

ABSTRACT
Papillomaviruses are small, double-stranded DNA viruses that encode the E2 protein, which controls transcription, replication, and genome maintenance in infected cells. Posttranslational modifications (PTMs) affecting E2 function and stability have been demonstrated for multiple types of papillomaviruses. Here we describe the first phosphorylation event involving a conserved tyrosine (Y) in the bovine papillomavirus 1 (BPV-1) E2 protein at amino acid 102. While its phosphodeficient phenylalanine (F) mutant activated both transcription and replication in luciferase reporter assays, a mutant that may act as a phosphomimetic, with a Y102-to-glutamate (E) mutation, lost both activities. The E2 Y102F protein interacted with cellular E2-binding factors and the viral helicase E1; however, in contrast, the Y102E mutant associated with only a subset and was unable to bind to E1. While the Y102F mutant fully supported transient viral DNA replication, BPV genomes encoding this mutation as well as Y102E were not maintained as stable episomes in murine C127 cells. These data imply that phosphorylation at Y102 disrupts the helical fold of the N-terminal region of E2 and its interaction with key cellular and viral proteins. We hypothesize that the resulting inhibition of viral transcription and replication in basal epithelial cells prevents the development of a lytic infection.
IMPORTANCE Papillomaviruses (PVs) are small, double-stranded DNA viruses that are responsible for cervical, oropharyngeal, and various genitourinary cancers. Although vaccines against the major oncogenic human PVs are available, there is no effective treatment for existing infections. One approach to better understand the viral replicative cycle, and potential therapies to target it, is to examine the posttranslational modification of viral proteins and its effect on function. Here we have discovered that the bovine papillomavirus 1 (BPV-1) transcription and replication regulator E2 is phosphorylated at residue Y102. While a phosphodeficient mutant at this site was fully functional, a phosphomimetic mutant displayed impaired transcription and replication activity as well as a lack of an association with certain E2-binding proteins. This study highlights the influence of posttranslational modifications on viral protein function and provides additional insight into the complex interplay between papillomaviruses and their hosts.
KEYWORDS: viral replication, papillomavirus, papillomavirus E2, tyrosine phosphorylation
INTRODUCTION
Papillomaviruses (PVs) are nonenveloped, double-stranded DNA viruses with circular genomes of ∼8 kb. This compact genome harbors 8 major genes, including 6 early genes (E1, E2, E4, E5, E6, and E7) and 2 late genes (L1 and L2). The early genes mediate all stages of the replicative cycle within the host cell, while L1 and L2 are capsid proteins involved in virus entry into and exit from the host. At present, more than 200 human and 100 nonhuman types have been identified (1). Bovine papillomavirus 1 (BPV-1) is a model organism for PV research due to its ability to transform and stably replicate as multicopy plasmids in mouse cell lines (2, 3) and the availability of antibodies specific for its gene products. Through the study of BPV-1 and other PVs, the viral replicative cycle has been characterized. In the initial stage, the genome replicates to a low level, with 1 to 10 copies, depending on the system used (4–6). Next, in the maintenance stage, the copy number is kept constant as viral DNA is replicated and partitioned in association with host chromosomes as the infected epithelial cell divides (7, 8). Finally, in the amplification stage, the differentiation of host cells produces as-yet-uncharacterized signals that prompt PV replication to thousands of copies per cell in preparation for virus assembly and release.
Despite significant advances in the understanding of papillomavirus biology, the mechanisms that control the transition from one stage of the viral replicative cycle to the next remain poorly defined. One viral gene product of interest is the E2 protein, which plays crucial roles in transcription and replication and interacts with multiple cellular factors. E2 DNA-binding sites occur throughout the PV genome at the consensus sequence ACC(N6)GGT, with several sites in the viral origin of replication (ori) and at individual promoters (9). Through binding to these sites, E2 controls PV transcription, recruits the viral DNA helicase E1 to the ori for replication initiation (10, 11), and tethers viral DNA to mitotic chromosomes to ensure genome maintenance in dividing host cells (12). E2 functions are mediated by the formation of complexes with cellular proteins, such as Brd4 (13–18), p300 (19), Gps2/AMF1 (19, 20), and Tax1BP1 (TXBP) (21), as well as viral proteins, including E1, E4, E6, and E7 (10, 22–26).
Posttranslational modification (PTM) of multifunctional proteins such as E2 can alter the protein's interaction with its cofactors, its localization within the cell, or its ability to be recognized by proteolytic machinery. The first reported E2 PTMs were serine (S) phosphorylation events in the hinge region of BPV-1 E2 at residues 290, 298, and 301 (27). Serine-to-alanine mutations at these sites resulted in increased copy numbers in mouse cells (28). Casein kinase II (CK2) was reported to phosphorylate these BPV-1 serines, leading to increased E2 degradation and decreased replication (29). Similar sites have been found in human papillomavirus 8 (HPV-8) at S253 (30) and in HPV-16 at S243 (31). We previously reported acetylation at lysine 111 (K111) and K112 of BPV-1 E2 and that mutation to arginine (R) at the former site inhibited viral transcription (32).
We describe the first tyrosine (Y) phosphorylation event in a PV E2 protein. Y102 is located in the BPV-1 E2 transactivation domain (TAD) and is conserved across multiple PV types, including cervical cancer-associated high-risk HPV-16 and -31. Point mutations at this site that prevent or mimic phosphorylation have differential effects on BPV-1 E2-mediated transcription, replication, and binding to known interacting partners. We hypothesize that tyrosine kinases act on E2 to restrict its function early in the replicative cycle.
RESULTS
Identification of phosphorylated tyrosine 102.
After having discovered in vitro lysine acetylation in BPV-1 E2 (32), we sought to determine whether other PTMs occurred on this protein in live cells. Two separate samples were prepared for mass spectrometric analysis. The first sample was obtained from C33a/E2 cells, derived from an HPV-negative human cervical cancer cell line but stably expressing the BPV-1 E2 gene (16). The second sample was obtained from unaltered C33a cells transfected with a full-length BPV-1 E2 (amino acids [aa] 1 to 410) expression plasmid. The E2 protein from both samples was purified by immunoprecipitation and PAGE separation and analyzed by tandem mass spectrometry using a high-resolution LTQ Orbitrap mass spectrometer. Data analysis included PTM identification as well as confidence scores. This approach detected previously reported phosphorylations at S298 and S301 (27) as well as acetylation at K112. Phosphorylation at Y102 was of particular interest since tyrosine phosphorylation has not been reported for any E2 protein. Furthermore, this PTM was assigned the highest confidence score of any PTM in a report generated by Thermo Proteome Discoverer software, including those at the previously reported S298 and S301 sites. While not identified in all PVs, Y102 (or the equivalent Y103 to -5) occurs in 28 HPVs for which complete genomes are available, including high-risk HPV-16 and HPV-31 (Table 1). Tyrosine residues are also observed at these positions in 44 nonhuman PVs. BPV-1 and HPV-16 E2 proteins share 80% similarity. Crystal structures of the E2 TADs from BPV-1 (Fig. 1A, dark blue) and HPV-16 (light blue) are highly conserved and depict Y102 as being located between the three alpha-helices and beta-sheet folds and oriented toward Brd4 (purple).
TABLE 1.
Appearance of tyrosine 102 in human papillomavirusesa
| HPV type | Genus | Position of tyrosine | Flanking sequence (±5 aa) |
|---|---|---|---|
| 8 | Betapapillomavirus | 102 | TSIETYKNAPE |
| 12 | Betapapillomavirus | 102 | TSAETYNNVPE |
| 16 | Alphapapillomavirus | 102 | VSLEVYLTAPT |
| 19 | Betapapillomavirus | 102 | TSAETYRSAPE |
| 25 | Betapapillomavirus | 102 | TSTETYKSPPE |
| 26 | Alphapapillomavirus | 102 | TSYEMYMTEPK |
| 31 | Alphapapillomavirus | 102 | TSLELYTLTAP |
| 35 | Alphapapillomavirus | 103 | TSIELYTTVPQ |
| 41 | Nupappilomavirus | 105 | TTKERYLAEPS |
| 49 | Betapapillomavirus | 102 | TSLETYNAPPA |
| 69 | Alphapapillomavirus | 102 | TCYELYVTEPK |
| 76 | Betapapillomavirus | 102 | TSLETYPTPPI |
| 85 | Alphapapillomavirus | 103 | TCQELYQTPPQ |
| 96 | Betapapillomavirus | 102 | TSLETYRAPPV |
| 98 | Betapapillomavirus | 102 | TSIETYKNAPE |
| 108 | Gammapapillomavirus | 102 | TSHELYTTPPE |
| 109 | Gammapapillomavirus | 103 | TSFELYNAPPQ |
| 115 | Betapapillomavirus | 102 | TSLETYRTPPS |
| 118 | Betapapillomavirus | 102 | TSLETYKNAPE |
| 123 | Gammapapillomavirus | 102 | TSFELYNSSPQ |
| 124 | Betapapillomavirus | 102 | TSLETYRNQPE |
| 134 | Gammapapillomavirus | 102 | TSLELYNTEPE |
| 139 | Gammapapillomavirus | 102 | TSFEVYNAAPF |
| 152 | Betapapillomavirus | 102 | TSLETYRNRPE |
| 155 | Gammapapillomavirus | 102 | TSYEIYSAAPK |
| 170 | Gammapapillomavirus | 102 | TSYEAYTSAPE |
| 202 | Gammapapillomavirus | 104 | TSADRYDSPPR |
| 204 | Mupapillomavirus | 102 | TSRERYTAAPG |
Viruses are listed by type, along with the genus and the presence of Y102 or an analogous site (e.g., Y103). The amino acid sequence surrounding the tyrosine of interest (in boldface type) is provided. Genus and sequence information was obtained from the Papillomavirus Episteme (https://pave.niaid.nih.gov/).
FIG 1.
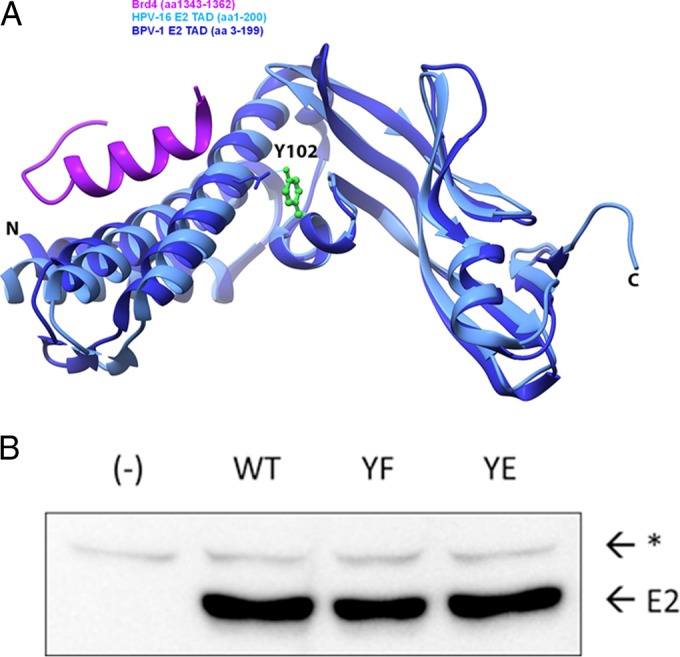
Location of Y102 in the TAD and mutagenesis. (A) A ribbon diagram of the BPV-1 E2 TAD crystal structure reported under PDB accession number 2JEU (60) (dark blue) overlapping the HPV-16 E2 TAD/Brd4 cocrystal structure reported under PDB accession number 2NNU (38) (light blue and purple, respectively) was created by using the UCSF Chimera package. The Y102 side chain, depicted in a green ball-and-stick model, is hydrogen bonded to E74, depicted in a wire frame model. N, amino terminus; C, carboxyl terminus. (B) Site-directed mutagenesis converts Y102 to phenylalanine (F) or glutamate (E) in pCG-E2. One microgram of WT and mutant constructs (YF and YE), or mRFP-GFP as negative control (−), was expressed in HEK293TT cells. The immunoblot was probed for BPV-1 E2 with mouse monoclonal antibody B201. E2, full-length BPV-1 E2; *, nonspecific band.
Transcriptional activities of mutations of BPV-1 E2 Y102.
To investigate the potential functional significance of this PTM, we used site-directed mutagenesis to generate E2 Y102 mutant constructs with phenylalanine (Y102F), which cannot be phosphorylated at this site, and glutamate (Y102E), which has a negative charge similar to that of phosphotyrosine. E2 protein expression levels of both mutants in HEK293TT cells was comparable to that of the wild type (WT) (Fig. 1B). We began activity studies with the tyrosine mutants by testing how they affected the ability of E2 to stimulate transcription. C33a cells were transfected with plasmids encoding WT BPV-1 E2 or the Y102F or Y102E mutant along with a firefly luciferase reporter (pGL2-E2BS-Luc) containing four E2-binding sites upstream of a simian virus 40 (SV40) promoter (33). While the Y102F mutant stimulated luciferase expression at levels comparable to those of the WT (38-fold above levels of the negative control), Y102E was inactive (Fig. 2A). However, in some experiments, Y102E protein levels in C33a cells were lower than those of the WT and Y102F proteins (Fig. 2B). To ensure these results were not due to insufficient expression or cell type-dependent differences, the experiments were repeated in CV-1 cells, an African green monkey kidney cell line. As in C33a cells, Y102F stimulated reporter transcription comparably to the WT (35-fold above values for the negative control), while Y102E failed to stimulate transcription (Fig. 2C); its luciferase readout was comparable to that of the truncated BPV-1 E2R protein (aa 162 to 410), which exhibits low levels of transcription activation (34). In these cells, overexpression of Y102E resulted in protein levels comparable to those of the WT and Y102F proteins but did not restore transcriptional activation (Fig. 2C and D).
FIG 2.
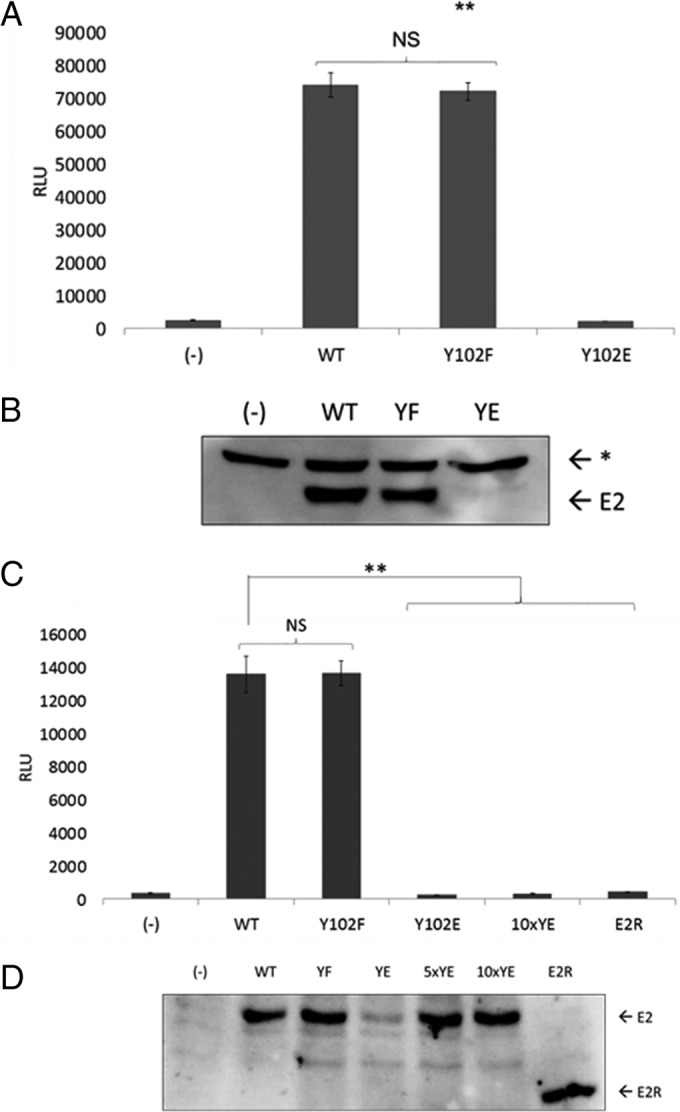
Y102E abrogates E2 transcriptional activation. (A) C33a cells were transfected in triplicate with 50 ng/well of the pCG-E2 wild type or mutants (Y102F and Y102E) or mRFP-GFP (−) as well as 75 ng/well pGL2-E2BS-Luc. Cells were lysed on-plate for luminescence detection using the PHERAstar system. RLU, relative light units; **, P < 0.01; NS, not significant. Values are expressed as means ± SEM. (B) One microgram each of the WT and mutant constructs (YF and YE) or mRFP-GFP (−) was expressed in C33a cells and probed for BPV-1 E2 with B201 antibodies. E2, full-length BPV-1 E2; *, nonspecific band. (C) CV-1 cells were transfected with 10 ng/well E2, pCG-E2R (E2R [aa 162 to 410]), or mRFP-GFP plus wells with 10 times the amount of Y102E (100 ng/well) (10×YE). **, P < 0.01. Values are expressed as means ± SEM. (D) CV-1 cells were transfected and prepared for Western blotting with the inclusion of samples with 5 times the amount of Y102E (5×YE), 10 times the amount of Y102E (10×YE), or E2R. The blot was probed for BPV-1 E2 with B201. E2, full-length BPV-1 E2; *, nonspecific band.
To determine whether the transcriptional defect of the Y102E mutant might be due to altered cofactor binding, coimmunoprecipitation (co-IP) experiments were performed with BPV-1 E2 WT and mutant proteins and different binding partners known to enhance E2-mediated transactivation. One of these binding partners, the chromatin modulator Brd4, has been shown in previous studies to interact via its extreme C-terminal domain (CTD) (also named the C-terminal motif [CTM]) (Fig. 3A) with E2 proteins from multiple PVs (13, 16, 35–37). BPV-1 E2 WT and Y102 mutant proteins were expressed in HEK293TT cells along with full-length FLAG-tagged Brd4, and protein complexes were immunoprecipitated with mouse monoclonal anti-FLAG (M2)-conjugated beads. While the WT and Y102F E2 proteins coimmunoprecipitated with Brd4, Y102E did not (Fig. 3B). This was surprising since the Brd4 CTM is known to bind to the N-terminal alpha-helices of the E2 TAD (38), and contacts with the Y102 region have not been described previously. To ascertain whether the observed effect was due to a lack of Y102E binding to the CTM, a glutathione S-transferase (GST)-tagged CTM was substituted for full-length Brd4 in transfections, and complexes were captured on glutathione beads. Again, the same pattern appeared, with Y102E failing to associate (Fig. 3C). It was recently reported that distinct domains in Brd4 undergo conformational changes to interact with p53 (39) and HPV E2 (40). We tested the basic residue-enriched interaction domain (BID) for an association with BPV-1 E2 (Fig. 3A). Notably, GST-BID pulled down BPV-1 E2 WT, Y102F, and Y102E proteins (Fig. 3D). Thus, Y102E is unable to interact with the established E2-binding CTM of Brd4 but can bind in vitro to the BID region, which is known to be mediated by the E2 C-terminal DNA-binding domain (DBD) (40).
FIG 3.
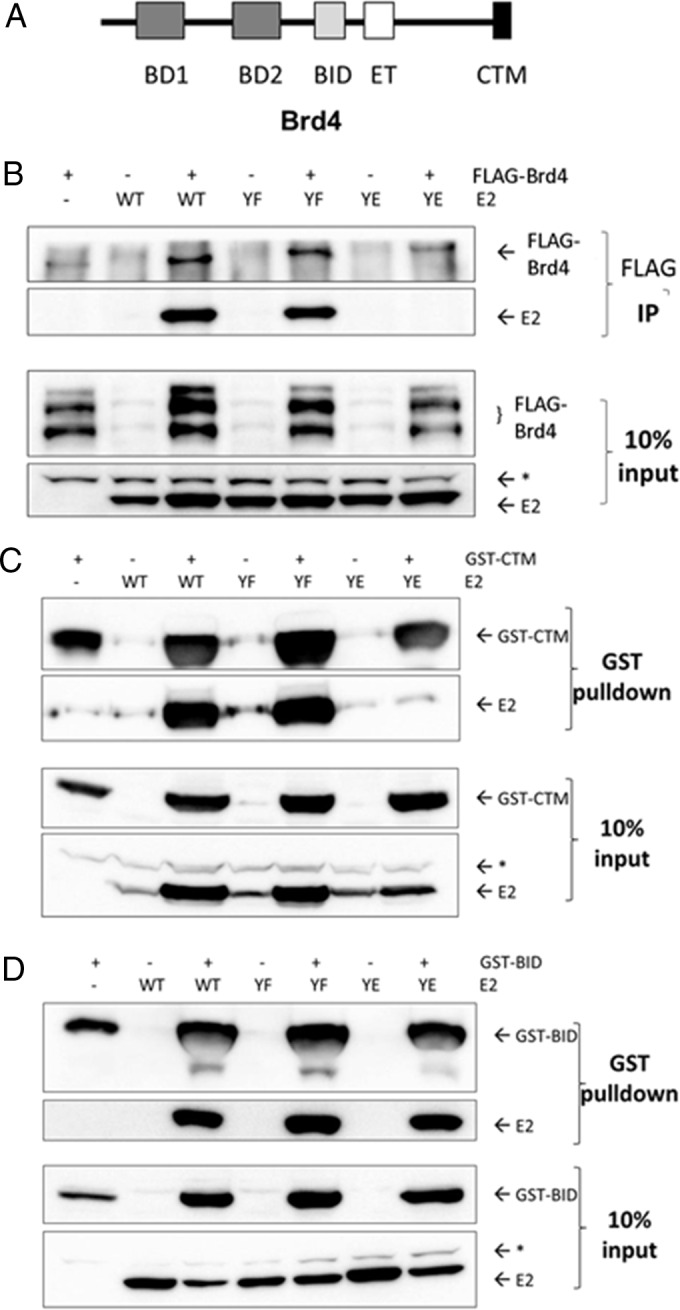
Y102E does not bind the Brd4 CTM but associates with an internal E2-binding region. (A) Domains of the Brd4 chromatin modulator protein (39). BD1, bromodomain 1; BID, basic interacting domain; ET, extraterminal domain. (B) HEK293TT cells were transfected with mRFP-GFP (−), the BPV-1 E2 WT and mutants (YF and YE), and FLAG-tagged full-length Brd4 (pVL-F:hBrd4) or pCI. Immunoprecipitates were collected with anti-FLAG mouse antibody M2 conjugated to agarose beads. Samples were probed by Western blotting for FLAG-Brd4 and BPV-1 E2 using M2 and B201, respectively. E2, full-length BPV-1 E2; *, nonspecific band. (C and D) GST-tagged CTM (C) and GST-BID (D). Complexes were collected with glutathione beads. Western blots were probed for GST with rabbit polyclonal antibody SD8 and for E2 with B201. *, nonspecific band.
To ensure that the inability of the Y102E mutant to stimulate transactivation was not due to a global disruption of the protein's structure, we examined complex formation with two other known E2-binding partners: Gps2 (also known as AMF1) and Tax1BP1. Gps2 has been found to stimulate the transcriptional activation of E2 and recruit it to form a complex with the cofactor p300 (19, 20). Tax1BP1 stabilizes protein levels of E2 and enhances its transactivation (21). BPV-1 E2 WT and mutant constructs were coexpressed with either hemagglutinin (HA)-tagged Gps2 or triple-FLAG (3×FLAG)-tagged Tax1BP1, and complexes were captured with an anti-BPV-1 E2 antibody. Both Y102F and Y102E coprecipitated HA-Gps2 and 3×FLAG-Tax1BP1 (Fig. 4). The E2R form was used as a negative control for Tax1BP1 associations (Fig. 4B).
FIG 4.
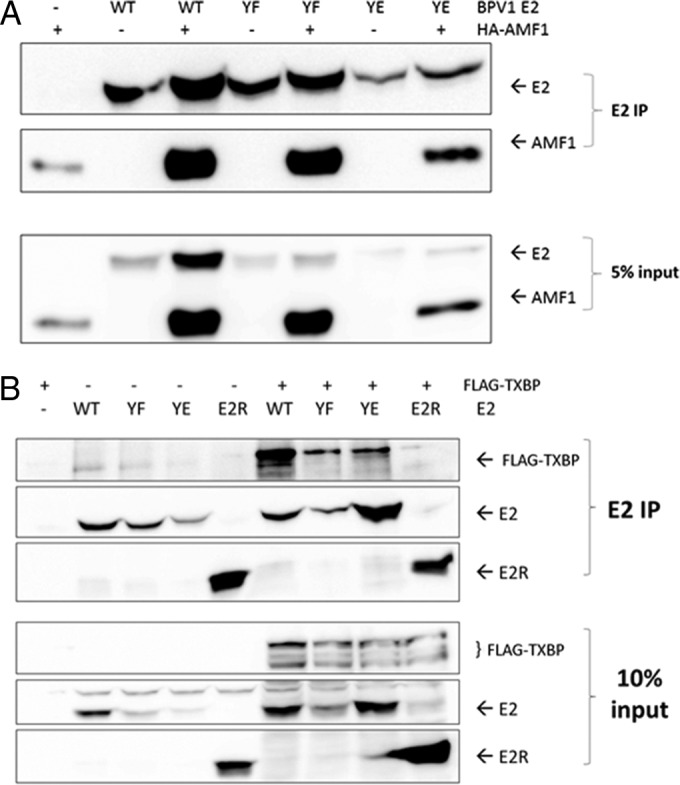
Y102 mutants associate with Gps2 and Tax1BP1. (A) HEK293TT cells were transfected with 1 μg each mRFP-GFP (−), WT BPV-1 E2 and mutants (YF and YE) plus E2R, and hemagglutinin-tagged Gps2 (HA-Gps2) (pHA:AMF1) or pCI. Western blots were probed for HA-Gps2 and BPV-1 E2 by using mouse monoclonal anti-HA antibodies HA-7 and B201, respectively. (B) Samples were prepared as described above for panel A by using p3XFLAG-CMV-7.1 Tax1BP1. Western blots were probed with FLAG-M2 and B201.
Transient DNA replication activities of BPV-1 E2 Y102 mutations.
E2 stimulates viral replication by recruiting the E1 monomer to the ori. The ability of the Y102 mutants to stimulate the transient replication of the viral ori was examined in a luciferase-based reporter assay. This method, developed by the Archambault laboratory for both BPV-1 and HPV-31 in C33a cells, requires the cotransfection of plasmids encoding E2, the homologous E1 helicase, Renilla luciferase (internal control), and firefly luciferase (target replicon) (41, 42). The firefly luciferase gene is constitutively expressed due to the presence of a cytomegalovirus (CMV) promoter. However, since the construct contains the BPV-1 ori, changes in plasmid copy number due to replication affect the amount of luciferase produced. Interestingly, Y102F-dependent luciferase activity was significantly increased above that of the WT (Fig. 5A). In contrast, Y102E did not generate a luminescence signal above baseline levels. Additional replication controls along with renilla luciferase levels are shown in Fig. 5B. The transfection of increased amounts of the Y102E expression vector to achieve levels comparable to or exceeding WT E2 levels in the transient-replication assays demonstrated that Y102E did not activate E1-dependent DNA replication (Fig. 5C). This led us to examine whether the Y102 mutants are capable of binding E1. BPV-1 E1 and E2 were expressed in HEK293TT cells and immunoprecipitated with anti-E2 antibodies. While Y102F coprecipitated E1 at levels comparable to those of the WT, Y102E did not (Fig. 5D).
FIG 5.

Y102E fails to stimulate transient BPV-1 replication. (A) C33a cells transfected in 8 replicate wells with 10 ng/well of the pCG-E2 WT or mutants (Y102F and Y102E), E2R, or mRFP-GFP (−) as well as 10 ng/well pCG-E1, 2.5 ng/well pFLORI-BPV1 (firefly luciferase reporter), and 0.5 ng/well pRL (Renilla luciferase reporter). Lysates were processed for luminescence. F/R, firefly output (in relative light units) divided by the Renilla output; **, P < 0.01; ****, P < 0.00001. Values are expressed as means ± SEM. (B) Replication assay completed as described above for panel A but with firefly and renilla luciferase levels being graphed separately. (C) Replication assay as described above for panel A. Each group had E1 transfected with either WT E2 or increasing amounts of Y102E. Fluc, firefly luciferase; Rluc, renilla luciferase (D) HEK293TT cells were transfected with 3 μg each mRFP-GFP (−), the BPV-1 E2 WT and mutants (YF and YE), and BPV-1 E1 or pCI. Lysates were immunoprecipitated with Sepharose A beads and BPV E2 antibody II-I. Western blots were probed for E1 and E2 by using rabbit anti-E1 peptide antibody 502-2 and antibody B201, respectively. *, nonspecific band.
E2 Y102 mutants localize to the nucleus.
The full-length E2 protein contains two putative nuclear localization signals, one in a basic region of the C-terminal DBD and another within the TAD (43, 44). Given the proximity of Y102 just amino terminally to the TAD nuclear localization site (NLS) (aa 111 to 120), we sought to determine the localization of the Y102F and Y102E proteins. Immunofluorescence experiments were performed in CV-1 cells transfected with WT BPV-1 E2, Y102F, Y102E, or E2R as well as BPV-1 E1. A3 cells, murine fibroblast cells stably carrying BPV-1 genomes at a high copy number (28), were used as a positive control. CV-1 cells transfected with enhanced green fluorescent protein (eGFP) in place of E2 were used as a negative control. While the E2 distribution pattern in CV-1 cells transfected with WT E2, Y102F, and E2R was almost exclusively nuclear (similar to that in A3 cells), the distribution of Y102E was mostly nuclear, with some cytoplasmic distribution (Fig. 6). Additionally, in the presence of E1, Y102F but not Y102E was able to form nuclear replication foci.
FIG 6.
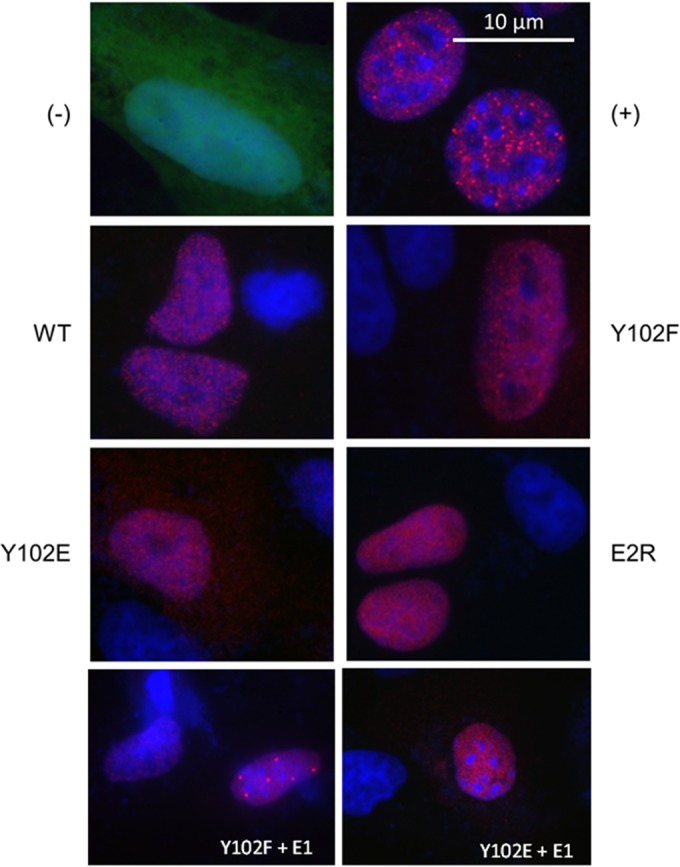
Y102 mutants localize to nuclei. CV-1 and A3 (positive control for E2 expression [+]) cells were cultured on 18-mm glass coverslips. CV-1 cells were transfected with 100 ng pCG-E1 and 100 ng pEGFP-C1 (eGFP [−]), WT E2, the Y102F or Y102E mutant, or E2R. Forty-eight hours after CV-1 cell transfection, cells were fixed and permeabilized, followed by incubation with B201. Cells were reacted with Alexa Fluor 594 anti-mouse antibody, washed, and mounted onto glass slides by using ProLong Gold with DAPI. Green, eGFP; red, BPV-1 E2; blue, DNA. Bar, 10 μm. All images were taken in all three channels (green, red, and blue) at a ×1,000 magnification.
Y102 mutant genomes are unable to maintain stable episomal replication.
To test whether the phosphomimetic Y102E and phosphodefective Y102F mutants are able to maintain stable episomal replication, C127 cells were cotransfected with Y102E, Y102F, or wild-type BPV genomes along with pBabePuro plasmids, and puromycin-resistant colonies were selected. Oncogenically transformed colonies were then obtained from puromycin-selected C127 cell lines by using a soft-agar assay and grown as monolayers. Previous studies demonstrated that BPV genomes carrying E1 or E2 mutations will integrate into and efficiently transform C127 cells (45, 46). Total DNA was isolated from multiple cell lines and subjected to Southern blot analysis. As shown in Fig. 7A, Y102E or Y102F mutant genomes were unable to maintain episomes, while wild-type genomes showed stable replication. However, total PCR amplification of the long control region (LCR) was seen in all cell lines (Fig. 7B), indicating the presence of integrated BPV genomes. Repeat experiments by using progressive passages of several different transformed cell clones, including Hirt-extracted DNA, confirmed our observations that both the Y102E and Y102F mutants were unable to maintain BPV genomes as episomes.
FIG 7.
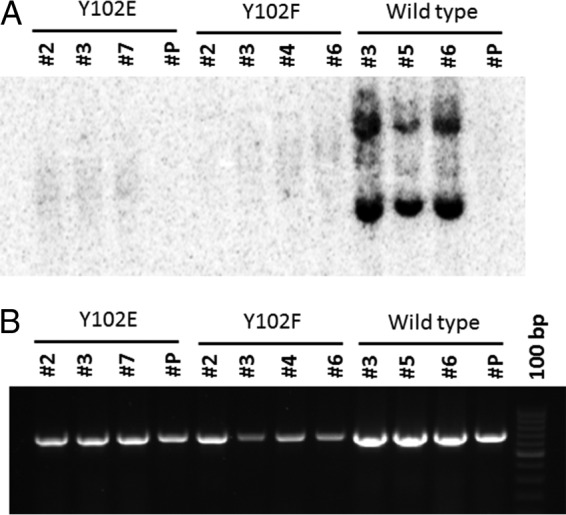
C127 cell lines do not maintain Y102F and Y102E episomes. (A) Total DNA from transformed C127 cells containing Y102E, Y102F, or wild-type BPV genomes was subjected to Southern blotting using a BPV LCR-specific probe. The number below each genome represents an individual clonal cell line. (B) A 677-bp PCR amplicon of the LCR region of the same cell lines represented in panel A demonstrates that each clonal population contains BPV DNA.
DISCUSSION
Here we report the identification of a novel tyrosine phosphorylation event in BPV-1 E2 and its functional characterization. Unfortunately, detection of E2 phosphorylation by mass spectroscopy is qualitative, not quantitative, so we cannot accurately calculate the fraction of E2 that is phosphorylated on Y102. This residue at position 102 is conserved in 72 of the known PV species, including several high-risk HPVs. The detection of phospho-Y102 in two separate samples from live cells, the high confidence score that this PTM received by mass spectrometry analysis, and the conservation of Y102 among diverse PVs indicate that phosphorylation at this residue is relevant for one or more functions of the protein in vivo. While Y102 is not conserved in all PV species and is not correlated with oncogenicity, it is likely that different PVs evolved other E2 phosphorylation sites that exert analogous functionality in regulating the viral replicative program.
To begin investigations of the biological significance of Y102, we generated phosphodefective phenylalanine (Y102F) and phosphomimetic glutamic acid (Y102E) forms. Although the use of glutamate as a tyrosine phosphomimetic is variably effective, there is precedent in tyrosine kinase studies where Tyr-to-Glu mutants successfully mimic the well-known effects of autophosphorylation of the enzyme (47, 48). The BPV-1 E2 Y102F mutant retains the ability to activate transcription and stimulate E2- and E1-dependent transient BPV ori replication. Y102F binds to E1 and reproducibly demonstrated activity that was somewhat greater than that of the WT. Our interpretation is that a fraction of the WT E2 protein may be phosphorylated on Y102, thereby reducing its activity, while Y102F cannot undergo this negatively acting PTM. Indeed, the difference in replication stimulation between the WT and the Y102F mutant in Fig. 5A may reflect the size of the WT phospho-Y102 pool; with this pool being eliminated by the Y102F mutation, E2 achieves its maximal activation. From these results, we conclude that phosphorylation of this tyrosine is not required for the transcription or replication activities of E2. In contrast, the Y102E mutant was completely defective for both transcriptional activation and transient viral DNA replication. The inability of Y102E to coimmunoprecipitate the BPV-1 E1 helicase likely explains its replication deficit. This mutant also failed to coimmunoprecipitate Brd4, which would impact transcription and replication activities (15, 37, 39). We also tested the functionality of Y102F and Y102E in the context of the viral genome. As expected due to the inability to activate transcription and stimulate replication, the Y102E genome was not maintained as a replicating episome in transformed C127 cells; transformation of these cells is dependent upon the expression of E6 and E5 (49). While the phosphorylation-deficient Y102F mutant was able to support transient BPV ori replication, the BPV genome with this mutation was found to be integrated into transfected C217 cells. While it is possible that Y102F genomes overreplicate and achieve toxic levels in host cells, further studies are needed to dissect the pathway and/or cellular partner that accounts for the inability of E2 Y102F to establish stable episomal BPV replicons.
The transcriptional defect of the Y102E mutant is more complex to unravel. Both the Y102E and Y102F mutants coimmunoprecipitated with the E2 transcription coactivators Gps2/AMF1 and Tax1BP1, which bind to the β-sheet region C-terminally to Y102 (20, 21) (Fig. 1A). The data for Gps2 and Tax1BP1 are consistent with our previous conclusion that associations with these two factors are necessary but not sufficient to stimulate E2-dependent transactivation. Unlike Y102F, Y102E was not associated with the full-length chromatin modulator Brd4 or the isolated Brd4 CTM. We believe that it is likely that the Y102E mutant disrupts the folding of the helical structures of the transactivation domain, thereby altering binding to both E1 and Brd4, which interact with opposing surfaces on these helices. These α-helices contain multiple residues important for E1 binding (50) and binding of transcriptional coactivators, including Brd4 (38), Brm (33), and TFIIB (aa 74 to 134) (51). This interpretation is consistent with the cocrystal structure of the HPV-16 E2 TAD in complex with a 20-amino-acid Brd4 CTM peptide (RCSB Protein Data Bank [PDB] accession number 2NNU), which shows that the hydroxyl group of Y102 makes hydrogen bonds with the side chain of E74 (Fig. 1) (38). The E1-binding surface of BPV-1 E2 spans the majority of the TAD (50, 52) and includes the small α-helical loop on which Y102 resides (Fig. 1A). The Y102E mutation prohibits the binding of BPV E2 to E1, perhaps preventing a critical contact with E1 or disrupting the conformation of this loop and/or the N-terminal α-helices in the TAD. It is also possible that the folding of the full-length Brd4 protein juxtaposes it in the vicinity of tyrosine 102, such that the phosphorylation of this residue directly destabilizes the interaction of Brd4 with E2.
The binding of E2 to the BID of Brd4 was unexpected, as the CTM was the only domain previously known to interact with E2. The BID binds to p53 (39) and was recently shown to interact with the PV E2 DNA-binding domain (40). In Brd4:p53 interaction studies, BID was found to undergo a conformational change induced by serine phosphorylation within a different Brd4 region termed the phosphorylation-dependent interaction domain (PDID) (39). Similarly, Brd4 uses a phosphorylation switch within the PDID to modulate binding between high-risk HPV and low-risk HPV (40). High-risk HPVs bind to phospho-PDID, CTM, and BID regions, whereas low-risk HPVs and BPV bind only to the CTM and BID regions of Brd4. While BPV E2 could potentially bind to the Brd4 BID, this interaction is normally undetectable in vivo, as the BID is usually masked by its intramolecular contact with the phospho-PDID and is thus unavailable for interactions with BPV-1 E2 (40).
Because the Y102E mutant failed to activate both transcription and replication and could not associate with key mediators of these processes, we infer that phosphorylation at Y102 is inhibitory. Altered subcellular localization preventing function can also be a consequence of PTMs, especially at or near an NLS. However, we did not observe phospho-Y102 mislocalization to the extent that it affected E2 function, since the Y102E mutant was predominately nuclear.
In keratinocytes, the PV replicative cycle is closely linked to the host cell life cycle, so kinase activity in these cells could be directly correlated with functional outcomes for a particular stage of viral infection. We expect that the kinase responsible for Y102 phosphorylation is most active in undifferentiated, basal keratinocytes, where viral replication and transcription occur at low levels. A study comparing the transcriptomes of keratinocytes in monolayer culture, skin, and reconstituted epidermis noted that growth factor receptors (such as the fibroblast growth factor receptors [FGFRs], c-Met, and insulin-like growth factor 1 receptor [IGF-1R]) and ephrin receptors (such as EphB2) were upregulated in skin and reconstituted epidermis (53). Bioinformatics analyses of E2 using GPS 3.0 (54), NetPhos 2.0 (55), KinasePhos 2.0 (56), and ScanSite 3 (57) predicted kinases that potentially phosphorylate Y102, including members of the FGFR family, IGF-1R, Tyk2, and BTK, many of which are expressed in skin. However, these predictions were inconsistent among these tools. Experiments to identify the specific kinase responsible for Y102 phosphorylation and the phosphatase that presumably removes this PTM are under way.
Our data led us to hypothesize the following scenario. Y102 phosphorylation accumulates early in infection to limit replication and viral gene expression and thereby prevent a lytic infection. As host cells differentiate, we predict that a tyrosine phosphatase acts on E2 Y102, thereby releasing its transcriptional and replication activities that allow viral genome amplification to proceed.
MATERIALS AND METHODS
Cells and transfections.
C33a/E2 cells from Peter Howley and Jianxin You; C127 BPV A3 cells from Michael Botchan; HEK293TT cells from John Schiller and Chris Buck; and C33a, CV-1, and ID13 cells from Douglas Lowy were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (Atlas Biologicals) and a 1% penicillin-streptomycin solution (Pen-Strep; Invitrogen). HEK293TT cells were transfected by using the calcium phosphate method. CV-1 and C33a cells were transfected by using Lipofectamine 2000 at a 2:1 Lipofectamine/DNA ratio. For transfections with Lipofectamine 2000, the DNA and transfection reagent were diluted in Opti-MEM serum-free medium (Invitrogen). The medium was replaced at ∼16 h posttransfection for all experiments.
Plasmids and mutagenesis.
Expression plasmids used include pCI, pEGFP-C1, monomeric RFP-GFP (mRFP-GFP), and pCG-E2 for full-length WT BPV-1 E2; pCG-E2R for the truncated form of BPV-1 E2; pCG-E1 for BPV-1 E1; pHA:AMF1 for HA-tagged Gps2/AMF1; and p3XFLAG-CMV-7.1 Tax1BP1 for Tax1BP1. Tyr-to-Phe (Y102F) and Tyr-to-Glu (Y102E) mutations in BPV-1 E2 were generated by using the QuikChange II site-directed mutagenesis system according to the manufacturer's instructions (Agilent Technologies). Primers used include 5′-CACAAGCTGGGACCGATTCATGTCAGAACCTAAAC-3′ (forward) and 5′-GTTTAGGTTCTGACATGAATCGGTCCCAGCTTGTG-3′ (reverse) for Y102F and 5′-CACAAGCTGGGACCGAGAGATGTCAGAACCTAAACG-3′ (forward) and 5′-CGTTTAGGTTCTGACATCTCTCGGTCCCAGCTTGTG-3′ (reverse) for Y102E. pVL-F:hBrd4 for full-length FLAG-tagged Brd4, pGEX:hBrd4(1224-1362) for GST-CTM, and pGEX:hBrd4(524-579) for GST-BID were previously described (39). Luciferase reporter constructs included pGL2-E2BS-Luc for transactivation assays and pFLORI-BPV1 (firefly luciferase with a long BPV-1 ori) as well as pRL (Renilla) for replication assays. The latter two plasmids were generously provided by J. Archambault, along with pCG-BPV-1 E1 Eag1235 (BPV-1 E1 with enhanced activity) for the E2 replication assays.
Sample preparations for mass spectrometry.
Two separate BPV-1 E2 samples were analyzed by mass spectrometry. One sample was derived from C33a/E2 cells, a cell line generated in the Howley laboratory that stably expresses BPV-1 E2. Cells were grown on 15-cm dishes, released from plates with a 0.05% trypsin solution, pelleted, and frozen at −80°C. A total of 15 frozen pellets were thawed, lysed in urea lysis buffer (8 M urea, 100 mM Tris-HCl [pH 8], 100 mM NaCl, 0.1% NP-40), pooled, and diluted in buffer minus urea for immunoprecipitation with equal volumes of Sepharose A and G beads (Invitrogen) cross-linked with two mouse monoclonal antibodies to BPV-1 E2, B201 and B202 (B201/2 A/G beads). After 4 h of rotation at 4°C, beads were washed 5 times with Tris-buffered saline (TBS), alternating between low and high concentrations of salt (150 and 500 mM). Beads were then suspended in Laemmli SDS-PAGE sample buffer plus 5% β-mercaptoethanol (BME), heated at 95°C, and run on a 12.5% SDS-PAGE minigel. After staining with InstantBlue Coomassie-based solution (Expedeon), a region spanning 50 to 65 kDa was excised and divided into 4 fragments to be analyzed separately. Additional samples were derived from C33a cells transfected with pCG-E2 and cultured on 10-cm dishes. Cells were lysed at 48 h posttransfection with NP-40 lysis buffer (0.5% NP-40, 50 mM Tris-HCl [pH 8], 150 mM NaCl, 2 mM Na3VO4, 10 mM NaF, protease inhibitor cocktail [Sigma]). B201/B202 and Sepharose A/G beads were used to immunoprecipitate E2. After low/high-salt TBS washes as described above, the beads were suspended in Laemmli buffer plus BME, heated, and loaded onto a 10% SDS-PAGE gel. After InstantBlue staining, bands spanning 45 to 50 kDa were excised.
The gel bands for mass spectrometric analysis were first reduced with 10 mM dithiothreitol (DTT) in 10 mM ammonium bicarbonate and then alkylated with 55 mM iodoacetamide (prepared freshly in 10 mM ammonium bicarbonate). Alkylated samples were digested by trypsin (Promega) overnight at 37°C. Tryptic peptides were first injected onto a C18 trapping column (NanoAcquity ultraperformance liquid chromatography [UPLC] trap column, 180 μm by 20 mm, 5 μm, Symmetry C18) before being injected onto an analytical column (NanoAcquity UPLC column, 100 μm by 100 mm, 1.7 μm, BEH130 C18). Peptides were eluted with a linear gradient from 3 to 40% acetonitrile in water with 0.1% formic acid and developed over 90 min at room temperature at a flow rate of 500 nl/min, and the effluent was electrosprayed into a Thermo-Fisher Scientific LTQ Orbitrap Velos Pro mass spectrometer (Thermo-Fisher Scientific) interfaced with a Waters Acquity UPLC system. Analysis of phosphopeptides was performed by using a data-dependent neutral loss scan. Blanks were run prior to the sample run to make sure that there were no significant background signals from solvents or the columns. A database search and data analysis were performed by using Thermo-Fisher Scientific Proteome Discoverer software (v1.3).
Luciferase reporter assays.
For transcriptional assays, C33a and CV-1 cells were cultured in 12-well dishes. Transfections were performed in triplicate. At 48 h posttransfection, cells were rinsed with PBS (Invitrogen) and lysed with the Steady-Glo luciferase substrate (Promega). Lysates were transferred to 96-well Opti-Plates, and luminescence was measured with the PHERAstar plate reader and software. Transient-replication assays were performed by using C33a cells on 96-well, white-walled, clear-bottom plates. At 72 h posttransfection, cells were lysed and treated with the Dual-Glo luciferase system (Promega). Firefly and Renilla luminescence was measured by using the PHERAstar system.
Statistical analysis.
All experiments were repeated at least 3 times. Student's t test was performed for luciferase assays comparing the function of the wild type to that of the mutant. For all experiments, data are expressed as means ± standard errors of the means (SEM). P values of ≤0.05 were considered significant.
Immunoprecipitation and immunoblotting.
Unless otherwise indicated, HEK293TT cells were used for immunoprecipitation and immunoblotting and were harvested for experiments at 48 h posttransfection. All cells were rinsed with cold DPBS before lysis on ice in NP-40 buffer, and lysates were cleared by centrifugation. Brd4 and E1 co-IP samples were also treated with Benzonase nuclease (Millipore) before centrifugation, while GST-Brd4 fragment co-IP samples were treated with ethidium bromide at 0.1 μg/μl, as previously described (39), during immunoprecipitation. For co-IP experiments, the total supernatant from each sample was used after setting aside 5 to 10% for the input; for non-IP immunoblots, volumes corresponding to equal protein levels were measured, as determined by a bicinchoninic acid (BCA) assay (Pierce Thermo Scientific). Co-IP samples were mixed with Sepharose A and G beads plus antibody, M2 affinity gel (Sigma), or glutathione beads (GE Healthcare) overnight as appropriate. Samples were washed with lysis buffer (co-IPs), run on SDS-PAGE gels, and transferred onto 0.45-μm polyvinylidene difluoride (PVDF) membranes (Millipore) in semidry transfer boxes (Bio-Rad). Membranes were blocked in 5% milk, incubated overnight at 4°C with the appropriate primary antibodies, washed with phosphate-buffered saline–Tween (PBST), and incubated at room temperature with either goat anti-mouse or goat anti-rabbit light chain-specific antibodies conjugated to horseradish peroxidase (Jackson Laboratory). Signals were detected with SuperSignal West Dura ECL solution (Pierce) and Amersham ECL Prime (GE Healthcare), using the ImageQuant LAS 4000 system (GE Healthcare). All experiments were repeated at least 3 times. Primary antibodies included B201 (mouse anti-BPV-1 E2), M2 (mouse anti-FLAG; Sigma), 502-2 (rabbit anti-BPV-1 E1), HA-7 (mouse anti-HA; Sigma), and SD8 (rabbit anti-GST).
Immunofluorescence.
Cells were grown on 12-well plates with 18-mm glass coverslips. At 48 h posttransfection, cells were rinsed with cold DPBS and fixed in a 4% paraformaldehyde solution. After permeabilization in antibody dilution buffer (5% normal goat serum and 0.1% Triton X-100 in DPBS), cells were incubated with dilution buffer plus B201 antibodies at 4°C overnight. Coverslips were incubated with dilution buffer plus Alexa Fluor 594 anti-mouse secondary antibody (Invitrogen) or Alexa Fluor 488 anti-rabbit secondary antibody (Invitrogen) as appropriate and mounted onto glass slides with ProLong Gold with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Slides were imaged with a Zeiss fluorescence microscope using QCapture Pro 6 software.
Viral genome detection by Southern blotting and PCR.
C127 cells were transfected with 5 μg of mutant or wild-type BPV genomic DNA (gDNA) along with 1 μg of pBabe-puro by using Lipofectamine reagent at a 1:1 DNA/reagent ratio. On the following day, cells were split and selected for 2 weeks in 1 μg/ml puromycin. Puromycin-selected colonies were isolated and cultured as monolayers. To obtain transformed C127 cells, the puromycin-selected cells were diluted in 0.3% agarose–DMEM supplemented with 10% fetal bovine serum (FBS) and 1× antibiotics (complete DMEM) at 2,500 cells per well and layered onto a 0.6% agarose gel (in complete DMEM) in a six-well plate. After selection for 2 weeks in agarose, large colonies were isolated and subsequently passaged as monolayer cultures in complete DMEM. Cellular total DNA was isolated from these clonal C127 cells by the phenol-chloroform-isoamyl alcohol method (58). About 10 μg of total DNA was digested with XhoI (which does not cut the BPV genome) and the single-cutter HindIII or BamHI restriction enzyme and run on a 1% agarose gel. The gel was treated with 0.1 N HCl and washed in water, and gDNA was transferred overnight with alkaline transfer buffer (0.4 N NaOH and 1 M NaCl) by capillary transfer onto a Hybond N+ nylon membrane. The DNA was UV cross-linked and probed by using a [32P]dCTP-labeled PCR fragment of the 677-bp BPV LCR region mentioned below in 2× SSC (0.3 M NaCl, 30 mM sodium citrate dehydrate, 1% SDS, 1× Denhardt's solution containing 10 mg Ficoll, 10 mg polyvinylpyrrolidone, and 10 mg bovine serum albumin [BSA]). Blots were washed with decreasing concentrations of a saline solution from 2× SSC–0.1% SDS to 0.5% SSC–0.1% SDS and exposed to a PhosphorImager screen. Images were captured after 48 to 72 h by using a Typhoon 9410 scanner (GE Healthcare). Genomic Southern blot analyses of different passages of cell lines were repeated three times. Multiple cell lines were analyzed for the mutant and the wild-type genomes. To probe for viral episomes, Hirt extracts were prepared (59), and a Southern blot procedure was carried out as described above.
Genomic DNA isolated from different cell lines that were used for Southern blotting were also subjected to PCR amplification of the LCR region. Briefly, 50 to 100 ng of gDNA (or Hirt DNA) was amplified by using primer set LCR_forward (ACACCCGGTACACATCCTGT) and LCR_reverse (CGGGAGCCAATCAAAATGCAGCATT) for GoTaq (Promega) polymerase PCR according to the manufacturer's instructions. PCR products (677 bp) were separated in a 1% agarose gel.
Software and Web resources.
The BPV-1 E2 (PDB accession number 2JEU) (60) and HPV16 E2/Brd4 (PDB accession number 2NNU) (38) ribbon structures were generated by using the UCSF Chimera package. Chimera was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (61). These structures are documented in the Papillomavirus Episteme (PaVE) (https://pave.niaid.nih.gov/) and the PDB. PaVE was used to derive information for Table 1.
ACKNOWLEDGMENTS
We thank the following colleagues who generously shared reagents: Jacques Archambault, Peter Howley, and Jianxin You. We appreciate the constructive feedback on the experimental design and reviews of the manuscript by members of the Androphy laboratory.
This project was supported by NIH grants R01CA58376 to E.J.A., F30AI114284 to S.P.C., T32AI060519 to M.D., and T32AI007637 and T32AR062495 to T.G. and by NIH grant R01CA103867, Cancer Prevention Research Institute of Texas grants RP110471 and RP140367, and Welch Foundation grant I-1805 to C.-M.C.
REFERENCES
- 1.Kocjan BJ, Bzhalava D, Forslund O, Dillner J, Poljak M. 21 May 2015. Molecular methods for identification and characterization of novel papillomaviruses. Clin Microbiol Infect doi: 10.1016/j.cmi.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 2.Thomas M, Boiron M, Tanzer J, Levy JP, Bernard J. 1964. In vitro transformation of mice cells by bovine papilloma virus. Nature 202:709–710. doi: 10.1038/202709a0. [DOI] [PubMed] [Google Scholar]
- 3.Law MF, Lowy DR, Dvoretzky I, Howley PM. 1981. Mouse cells transformed by bovine papillomavirus contain only extrachromosomal viral DNA sequences. Proc Natl Acad Sci U S A 78:2727–2731. doi: 10.1073/pnas.78.5.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chesters PM, McCance DJ. 1985. Human papillomavirus type 16 recombinant DNA is maintained as an autonomously replicating episome in monkey kidney cells. J Gen Virol 66(Part 3):615–620. [DOI] [PubMed] [Google Scholar]
- 5.Botchan M, Berg L, Reynolds J, Lusky M. 1986. The bovine papillomavirus replicon. Ciba Found Symp 120:53–67. [DOI] [PubMed] [Google Scholar]
- 6.Geimanen J, Isok-Paas H, Pipitch R, Salk K, Laos T, Orav M, Reinson T, Ustav M Jr, Ustav M, Ustav E. 2011. Development of a cellular assay system to study the genome replication of high- and low-risk mucosal and cutaneous human papillomaviruses. J Virol 85:3315–3329. doi: 10.1128/JVI.01985-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sekhar V, Reed SC, McBride AA. 2010. Interaction of the betapapillomavirus E2 tethering protein with mitotic chromosomes. J Virol 84:543–557. doi: 10.1128/JVI.01908-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McBride AA, Sakakibara N, Stepp WH, Jang MK. 2012. Hitchhiking on host chromatin: how papillomaviruses persist. Biochim Biophys Acta 1819:820–825. doi: 10.1016/j.bbagrm.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Androphy EJ, Lowy DR, Schiller JT. 1987. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature 325:70–73. doi: 10.1038/325070a0. [DOI] [PubMed] [Google Scholar]
- 10.Ustav M, Stenlund A. 1991. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J 10:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. 1990. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 250:1694–1699. doi: 10.1126/science.2176744. [DOI] [PubMed] [Google Scholar]
- 12.Piirsoo M, Ustav E, Mandel T, Stenlund A, Ustav M. 1996. cis and trans requirements for stable episomal maintenance of the BPV-1 replicator. EMBO J 15:1–11. [PMC free article] [PubMed] [Google Scholar]
- 13.Lee AY, Chiang CM. 2009. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J Biol Chem 284:2778–2786. doi: 10.1074/jbc.M805835200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schweiger MR, You J, Howley PM. 2006. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J Virol 80:4276–4285. doi: 10.1128/JVI.80.9.4276-4285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Helfer CM, Pancholi N, Bradner JE, You J. 2013. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J Virol 87:3871–3884. doi: 10.1128/JVI.03068-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.You J, Croyle JL, Nishimura A, Ozato K, Howley PM. 2004. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 117:349–360. doi: 10.1016/S0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- 17.Ilves I, Maemets K, Silla T, Janikson K, Ustav M. 2006. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J Virol 80:3660–3665. doi: 10.1128/JVI.80.7.3660-3665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. 2006. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev 20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng YC, Breiding DE, Sverdrup F, Richard J, Androphy EJ. 2000. AMF-1/Gps2 binds p300 and enhances its interaction with papillomavirus E2 proteins. J Virol 74:5872–5879. doi: 10.1128/JVI.74.13.5872-5879.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breiding DE, Sverdrup F, Grossel MJ, Moscufo N, Boonchai W, Androphy EJ. 1997. Functional interaction of a novel cellular protein with the papillomavirus E2 transactivation domain. Mol Cell Biol 17:7208–7219. doi: 10.1128/MCB.17.12.7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Naidu SR, Sverdrup F, Androphy EJ. 2009. Tax1BP1 interacts with papillomavirus E2 and regulates E2-dependent transcription and stability. J Virol 83:2274–2284. doi: 10.1128/JVI.01791-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambert PF. 1991. Papillomavirus DNA replication. J Virol 65:3417–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen G, Stenlund A. 2000. Two patches of amino acids on the E2 DNA binding domain define the surface for interaction with E1. J Virol 74:1506–1512. doi: 10.1128/JVI.74.3.1506-1512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gammoh N, Grm HS, Massimi P, Banks L. 2006. Regulation of human papillomavirus type 16 E7 activity through direct protein interaction with the E2 transcriptional activator. J Virol 80:1787–1797. doi: 10.1128/JVI.80.4.1787-1797.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grm HS, Massimi P, Gammoh N, Banks L. 2005. Crosstalk between the human papillomavirus E2 transcriptional activator and the E6 oncoprotein. Oncogene 24:5149–5164. doi: 10.1038/sj.onc.1208701. [DOI] [PubMed] [Google Scholar]
- 26.Davy C, McIntosh P, Jackson DJ, Sorathia R, Miell M, Wang Q, Khan J, Soneji Y, Doorbar J. 2009. A novel interaction between the human papillomavirus type 16 E2 and E1-E4 proteins leads to stabilization of E2. Virology 394:266–275. doi: 10.1016/j.virol.2009.08.035. [DOI] [PubMed] [Google Scholar]
- 27.McBride AA, Bolen JB, Howley PM. 1989. Phosphorylation sites of the E2 transcriptional regulatory proteins of bovine papillomavirus type 1. J Virol 63:5076–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McBride AA, Howley PM. 1991. Bovine papillomavirus with a mutation in the E2 serine 301 phosphorylation site replicates at a high copy number. J Virol 65:6528–6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Penrose KJ, Garcia-Alai M, de Prat-Gay G, McBride AA. 2004. Casein kinase II phosphorylation-induced conformational switch triggers degradation of the papillomavirus E2 protein. J Biol Chem 279:22430–22439. doi: 10.1074/jbc.M314340200. [DOI] [PubMed] [Google Scholar]
- 30.Sekhar V, McBride AA. 2012. Phosphorylation regulates binding of the human papillomavirus type 8 E2 protein to host chromosomes. J Virol 86:10047–10058. doi: 10.1128/JVI.01140-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang SW, Liu WC, Liao KY, Tsao YP, Hsu PH, Chen SL. 2014. Phosphorylation of HPV-16 E2 at serine 243 enables binding to Brd4 and mitotic chromosomes. PLoS One 9:e110882. doi: 10.1371/journal.pone.0110882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quinlan EJ, Culleton SP, Wu SY, Chiang CM, Androphy EJ. 14 November 2012. Acetylation of conserved lysines in bovine papillomavirus E2 by p300. J Virol doi: 10.1128/JVI.02771-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar RA, Naidu SR, Wang X, Imbalzano AN, Androphy EJ. 2007. Interaction of papillomavirus E2 protein with the Brm chromatin remodeling complex leads to enhanced transcriptional activation. J Virol 81:2213–2220. doi: 10.1128/JVI.01746-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lace MJ, Ushikai M, Yamakawa Y, Anson JR, Ishiji T, Turek LP, Haugen TH. 2012. The truncated C-terminal E2 (E2-TR) protein of bovine papillomavirus (BPV) type-1 is a transactivator that modulates transcription in vivo and in vitro in a manner distinct from the E2-TA and E8∧E2 gene products. Virology 429:99–111. doi: 10.1016/j.virol.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 35.You J, Schweiger MR, Howley PM. 2005. Inhibition of E2 binding to Brd4 enhances viral genome loss and phenotypic reversion of bovine papillomavirus-transformed cells. J Virol 79:14956–14961. doi: 10.1128/JVI.79.23.14956-14961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng G, Schweiger MR, Martinez-Noel G, Zheng L, Smith JA, Harper JW, Howley PM. 2009. Brd4 regulation of papillomavirus protein E2 stability. J Virol 83:8683–8692. doi: 10.1128/JVI.00674-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. 2006. Brd4 is required for E2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol 80:9530–9543. doi: 10.1128/JVI.01105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abbate EA, Voitenleitner C, Botchan MR. 2006. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol Cell 24:877–889. doi: 10.1016/j.molcel.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. 2013. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell 49:843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu SY, Nin DS, Lee AY, Simanski S, Kodadek T, Chiang CM. 2016. BRD4 phosphorylation regulates HPV E2-mediated viral transcription, origin replication, and cellular MMP-9 expression. Cell Rep 16:1733–1748. doi: 10.1016/j.celrep.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. 2010. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology 399:65–76. doi: 10.1016/j.virol.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gagnon D, Senechal H, D'Abramo CM, Alvarez J, McBride AA, Archambault J. 2013. Genetic analysis of the E2 transactivation domain dimerization interface from bovine papillomavirus type 1. Virology 439:132–139. doi: 10.1016/j.virol.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou N, Lin BY, Duan F, Lee KY, Jin G, Guan R, Yao G, Lefkowitz EJ, Broker TR, Chow LT. 2000. The hinge of the human papillomavirus type 11 E2 protein contains major determinants for nuclear localization and nuclear matrix association. J Virol 74:3761–3770. doi: 10.1128/JVI.74.8.3761-3770.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skiadopoulos MH, McBride AA. 1996. The bovine papillomavirus type 1 E2 transactivator and repressor proteins use different nuclear localization signals. J Virol 70:1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schiller JT, Kleiner E, Androphy EJ, Lowy DR, Pfister H. 1989. Identification of bovine papillomavirus E1 mutants with increased transforming and transcriptional activity. J Virol 63:1775–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang YC, Spalholz BA, Rabson MS, Howley PM. 1985. Dissociation of transforming and trans-activation functions for bovine papillomavirus type 1. Nature 318:575–577. doi: 10.1038/318575a0. [DOI] [PubMed] [Google Scholar]
- 47.Zisch AH, Pazzagli C, Freeman AL, Schneller M, Hadman M, Smith JW, Ruoslahti E, Pasquale EB. 2000. Replacing two conserved tyrosines of the EphB2 receptor with glutamic acid prevents binding of SH2 domains without abrogating kinase activity and biological responses. Oncogene 19:177–187. doi: 10.1038/sj.onc.1203304. [DOI] [PubMed] [Google Scholar]
- 48.Kassenbrock CK, Anderson SM. 2004. Regulation of ubiquitin protein ligase activity in c-Cbl by phosphorylation-induced conformational change and constitutive activation by tyrosine to glutamate point mutations. J Biol Chem 279:28017–28027. doi: 10.1074/jbc.M404114200. [DOI] [PubMed] [Google Scholar]
- 49.Mansur CP, Androphy EJ. 1993. Cellular transformation by papillomavirus oncoproteins. Biochim Biophys Acta 1155:323–345. [DOI] [PubMed] [Google Scholar]
- 50.Baxter MK, McBride AA. 2005. An acidic amphipathic helix in the bovine papillomavirus E2 protein is critical for DNA replication and interaction with the E1 protein. Virology 332:78–88. doi: 10.1016/j.virol.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 51.Yao JM, Breiding DE, Androphy EJ. 1998. Functional interaction of the bovine papillomavirus E2 transactivation domain with TFIIB. J Virol 72:1013–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abbate EA, Berger JM, Botchan MR. 2004. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes Dev 18:1981–1996. doi: 10.1101/gad.1220104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gazel A, Ramphal P, Rosdy M, De Wever B, Tornier C, Hosein N, Lee B, Tomic-Canic M, Blumenberg M. 2003. Transcriptional profiling of epidermal keratinocytes: comparison of genes expressed in skin, cultured keratinocytes, and reconstituted epidermis, using large DNA microarrays. J Invest Dermatol 121:1459–1468. doi: 10.1111/j.1523-1747.2003.12611.x. [DOI] [PubMed] [Google Scholar]
- 54.Cheng Z, Ke Y, Ding X, Wang F, Wang H, Wang W, Ahmed K, Liu Z, Xu Y, Aikhionbare F, Yan H, Liu J, Xue Y, Yu J, Powell M, Liang S, Wu Q, Reddy SE, Hu R, Huang H, Jin C, Yao X. 2008. Functional characterization of TIP60 sumoylation in UV-irradiated DNA damage response. Oncogene 27:931–941. doi: 10.1038/sj.onc.1210710. [DOI] [PubMed] [Google Scholar]
- 55.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. 2004. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4:1633–1649. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 56.Wong YH, Lee TY, Liang HK, Huang CM, Wang TY, Yang YH, Chu CH, Huang HD, Ko MT, Hwang JK. 2007. KinasePhos 2.0: a Web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res 35:W588–W594. doi: 10.1093/nar/gkm322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Obenauer JC, Cantley LC, Yaffe MB. 2003. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kanginakudru S, DeSmet M, Thomas Y, Morgan IM, Androphy EJ. 2015. Levels of the E2 interacting protein TopBP1 modulate papillomavirus maintenance stage replication. Virology 478:129–135. doi: 10.1016/j.virol.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 60.Sanders CM, Sizov D, Seavers PR, Ortiz-Lombardia M, Antson AA. 2007. Transcription activator structure reveals redox control of a replication initiation reaction. Nucleic Acids Res 35:3504–3515. doi: 10.1093/nar/gkm166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]