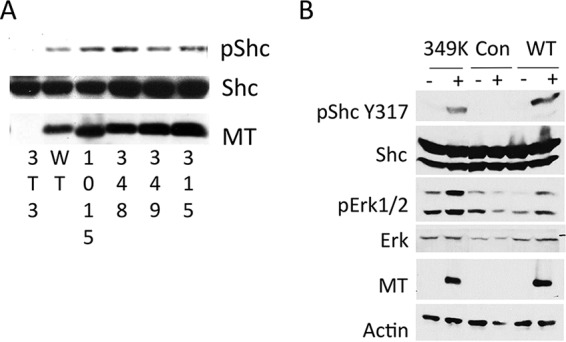
E349K MT activates Shc. (A) Expression of WT and mutant MTs leads to the phosphorylation of Shc. Cell lines expressing either WT or mutant middle T antigen (MT) or normal NIH/3T3 cells were grown under serum-starved conditions for 24 h. Equal amounts of extract were immunoprecipitated with an antibody directed against pTyr (4G10). The washed immunoprecipitates were resolved on a 10% acrylamide gel, along with cell extracts for blotting with total Shc or MT. (B) Expression of WT and mutant MTs leads to the phosphorylation of Shc and Erk. NIH/3T3 cells were induced to express EGFP, WT MT, or E349K MT upon exposure to doxycycline for 24 h, the last 16 h of which took place without serum. Western blotting of whole-cell extracts shows wild-type activation of Shc (Y317) and Erk1/2 (pErk, T202 Y204).
A very different picture emerges when PI3K signaling is examined in vivo. Two principal pathways after PI3K activation lead to the activation of Akt and Rac1. These two targets are clearly relevant for MT. Rac1 is known to be involved in MT activation of c-fos transcription (58). Expression of dominant-negative forms of Rac1 or its downstream effectors blocks PI3K-mediated signaling by MT (59, 60). MT is also an activator of Akt, and this activation is dependent on association with PI3K (61–63). Knockout experiments have confirmed the importance of Akt for MT tumorigenesis (64).
Activation of Akt was tested first. Akt binds PIP3 (or PI-3,4P2) produced by PI3K. Phospholipid binding causes Akt to be relocalized to the plasma membrane, where it undergoes a conformational change, making it more accessible as a substrate (65). Akt is activated by activation loop phosphorylation at Thr308 by PDK1 (66, 67) and by phosphorylation near the C terminus at Ser473 by mTor2 (68). Phosphospecific antibodies can be used to assess Akt activation. Figure 7A shows, as previously demonstrated (61–63), that wild-type MT activates Akt at the 473 site. However, dl1015, W348R, and E349K MTs failed to do so. Figure 7B shows that the wild-type, but not E349K, MT activates Akt at the PDK1 site at T308. To test whether E349K MT might have a dominant-negative effect on Akt activation, constitutively activatable mutant Akt2 or Akt3 was introduced into control and E349K cells. It is clear that both Akt isoforms can be activated as assessed by S473 phosphorylation. When put into soft agar, E349K MT-expressing cells expressing Akt3, but not Akt2, showed significant transforming ability. This indicates that part of the transformation defect in E349K MT is from failure to activate Akt. The failure of Akt2 to give transformation is not surprising, since Akt2 is known to be unimportant for MT tumorigenesis (64).
FIG 7.

The wild-type MT, but not dl1015, W348R, or E349K MT, activates Akt. (A) Control NIH/3T3 cells or lines stably expressing either wild-type or mutant MT were grown under serum-starved conditions for 24 h. Extracts were resolved by PAGE and blotted for pan-serine 473 phosphorylated Akt, total Akt, or MT. (B) E349K MT is defective in both S473 and T308 Akt phosphorylation. Control cells or cells expressing wild-type or E349K MT were treated with doxycycline for 24 h, of which the last 16 h were serum free. Extracts were made and Western blotting was carried out after SDS-PAGE using phosphospecific antibodies for pT308 and pS473 as well as antibody for total Akt and MT. (C) E349K MT does not prevent Akt activation. 3T3 cells stably expressing EGFP (control [CON]) or E349K MT were infected with empty vector (EV) pBABE retrovirus or pBABE retrovirus encoding constitutively activatable Akt2 or Akt3 and selected in puromycin. Cells were induced to express EGFP or E349K MT by exposure to doxycycline for 24 h, the last 16 h of which took place in the absence of serum. Blotting of cell extracts was carried with antibody recognizing S473 phosphorylation, total Akt, PI3K p85 subunit or MT. (D) Activated Akt3 partially restores E349K MT transformation. Cells with or without mutant Akt expression were induced to express EGFP (CON), WT MT, or E349K MT by exposure to doxycycline for 24 h, after which the cells were seeded into soft agar in the continued presence of doxycycline. Colonies were counted after 4 weeks.
The activation of Rac1 was tested next. Activated GTP-bound Rac1, but not GDP-bound Rac1, will bind to a domain on its downstream effector, p21-activated kinase 1 (PAK1). This Rac1 binding domain of PAK can be expressed as a glutathione S-transferase (GST) fusion. Figure 8 shows total Rac from control and MT-expressing cells as well as the activated GTP-bound Rac brought down by GST-PAK beads. It is clear that wild-type MT, but not dl1015, W348R, or E349K MT, stimulates the activation of Rac1.
FIG 8.
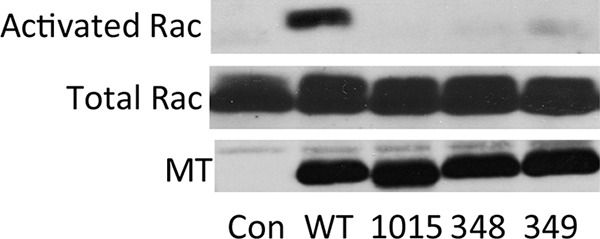
The wild-type MT, but not dl1015, W348R, or E349K MT, activates Rac. Cell lines expressing either WT or mutant middle T antigen (MT) or control NIH/3T3s were grown under serum-starved conditions for 24 h. Equal amounts of extract were incubated with a GST-PAK-Rac1-binding domain fusion, bound to glutathione (GSH) beads. The washed GST pulldowns were resolved on a 10% acrylamide gel, transferred to nitrocellulose, then immunoblotted with an antibody directed against Rac1. For total Rac1 and MT, equal protein concentrations of cell extract were immunoblotted for either Rac1 or MT.
Akt is activated by direct binding of PIP3. Rac is likely to be activated because PIP3 binds to Rac exchange factors such as Tiam1 (69). Enzyme-linked immunosorbent assays (ELISAs) were used to assess the levels of PIP3 in control and MT-expressing cells (Fig. 9A). As shown previously (70), wild-type MT elevates the levels of PIP3 in cells. However, E349K MT does not increase PIP3 levels significantly above those in control cells. A similar conclusion was observed by immunofluorescence for PIP3. Control cells responded to insulin signaling with an elevation of PIP3 (Fig. 9B). Wild-type MT-expressing cells, but not E349K MT-expressing cells showed constitutive PIP3 expression. This defect is sufficient to explain why the mutants activate neither Akt nor Rac1. Since the tumor suppressor PTEN functions to dephosphorylate PIP3 (71, 72), its levels were compared in control or MT-expressing cells (Fig. 9C). No differences were observed.
FIG 9.

E349K cells lack PIP3. (A) 3T3 cells were exposed to doxycycline for 24 h to induce expression of EGFP (CON), wild-type, or E349K MT. The last 16 h of the induction took place in the absence of serum. Lipids were extracted, and PIP3 was measured by ELISA (top). Control Western blotting of duplicate samples shows MT in cell extracts and confirms that E349K MT was not activating Akt (bottom). (B) Control cells and those expressing either wild-type or E349K MT were grown under serum-starved conditions for 24 h. A subset of the controls were treated with insulin (100 ng/ml) for 30 min. Cells were then stained with antibody to PIP3 (Echelon). (C) PTEN levels are unaffected by MT. Induction of MT was carried out as in panel A. Cell extracts were analyzed for PTEN by Western blotting.
Membrane targeting and E349K.
Another possibility is that the E349K MT is somehow mislocalized. PIP3 is largely generated in plasma membranes (reviewed in reference 73). Wild-type MT associates with membranes through a hydrophobic domain at the C terminus (36) well distal to the E349K mutation. Fractionation experiments confirmed that E349K MT is associated with membranes like the wild type (Fig. 10A). We further considered the possibility that the E349K mutation might affect the usual mechanism of membrane association. To do this, we tested whether a surrogate targeting signal could overcome the E349K defect. We took advantage of the observation that MT can be directed to membranes using the H-ras CAAX-box localization signal (74). However, attempts to localize wild-type or E349K MT to membranes by using the H-ras membrane localization sequence failed to restore transformation (Fig. 10B).
FIG 10.
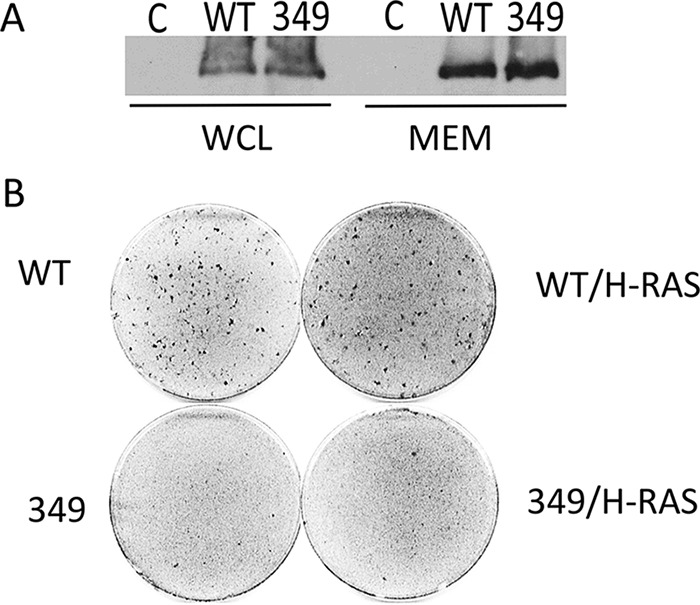
E349K MT and membrane localization. (A) E349K is associated with membranes: NIH/3T3 cells were induced to express EGFP (C) or wild-type or E349K MT by exposure to doxycycline for 24 h. Hypotonic cell fractionation followed by centrifugation was conducted to isolate the membrane fraction. After SDS-PAGE, Western blotting was carried out to detect MT in whole-cell lysates or the membrane fraction. The relative amount of whole-cell lysate was 1/15 that of the purified membranes. (B) Using H-RAS targeting of E349K MT to membranes fails to restore transformation. Rat1 cells were transfected with either wild-type, wild type-CAAX, E349K, or E349K-CAAX MT. Foci were stained as described in the legend to Fig. 1B.
DISCUSSION
This work describes mutations in the proline-rich region of MT. Originally motivated by the transformation-defective mutant dl1015, we tested the hypothesis that the region might be a site for interactions with SH3 domains. While wild-type MT can associate in vitro with the N-terminal Grb2 SH3 domain, mutation of the PXXP at the residue 338–341 motif prevents binding with no effect on the transforming phenotype. We therefore conclude that the SH3 binding observed here in vitro either represents an artifact, or if SH3 binding occurs, it is not relevant to transformation here.
Two point mutants, W348R and E349K, were uncovered in the course of our mutagenesis. Despite not affecting the proline sequences, these mutants were highly defective in transformation. We characterized them, especially the E349K mutant in some detail. As for the wild type, they associate with MT targets known to be important in transformation: PP2A, Src, Shc, PI3 kinase, PLCγ1, and YAP. In vitro activity in the tyrosine kinase assays appeared to be normal, with phosphorylation at the usual sites. This is consistent with the observation that E349K MT associates with Shc, PI3K, and PLCγ1 in vivo. All of these associations are known to be dependent on tyrosine phosphorylation.
The E349K mutant, unlike mutants defective in Src association, retains some biological activity. Shc is phosphorylated normally. This signaling appears to result in Erk activation as well (Fig. 6B). Also, previous work showed that it is necessary to knockout both 250 and 315 binding to abrogate completely the ability of MT to promote cell cycle progression (75). Experiments not shown indicate that E349K MT retains partial ability to promote cell cycle progression in serum-starved cells. This is again consistent with the notion that Shc activation is normal.
Unlike Shc, the situation with PI3K and its signaling was clearly different for E349K MT than for the wild type. E349K MT did not activate Akt measured by T308 or S473 phosphorylation, nor did it enhance the amount of GTP-bound Rac1. The observed failure in PI3 kinase signaling to activate Akt and Rac1 would explain the transformation defect. Superficially, this might have seemed to be like the situation with the Y315F mutant that fails to bind PI3 kinase (26) and therefore to signal to Akt (61). However, the actual situation is quite different. In contrast to the Y315F mutant, the E349K mutant seems to be perfectly capable of recruiting PI3 kinase into MT complexes. Both the α and β isoforms of the p110 catalytic subunit were detected. p85 regulatory subunit binding is shown in Fig. 3. Given all these results, it is not surprising that PI3 kinase enzymatic activity associated with E349K MT appeared to be normal (Fig. 5A). In MT complexes, the p85 subunit is phosphorylated (53, 76). Peptide mapping indicated that E349K phosphorylation in vitro was also normal.
Observations showing that the association of E349K MT with PI3 kinase was normal but that no PI3 kinase signaling seemed to be occurring define an entirely new kind of MT mutant. We considered and ruled out the possibility that E349K MT is somehow dominant negative for PI3 kinase signaling. Experiments with transfection of activated Akt showed that the mutant Akt was phosphorylated in E349K-expressing cells and that transformation was substantially restored.
The most telling result is that E349K MT does not elevate the level of PIP3 in cells in the same way as the wild type (Fig. 9). This was confirmed by immunofluorescence of wild-type and MT-expressing cells (not shown). The lack of PIP3 response would account for the Akt/Rac1 and transformation defects. The question is how could this be possible given that association of PI3 kinase with E349K seems to be normal? One possibility is that E349K mutant MT, but not wild type MT, increases PIP3 phosphatase activity. The tumor suppressor PTEN is well known to downregulate PI3 kinase activity (71, 72). However, PTEN has never been found in association with MT complexes. Nor is there any increase in the amount of PTEN in E349K MT-expressing cells compared to the wild type (Fig. 9B). There is some reason to believe that submembrane localization such as in lipid rafts is important for PI3 kinase signaling to Akt (77). Simple membrane localization does not appear to offer any explanation for the transformation defect. In addition, preliminary characterization suggests that wild-type MT can be found in rafts, but that the E349K mutation does not change this.
Since the MT story has always been that of associations with cellular targets, it seems most likely that the E349K mutant has lost an association that might, for example, affect localization to some specific cell complex. In the recent proteomic work that led to the MT-YAP story, a number of novel interactors were uncovered (8). Detailed proteomics will be needed to determine whether these or other interactions could explain the E349K phenotype.
MATERIALS AND METHODS
Cells.
The normal mouse fibroblast cell line NIH/3T3, normal mouse mammary gland epithelial cell line NMuMG, normal rat fibroblast cell line Rat-1, and human 293, 293T, 293FT, and 293 Phoenix cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA). NIH/3T3 cells were propagated in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO) supplemented with 10% calf serum (CS; Invitrogen, Grand Island, NY). NMuMG, Rat-1, 293T, 293FT, and 293 Phoenix cells were propagated in DMEM supplemented with 10% fetal bovine serum (FBS; Invitrogen). All media were supplemented with 1% glutamine and 1% penicillin-streptomycin (Invitrogen). 293FT cells were propagated in the presence of 500 μg/ml G418 (Invitrogen). All cells were propagated at 37°C in a humidified atmosphere composed of 5% CO2.
Constructs and stable expression.
Plasmid for wild-type MT expressed from the cytomegalovirus (CMV) promoter has been described previously (35). Mutants were constructed by site-directed PCR mutagenesis using the following oligonucleotides:
Dl 1015 (5′ GTA CCC CAG CTC ATC CCA CCA TGG GAG GGT CGT ATT CTT 3′ and 5′ CCG AAG AAT CAG ACC CTC CCA TGG TGG GAT GAG CTG GGG TAC 3′), P338A/P341A (2XP) [5′ AGC TCA TCC CCC CC(G/C) CTA TCA TT(G/C) CCA GGG CGG GTC TG 3′ and 5′ CAG ACC CGC CCT GG(G/C) AAT GAT AG(G/C) GG GG GAT GAG CT 3′], P347A (5′ GCG GGT CTG AGT GCA TGG GAG GGT C 3′ and 5′ G ACC CTC CCA TGC ACT CAG ACC CG), W348R (5′ GGT CTG AGT CCA CGG GAG GGT CTG 3′ and 5′ CAG ACC CTC CCG TGG ACT CAG ACC 3′), and E349K (5′ CTG AGT CCA TGG AAG GGT CTG ATT 3′ and 5′ G AAT CAG ACC CTT CCA TGG ACT CAG 3′).
MT–H-Ras chimeras were created with the oligonucleotides 5′ GCT CAT TCA ATG TCC GGC TGC ATG AGC TGC AAG TGT GTG CTC TCC TGA GGA TCC GCG 3′ and 5′ CGC GGA TCC TCA GGA GAG CAC ACA CTT GCA GCT CAT GCA GCC GGA CAT TGA ATG AGC 3′.
The resulting fusion creates a chimera with the first 383 amino acids of MT, a single serine spacer, and the 10 amino acids from the C terminus of H-ras that is responsible for membrane localization.
pBABE vectors expressing constitutively activatable Akt2 and Akt3 were a gift from Philip Tsichlis.
Cell lines that stably expressed the CMV wild type or MT mutants were made by cotransfection of the desired CMV expression plasmid and pBabe puro (10:1 ratio of expression vector to selection vector) with HEPES-buffered saline (HBS)/calcium chloride cotransfection. After 48 h, cells were placed in selection medium (10% CS-DMEM with 5 μg/ml puromycin). At approximately 2 weeks posttransfection, colonies were isolated and amplified to test for protein expression.
pInducer 20 constructs for inducible expression of wild-type MT and wild-type MT that is hemagglutinin (HA) and FLAG tagged at the C terminus have been described previously (8). pInducer MT-expressing cells were isolated after lentivirus infection and G418 selection as described previously (8).
Transformation assays. (i) Focus formation.
Focus-forming assays in Rat-1 fibroblasts or mouse NIH/3T3 cells were performed after transfection with HBS/calcium chloride or FuGENE 6. On the third day, one plate of the three was harvested, and extracts were used to check protein expression; the other two plates were maintained until foci were readily detectable (10 days to 2 weeks). Plates were then fixed in 3.7% formaldehyde–phosphate-buffered saline (PBS) for 30 min. at room temperature, and stained with 0.1% crystal violet in 3.7% formaldehyde–PBS.
(ii) Soft agar transformation.
NIH/3T3 or NMuMG cells containing pInducer constructs were induced to express MT protein by exposure to doxycycline for 24 h prior to being suspended in agar. Doxycycline was maintained in the agar throughout the duration of the assay. The assay was set up in 6-well plates. The bottom of each well was coated with 1 ml propagation medium containing 0.6% agar. Once the bottom coat was polymerized, cells (1 × 105) were suspended in 1 ml propagation medium containing 0.4% agar. Colonies were allowed to form for 3 to 4 weeks. At the completion of the assay, colonies were imaged using an AlphaImager (Alpha Innotech, San Jose, CA) and quantified using the open source software ImageJ (78).
Antibodies.
The antibody to PP2A A subunit recognizing both Aα and Aβ was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The antibodies to Src, Shc, phospho-Shc, phospho-Akt, phospho-MEK, phospho-extracellular signal-regulated kinase (phospho-ERK), YAP, phospho-YAP, and TAZ were purchased from Cell Signaling Technology (Danvers, MA). MT-1 is an antipeptide antibody recognizing MT between residues 282 and 300, a region known to be dispensable for virus growth and transformation (79); monoclonal antibody (MAb) PN116 recognizing the domain common to large T antigen (LT), MT, and small t antigen (ST) was described previously (80). The polyclonal antibody to p85, the PI3K regulatory subunit, used in this study was generated in our laboratories. FLAG-tagged proteins were immunoprecipitated using anti-FLAG resin (Sigma-Aldrich). The anti-FLAG antibody used for Western blotting was purchased from Clontech (Mountain View, CA). HA-tagged proteins were immunoprecipitated with anti-HA resin (Sigma-Aldrich). The anti-HA antibody used for Western blotting was purchased from Covance (Princeton, NJ). Anti-Rac-1 was from Upstate Biotechnologies.
Immunoprecipitation and Western blotting.
MT was immunoprecipitated using MT-1, HA-tagged proteins were immunoprecipitated using anti-HA resin, and FLAG-tagged proteins were immunoprecipitated using anti-FLAG resin according to methods published previously (81). SDS-PAGE (10%) (82) was used. Western blotting was carried out by standard methods (83). The procedure for recovering MT or PI3K p85 from immunoprecipitates was described previously (84).
In vitro protein kinase reactions.
Immunoprecipitates were washed and resuspended in kinase buffer (20 mM Tris-HCI [pH 7.5], 5 mM MnCl2), and then incubated in 200 μl kinase buffer and 50 μCi [γ-32P]ATP (2,000 Ci/mmol)) for 15 min at room temperature. At the end of the incubation, the beads were washed three times with PBS and once with water and boiled in SDS dissociation buffer.
Proteolytic mapping.
Partial proteolysis has been described previously (21). Immunoprecipitated samples were electrophoresed on a 7.5% acrylamide cylindrical gel. Relevant regions for MT or PI3K p85 were excised and placed horizontally onto 12.5 to 15% acrylamide gels and overlaid with 2 ml of enzymatic digestion solution (0.0125 M Tris-HCl [pH 6.8], 0.01 M EDTA, 20% glycerol [vol/vol], 0.015% [wt/vol] bromophenol blue, 50 μg/ml bovine serum albumin, and 40 μg/ml Staphylococcus aureus V8 protease [USB] or chymotrypsin [Sigma]).
Lipid assays.
In vitro PI3K activity assays on immunoprecipitates were carried out as previously described (25). After 10 min, the reaction was stopped with 1 N HCl. Phosphorylated lipids were extracted with chloroform-methanol (1:1), spotted onto thin-layer chromatography (TLC) plates, and separated by chromatography in N-propanol–2 N acetic acid (65:35).
To analyze PIP3, NIH/3T3 cells were exposed to doxycycline for 24 h to induce expression of enhanced green fluorescent protein (EGFP), MT, or E349K MT. The last 16 h of the induction took place in the absence of serum. Lipids were extracted and PIP3 was measured by ELISA using the assay kit from Echelon Biosciences, Inc. (Salt Lake City, UT).
Rac activation assay.
GST PAK-Rac-binding domain, described and provided by R. Buchsbaum (85), was used to produce GST-Pak Rac1-binding domain bound to glutathione-Sepharose beads. Cells to be tested for activated Rac were harvested and extracted in 100 μl lysis buffer (50 mM Tris-HCI [pH 7.5], 10 mM MgCl2, 120 mM NaCl, 10% glycerol, 2% NP-40, l mM dithiothreitol with 5 μg/ml leupeptin, aprotinin, and pepstatin, and 0.1 mg/ml phenylmethylsulfonyl fluoride [PMSF]) for 30 min on ice. Lysates were cleared by spinning in an Eppendorf microcentrifuge at 10,000 rpm for 10 min at 4°C. Cleared lysates were incubated with GST-Pak bound to glutathione-Sepharose beads for 30 min at 4°C. Washed beads were treated with SDS dissociation buffer and separated by gel electrophoresis. Blotting for Rac was then carried out.
Cell fractionation.
Cytoplasmic and membrane fractions were extracted by hypotonic fractionation (86).
ACKNOWLEDGMENTS
We thank Nathalie Faure for technical assistance. We thank Larry Feig, Ole Gjoerup, Karl Munger, and Tom Roberts for critical readings of the manuscript.
This work was supported by HHS NIH National Cancer Institute grants CA50661 and CA34722 to B.S.S.
REFERENCES
- 1.Dawe CJ, Freund R, Mandel G, Ballmer-Hofer K, Talmage DA, Benjamin TL. 1987. Variations in polyoma virus genotype in relation to tumor induction in mice. Characterization of wild type strains with widely differing tumor profiles. Am J Pathol 127:243–261. [PMC free article] [PubMed] [Google Scholar]
- 2.Eddy B. 1969. Polyoma virus. Virol Monogr 7:1–114. [Google Scholar]
- 3.Fluck MM, Haslam SZ. 1996. Mammary tumors induced by polyomavirus. Breast Cancer Res Treat 39:45–56. doi: 10.1007/BF01806077. [DOI] [PubMed] [Google Scholar]
- 4.Schaffhausen BS, Roberts TM. 2009. Lessons from polyoma middle T antigen on signaling and transformation: a DNA tumor virus contribution to the war on cancer. Virology 384:304–316. doi: 10.1016/j.virol.2008.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fluck MM, Schaffhausen BS. 2009. Lessons in signaling and tumorigenesis from polyomavirus middle T antigen. Microbiol Mol Biol Rev 73:542–563. doi: 10.1128/MMBR.00009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eckhart W, Hutchinson MA, Hunter T. 1979. An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 18:925. doi: 10.1016/0092-8674(79)90205-8. [DOI] [PubMed] [Google Scholar]
- 7.Whitman M, Downes CP, Keeler M, Keller T, Cantley L. 1988. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 332:644–646. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- 8.Rouleau C, Pores Fernando AT, Hwang JH, Faure N, Jiang T, White EA, Roberts TM, Schaffhausen BS. 2016. Transformation by polyomavirus middle T antigen involves a unique bimodal interaction with the Hippo effector YAP. J Virol 90:7032–7045. doi: 10.1128/JVI.00417-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang JH, Jiang T, Kulkarni S, Faure N, Schaffhausen BS. 2013. Protein phosphatase 2A isoforms utilizing Abeta scaffolds regulate differentiation through control of Akt protein. J Biol Chem 288:32064–32073. doi: 10.1074/jbc.M113.497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Utermark T, Rao T, Cheng H, Wang Q, Lee SH, Wang ZC, Iglehart JD, Roberts TM, Muller WJ, Zhao JJ. 2012. The p110alpha and p110beta isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev 26:1573–1586. doi: 10.1101/gad.191973.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, Roberts TM. 1990. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 60:167–176. doi: 10.1016/0092-8674(90)90726-U. [DOI] [PubMed] [Google Scholar]
- 12.Walter G, Ruediger R, Slaughter C, Mumby M. 1990. Association of protein phosphatase 2A with polyoma virus medium tumor antigen. Proc Natl Acad Sci U S A 87:2521–2525. doi: 10.1073/pnas.87.7.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogris E, Mudrak I, Mak E, Gibson D, Pallas DC. 1999. Catalytically inactive protein phosphatase 2A can bind to polyomavirus middle tumor antigen and support complex formation with pp60(c-src). J Virol 73:7390–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Courtneidge S, Smith AE. 1983. Polyoma virus transforming protein associates with the product of the c-src cellular gene. Nature 303:435–439. doi: 10.1038/303435a0. [DOI] [PubMed] [Google Scholar]
- 15.Cheng SH, Harvey R, Espino PC, Semba K, Yamamoto T, Toyoshima K, Smith AE. 1988. Peptide antibodies to the human c-fyn gene product demonstrate pp59c-fyn is capable of complex formation with the middle-T antigen of polyomavirus. EMBO J 7:3845–3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horak ID, Kawakami T, Gregory F, Robbins KC, Bolen JB. 1989. Association of p60fyn with middle tumor antigen in murine polyomavirus-transformed rat cells. J Virol 63:2343–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kornbluth S, Sudol M, Hanafusa H. 1987. Association of the polyomavirus middle-T antigen with c-yes protein. Nature 325:171–173. doi: 10.1038/325171a0. [DOI] [PubMed] [Google Scholar]
- 18.Cartwright CA, Hutchinson MA, Eckhart W. 1985. Structural and functional modification of pp60c-src associated with polyoma middle tumor antigen from infected or transformed cells. Mol Cell Biol 5:2647–2652. doi: 10.1128/MCB.5.10.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunter T, Hutchinson MA, Eckhart W. 1984. Polyoma middle-T antigen can be phosphorylated on tyrosine at multiple sites in vitro. EMBO J 3:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey R, Oostra GL, Belsham GJ, Gillett P, Smith AE. 1984. An antibody to a synthetic peptide recognizes polyoma virus middle T antigen and reveals multiple in vitro tyrosine phosphorylation sites. Mol Cell Biol 4:1334–1342. doi: 10.1128/MCB.4.7.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaffhausen B, Benjamin TL. 1981. Comparison of phosphorylation of two polyomavirus in vivo and in vitro. J Virol 40:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carmichael G, Schaffhausen BS, Mandel G, Liang TJ, Benjamin TL. 1984. Transformation by polyoma virus is drastically reduced by substitution of phenylalanine for tyrosine at residue 315 of middle-sized tumor antigen. Proc Natl Acad Sci U S A 81:679–683. doi: 10.1073/pnas.81.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell KS, Ogris E, Burke B, Su W, Auger KR, Druker BJ, Schaffhausen BS, Roberts TM, Pallas DC. 1994. Polyoma middle tumor antigen interacts with SHC protein via the NPTY (Asn-Pro-Thr-Tyr) motif in middle tumor antigen. Proc Natl Acad Sci U S A 91:6344–6348. doi: 10.1073/pnas.91.14.6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dilworth SM, Brewster CE, Jones MD, Lanfrancone L, Pelicci G, Pelicci PG. 1994. Transformation by polyoma virus middle T-antigen involves the binding and tyrosine phosphorylation of Shc. Nature 367:87–90. doi: 10.1038/367087a0. [DOI] [PubMed] [Google Scholar]
- 25.Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM. 1985. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315:239–242. doi: 10.1038/315239a0. [DOI] [PubMed] [Google Scholar]
- 26.Kaplan DR, Whitman M, Schaffhausen B, Raptis L, Garcea RL, Pallas D, Roberts TM, Cantley L. 1986. Phosphatidylinositol metabolism and polyoma-mediated transformation. Proc Natl Acad Sci U S A 83:3624–3628. doi: 10.1073/pnas.83.11.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su W, Liu W, Schaffhausen B, Roberts T. 1995. Association of polyomavirus middle tumor antigen with phospholipase C-gamma 1. J Biol Chem 270:12331–12335. doi: 10.1074/jbc.270.21.12331. [DOI] [PubMed] [Google Scholar]
- 28.Schaffhausen BS, Liang TJ, Carmichael GG, Benjamin TL. 1985. Residual transforming activity of PY1178T, a mutant lacking the principal in vitro phosphorylation site is not affected by removal of the secondary tyrosine phosphorylation site at residue 322. Virology 143:671–675. doi: 10.1016/0042-6822(85)90410-6. [DOI] [PubMed] [Google Scholar]
- 29.Chen L, Wang X, Fluck MM. 2006. Independent contributions of polyomavirus middle T and small T to the regulation of early and late gene expression and DNA replication. J Virol 80:7295–7307. doi: 10.1128/JVI.00679-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian Y, Li D, Dahl J, You J, Benjamin T. 2004. Identification of TAZ as a binding partner of the polyomavirus T antigens. J Virol 78:12657–12664. doi: 10.1128/JVI.78.22.12657-12664.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shanzer M, Ricardo-Lax I, Keshet R, Reuven N, Shaul Y. 2014. The polyomavirus middle T-antigen oncogene activates the Hippo pathway tumor suppressor Lats in a Src-dependent manner. Oncogene 34:4190–4198. doi: 10.1038/onc.2014.347. [DOI] [PubMed] [Google Scholar]
- 32.Freund R, Dawe CJ, Carroll JP, Benjamin TL. 1992. Changes in frequency, morphology, and behavior of tumors induced in mice by a polyoma virus mutant with a specifically altered oncogene. Am J Pathol 141:1409–1425. [PMC free article] [PubMed] [Google Scholar]
- 33.Bronson R, Dawe C, Carroll J, Benjamin T. 1997. Tumor induction by a transformation-defective polyoma virus mutant blocked in signaling through shc. Proc Nat Acad Sci U S A 94:7954–7958. doi: 10.1073/pnas.94.15.7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yi X, Peterson J, Freund R. 1997. Transformation and tumorigenic properties of a mutant polyoma virus containing a middle T antigen defective in shc binding. J Virol 71:6279–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cullere X, Rose P, Thathamangalam U, Chatterjee A, Mullane KP, Pallas DC, Benjamin TL, Roberts TM, Schaffhausen BS. 1998. Serine 257 phosphorylation regulates association of polyomavirus middle T antigen with 14-3-3 proteins. J Virol 72:558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carmichael G, Schaffhausen B, Dorsky D, Oliver D, Benjamin T. 1982. The role of the carboxy terminus of middle T antigen of polyoma virus. Proc Natl Acad Sci U S A 79:3579–3583. doi: 10.1073/pnas.79.11.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ichaso N, Dilworth SM. 2001. Cell transformation by the middle T-antigen of polyoma virus. Oncogene 20:7908–7916. doi: 10.1038/sj.onc.1204859. [DOI] [PubMed] [Google Scholar]
- 38.Magnusson G, Berg P. 1979. Construction and analysis of viable deletion mutants of polyoma virus. J Virol 32:523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Magnusson G, Nilsson M, Dilworth S, Smolar N. 1981. Characterization of polyoma mutants with altered middle T and large T antigens. J Virol 39:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yi X, Freund R. 1998. Deletion of proline-rich domain in polyomavirus T antigens results in virus partially defective in transformation and tumorigenesis. Virology 248:420–431. doi: 10.1006/viro.1998.9246. [DOI] [PubMed] [Google Scholar]
- 41.Saksela K, Permi P. 2012. SH3 domain ligand binding: what's the consensus and where's the specificity? FEBS Lett 586:2609–2614. doi: 10.1016/j.febslet.2012.04.042. [DOI] [PubMed] [Google Scholar]
- 42.Thorpe LM, Yuzugullu H, Zhao JJ. 2015. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samuels Y, Velculescu VE. 2004. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 3:1221–1224. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 44.Kang S, Bader AG, Vogt PK. 2005. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A 102:802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samuels Y, Diaz LA Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. 2005. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 7:561–573. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 46.Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. 2005. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A 102:18443–18448. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. 2005. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res 65:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 48.Utermark T, Schaffhausen BS, Roberts TM, Zhao JJ. 2007. The p110alpha isoform of phosphatidylinositol 3-kinase is essential for polyomavirus middle T antigen-mediated transformation. J Virol 81:7069–7076. doi: 10.1128/JVI.00115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cicchetti P, Mayer BJ, Thiel G, Baltimore D. 1992. Identification of a protein that binds to the SH3 region of Abl and is similar to Bcr and GAP-rho. Science 257:803–806. doi: 10.1126/science.1379745. [DOI] [PubMed] [Google Scholar]
- 50.Ren R, Mayer BJ, Cicchetti P, Baltimore D. 1993. Identification of a ten-amino acid proline-rich SH3 binding site. Science 259:1157–1161. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- 51.Macias MJ, Wiesner S, Sudol M. 2002. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett 513:30–37. doi: 10.1016/S0014-5793(01)03290-2. [DOI] [PubMed] [Google Scholar]
- 52.Ong SH, Dilworth S, Hauck-Schmalenberger I, Pawson T, Kiefer F. 2001. ShcA and Grb2 mediate polyoma middle T antigen-induced endothelial transformation and Gab1 tyrosine phosphorylation. EMBO J 20:6327–6336. doi: 10.1093/emboj/20.22.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen B, Yoakim M, Piwnica-Worms H, Roberts T, Schaffhausen B. 1990. Tyrosine phosphorylation is a signal for the trafficking of pp85, a polypeptide associated with phosphatidylinositol kinase activity. Proc Nat Acad Sci U S A 87:4458–4462. doi: 10.1073/pnas.87.12.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cohen B, Liu YX, Druker B, Roberts TM, Schaffhausen BS. 1990. Characterization of pp85, a target of oncogenes and growth factor receptors. Mol Cell Biol 10:2909–2915. doi: 10.1128/MCB.10.6.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salcini AE, McGlade J, Pelicci G, Nicoletti I, Pawson T, Peilicci P. 1994. Formation of Shc-Grb2 complexes is necessary to induce neoplastic transformation by overexpression of Shc proteins. Oncogene 9:2827–2836. [PubMed] [Google Scholar]
- 56.Gotoh N, Tojo A, Shibuya M. 1996. A novel pathway from phosphorylation of tyrosine residues 239/240 of Shc, contributing to suppress apoptosis by IL-3. EMBO J 15:6197–6204. [PMC free article] [PubMed] [Google Scholar]
- 57.van der Geer P, Wiley S, Gish GD, Pawson T. 1996. The Shc adaptor protein is highly phosphorylated at conserved, twin tyrosine residues (Y239/240) that mediate protein-protein interactions. Curr Biol 6:1435–1444. doi: 10.1016/S0960-9822(96)00748-8. [DOI] [PubMed] [Google Scholar]
- 58.Urich M, Senften M, Shaw PE, Ballmer-Hofer K. 1997. A role for the small GTPase Rac in polyomavirus middle-T antigen-mediated activation of the serum response element and in cell transformation. Oncogene 14:1235–1241. doi: 10.1038/sj.onc.1200982. [DOI] [PubMed] [Google Scholar]
- 59.Connolly JO, Soga N, Guo XL, Alvarez U, Hruska KA. 2000. Rac is essential in the transformation of endothelial cells by polyoma middle T. Cell Adhes Commun 7:409–422. doi: 10.3109/15419060009109022. [DOI] [PubMed] [Google Scholar]
- 60.Chen Y, Freund R, Listerud M, Wang Z, Talmage DA. 1999. Retinoic acid inhibits transformation by preventing phosphatidylinositol 3-kinase dependent activation of the c-fos promoter. Oncogene 18:139–148. doi: 10.1038/sj.onc.1202272. [DOI] [PubMed] [Google Scholar]
- 61.Dahl J, Jurczak A, Cheng L, Baker D, Benjamin T. 1998. Evidence of a role for phosphatidylinositol 3-kinase activation in the blocking of apoptosis by polyomavirus middle T antigen. J Virol 72:3221–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meili R, Cron P, Hemmings B, Ballmer-Hofer K. 1998. Protein kinase B/Akt is activated by polyomavirus middle-T antigen via a phosphatidylinositol 3-kinase-dependent mechanism. Oncogene 16:903–907. doi: 10.1038/sj.onc.1201605. [DOI] [PubMed] [Google Scholar]
- 63.Summers SA, Lipfert L, Birnbaum MJ. 1998. Polyoma middle T antigen activates the Ser/Thr kinase Akt in a PI3-kinase-dependent manner. Biochem Biophys Res Commun 246:76–81. doi: 10.1006/bbrc.1998.8575. [DOI] [PubMed] [Google Scholar]
- 64.Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. 2007. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res 67:167–177. doi: 10.1158/0008-5472.CAN-06-3782. [DOI] [PubMed] [Google Scholar]
- 65.Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. 1998. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene 17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 66.Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. 1995. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 67.Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter G, Holmes A, Gaffney P, Reese C, McCormick F, Tempst P, Coadwell J, Hawkins P. 1998. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- 68.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 69.Boissier P, Huynh-Do U. 2014. The guanine nucleotide exchange factor Tiam1: a Janus-faced molecule in cellular signaling. Cell Signal 26:483–491. doi: 10.1016/j.cellsig.2013.11.034. [DOI] [PubMed] [Google Scholar]
- 70.Ling LE, Druker BJ, Cantley LC, Roberts TM. 1992. Transformation-defective mutants of polyomavirus middle T antigen associate with phosphatidylinositol 3-kinase (PI 3-kinase) but are unable to maintain wild-type levels of PI 3-kinase products in intact cells. J Virol 66:1702–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leslie NR, Kriplani N, Hermida MA, Alvarez-Garcia V, Wise HM. 2016. The PTEN protein: cellular localization and post-translational regulation. Biochem Soc Trans 44:273–278. doi: 10.1042/BST20150224. [DOI] [PubMed] [Google Scholar]
- 72.Bermudez Brito M, Goulielmaki E, Papakonstanti EA. 2015. Focus on PTEN regulation. Front Oncol 5:166. doi: 10.3389/fonc.2015.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carlton JG, Cullen PJ. 2005. Coincidence detection in phosphoinositide signaling. Trends Cell Biol 15:540–547. doi: 10.1016/j.tcb.2005.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Elliott J, Jones MD, Griffin BE, Krauzewicz N. 1998. Regulation of cytoskeletal association by a basic amino acid motif in polyoma virus middle T antigen. Oncogene 17:1797–1806. doi: 10.1038/sj.onc.1202083. [DOI] [PubMed] [Google Scholar]
- 75.Mullane KP, Ratnofsky M, Cullere X, Schaffhausen B. 1998. Signaling from polyomavirus middle T and small T defines different roles for protein phosphatase 2A. Mol Cell Biol 18:7556–7565. doi: 10.1128/MCB.18.12.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaplan DR, Whitman M, Schaffhausen B, Pallas DC, White M, Cantley L, Roberts TM. 1987. Common elements in growth factor stimulation and oncogenic transformation: 85 kd phosphoprotein and phosphatidylinositol kinase activity. Cell 50:1021–1029. doi: 10.1016/0092-8674(87)90168-1. [DOI] [PubMed] [Google Scholar]
- 77.Gao X, Lowry PR, Zhou X, Depry C, Wei Z, Wong GW, Zhang J. 2011. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc Natl Acad Sci U S A 108:14509–14514. doi: 10.1073/pnas.1019386108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bendig M, Thomas T, Folk W. 1980. Viable deletion mutant in the medium and large T antigen coding sequences of the polyoma virus genome. J Virol 33:1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Holman P, Gjoerup O, Davin T, Schaffhausen B. 1994. Characterization of an immortalizing N-terminal domain of polyomavirus large T antigen. J Virol 68:668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hwang JH, Pores Fernando AT, Faure N, Andrabi S, Hahn WC, Schaffhausen BS, Roberts TM. 2014. Polyoma small T antigen interacts with YES-associated protein (YAP) to regulate cell survival and differentiation. J Virol 88:12055–12064. doi: 10.1128/JVI.01399-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 83.Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoakim M, Hou W, Liu Y, Carpenter CL, Kapeller R, Schaffhausen BS. 1992. Interactions of polyomavirus middle T with the SH2 domains of the pp85 subunit of phosphatidylinositol-3-kinase. J Virol 66:5485–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Buchsbaum RJ, Connolly BA, Feig LA. 2002. Interaction of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. Mol Cell Biol 22:4073–4085. doi: 10.1128/MCB.22.12.4073-4085.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Silver J, Schaffhausen B, Benjamin T. 1978. Tumor antigens induced by nontransforming mutants of polyoma virus. Cell 15:485–496. doi: 10.1016/0092-8674(78)90018-1. [DOI] [PubMed] [Google Scholar]