

ABSTRACT
Herpes simplex virus (HSV) establishes a latent reservoir in neurons of human peripheral nerves. In this quiescent state, the viral genome persists as a circular, histone-associated episome, and transcription of viral lytic cycle genes is largely suppressed through epigenetic processes. Periodically, latent virus undergoes reactivation whereby lytic genes are activated and viral replication occurs. In this Gem, we review recent evidence that mechanisms governing the initial transcription of lytic genes are distinct from those of de novo infection and directly link reactivation to neuronal stress response pathways. We also discuss evidence that lytic cycle gene expression can be uncoupled from the full reactivation program, arguing for a less sharply bimodal definition of latency.
KEYWORDS: JNK signaling, episome, herpes simplex virus, heterochromatin, histone methylation, latency, neurotropic viruses, reactivation, transcriptional regulation
INTRODUCTION
Often we think of viruses as self-serving agents that aggressively replicate to the detriment of the infected cell, but in truth many establish a long-term relationship with their hosts, thereby ensuring a continuous presence and prolonged transmission. Herpesviruses offer a clear example, having evolved a dedicated strategy, termed latency, that limits the pathogenic consequences of infection and avoids immune clearance. For herpes simplex virus (HSV), latency is restricted to postmitotic neurons in the peripheral nervous system (1). Latently infected neurons serve as lifelong reservoirs from which infectious particles can emerge (reactivate) from time to time. In some individuals, repeated reactivation gives rise to painful oral or genital lesions, corneal scarring, nerve inflammation, and even life-threatening encephalitis.
WHAT IS LATENCY?
Herpesvirus latency is defined as the persistence of viral DNA in the absence of infectious viral particles with the potential for these latent genomes to reactivate. For HSV, this refers to latency at the level of individual ganglia because in reality only a fraction of individual infected neurons will reactivate, even in response to a strong stimulus. The clearest molecular hallmark of HSV latency is the expression of several noncoding RNAs known as the latency-associated transcripts (LATs). However, levels of LAT expression also vary between neurons, and HSV latency can be established in the absence of LAT expression, indicating that the transcripts themselves are not required for latency but do contribute to neuronal survival and modulate the efficiency of reactivation (2–4).
In the neuronal nucleus, the 150-kb double-stranded DNA HSV genome exists as a closed circle loaded with regularly spaced nucleosomes. Histones assembled on the viral lytic gene promoters are decorated with posttranslational modifications associated with transcriptional repression (Fig. 1), specifically histone H3 trimethylation at lysine 27 (H3K27me3) and histone H3 di- and trimethylation at lysine 9 (H3K9me2/3) (5–7). Expression of the LATs is thought to modify the chromatin on the viral genome to promote stable but ultimately reversible silencing (8). Repressive chromatin can act as a barrier to positively acting transcription factors, and RNA polymerase II and silencing are augmented by viral microRNAs expressed from the LAT locus that act cooperatively with neuronal miR-138 to suppress translation of key viral mRNAs (9, 10). These blocks result in very reduced levels of viral antigen and prevent viral replication, protecting infected neurons from both intrinsic and acquired immune defenses.
FIG 1.
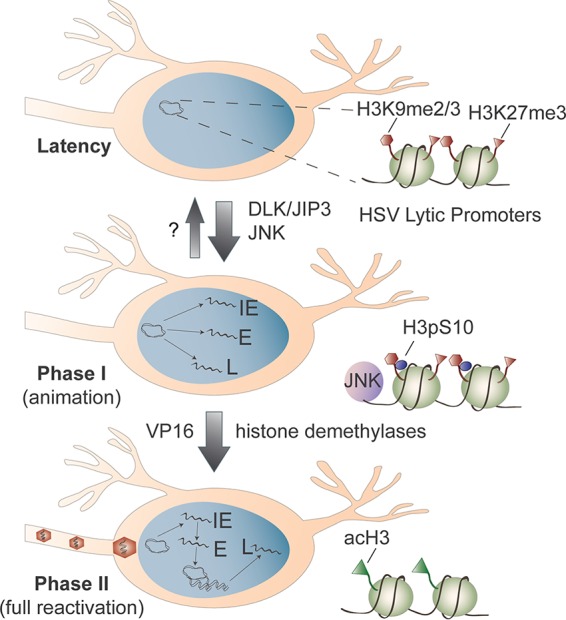
Stepwise reversal of host-mediated epigenetic silencing of the HSV latent genome. During latency, the histones associated with HSV lytic promoters, such as histone H3 trimethylation at lysine 27 (H3K27me3) and histone H3 di- and trimethylation at lysine 9 (H3K9me2/3), are enriched for epigenetic silencing modifications, resulting in a stable chromatin state refractory to transcription by RNA polymerase II. Activation of neuronal stress response pathways triggers DLK/JIP-3-mediated activation of JNK and phosphorylation of histone H3 serine 10 (H3pS10) adjacent to the H3K9me2/3 mark. This dual modification (the methyl-phospho switch) displaces repressive factors and renders the chromatin permissive for simultaneous transcription of the viral IE, E, and L gene mRNAs characteristic of phase I (animation). Viral regulatory factors, such as VP16, synthesized during phase I can then associate with the viral IE promoters and recruit cellular demethylases to remove heterochromatic marks along with histone acetyltransferases that promote hyperacetylation of the viral chromatin. This increases the transcriptional capabilities of viral genes, achieving sufficient levels to initiate HSV DNA replication and assembly of new infectious particles.
MECHANISMS OF HSV LYTIC GENE EXPRESSION DURING THE FIRST PHASE OF HSV REACTIVATION ARE DISTINCT FROM THOSE OF DE NOVO INFECTION
Although the endpoint of reactivation is similar to that of a de novo infection, the start points are very different. When HSV infects a permissive cell, it delivers the viral genome together with tegument proteins that ensure high levels of viral gene expression. The tegument factors include VP16, a transcriptional activator that is corecruited with cellular transcription factors HCF-1 and Oct-1/POU2F1 to a VP16-response sequence found in the promoters of the viral immediate early (IE) gene (11). Synthesis of the IE proteins is necessary for early (E) gene expression, which in turn allows HSV DNA replication and late (L) gene expression. Expression of viral genes at each stage requires both viral and cellular proteins, including histone demethylase enzymes that remove repressive chromatin modifications deposited into the genome immediately following infection (12–15).
Reactivation incurs two major differences from de novo infection. First, the viral genome is associated with a more compact chromatin structure (16), and second, tegument factors such as VP16 are likely absent. Even if transcription occurs, host and viral microRNAs suppress translation of stimulatory viral factors, such as ICP0. So how is latency reversed, and how is the switch from one epigenetic state to the other linked to environmental inputs? Our recent work has uncovered a conceptually simple answer in the form of a two-stage reactivation program initiated by a preexisting epigenetic switch (14, 17). The viral response begins with a generalized burst of HSV gene transcription, termed phase I or animation, resulting in the de novo synthesis of many viral proteins, including the missing tegument factors (17). This discovery came from in vitro studies using primary neurons isolated from peripheral ganglia of prenatal rats or postnatal mice (18, 19). In these models, reactivation can be deliberately induced by interruption of signaling by neurotrophic factors, such as nerve growth factor (NGF), through inhibition of phosphoinositide 3-kinase (PI3K), AKT, mammalian target of rapamycin complex 1 (mTORC1), and/or cap-dependent protein synthesis (20, 21).
Phase I corresponds to a transient wave of viral lytic gene transcription not observed during acute infection. In phase I, IE gene expression is not dependent on the viral transactivator VP16 and the expression of both viral E and L genes occurs even when viral protein synthesis is inhibited, indicating that synthesis of the IE proteins is not required. Likewise, L gene expression is not affected by inhibition of viral DNA replication. Similar disordered patterns of gene expression occur during ex vivo reactivation triggered by axotomy in combination with neurotrophin deprivation (22), cementing the notion of a unique mechanism for transcriptional activation from latent HSV genomes. Activation of the late VP16 promoter has also been found to occur independently of IE gene expression in an in vivo model of HSV reactivation (23). Finally, phase I gene expression occurs in the presence of histone demethylase inhibitors (14), indicating that viral transcription take places without removal of repressive histone modifications.
CO-OPTION OF A NEURONAL STRESS PATHWAY FOR PHASE I OF REACTIVATION
At the onset of reactivation, viral proteins are absent and activation of HSV genes must rely on cellular activities. In the studies described above, reactivation was triggered by inhibiting PI3K signaling. This and other triggers, such as withdrawal of trophic support, axotomy, and heat shock, are known inducers of c-Jun N-terminal kinase (JNK) signaling. In most cells, JNKs play an important role in stress response pathways, but in neurons they also control dendritic arborization and synaptic plasticity (24). In response to nerve injury, JNKs are redirected through mobilization of JNK-interacting protein 3 (JIP3) and dual-leucine-zipper kinase (DLK). This can be induced by inactivation of AKT, a negative regulator of DLK, which occurs when neurotrophin signaling is interrupted. Recent studies showed that activation of JNK by DLK/JIP3 is required for phase I, indicating that activation of the JNK stress response can initiate HSV reactivation (14). Although JNK signaling is important in HSV lytic replication (25), the mechanism by which JNK functions to initiate phase I is distinct because JNK activation by DLK/JIP3 is not required during de novo infection.
Induction of the JNK stress response pathway results in activation of multiple transcription factors that might be involved in HSV reactivation. Less easily understood is how activation permits transcription of lytic genes associated with histones that carry repressive lysine modifications. Although many studies focusing on histone modifications use antibodies generated against individual modifications, the reality is much more complex, with multiple combinations of histone modifications acting together to control gene expression (26). One way in which gene expression can still be initiated even when histones carry repressive lysine modifications is through phosphorylation on the neighboring serine residue, a process known as a histone methyl/phospho switch (27, 28). This switch is used during phase I and is dependent upon JNK activity and correlated with JNK recruitment to viral promoters (14). Therefore, activation of the JNK pathway results in a chromatin state permissive for transcription, even though repressive histone lysine modifications are maintained.
THE SECOND PHASE OF REACTIVATION CLOSELY RESEMBLES DE NOVO INFECTION
Even if all latently infected neurons are exposed to the stress, only a subset will undergo productive reactivation. One idea is that in the reactivating population, sufficient quantities of key viral proteins are made in phase I to cross a threshold that allows a second wave of viral gene expression termed phase II or the synthesis phase, which culminates in the amplification of viral DNA and production of infectious virus (17). Viral gene expression in phase II resembles the cascade of viral gene expression observed during de novo infection. Expression of the L genes is dependent on viral DNA replication, and the stimulatory effect of VP16 is evident. As already mentioned, VP16 is recruited together with host cofactors, such as HCF-1, to the IE promoters and facilitates the recruitment of additional chromatin-remodeling and histone-modifying proteins (29). The requirement for VP16 in phase II argues that chromatin remodeling is critical for full reactivation. Likewise, the activities of cellular H3K27 demethylases (UTX/KDM6A and JMJD3/KDM6B) and the H3K9 demethylase (LSD1/KDM1) are also required for the transition to full reactivation (14, 29–31). Interestingly, explant reactivation, which involves significant physical trauma (axotomy) to the neurons, produces new virus much faster than reactivation through PI3K inhibition (32). Explant/axotomy activates multiple stress responses simultaneously (33), and this might compress the biphasic program, accelerating the onset of phase II.
LATENT INFECTION IN VIVO IS DYNAMIC
Early studies implied that the outcome of neuronal infection is essentially binary: viruses either express lytic mRNAs and attempt to replicate or express the LATs and become latent (34, 35). However, new findings argue for a less rigid distinction. Use of a tracing method to permanently mark neurons that have expressed lytic genes revealed latently infected neurons with evidence of prior lytic promoter activation (36). Likewise, low levels of lytic mRNA can be detected in the ganglia of latently infected mice, and a recent study found lytic mRNAs in more than two-thirds of latently infected neurons (37). These and other observations challenge the either/or view of HSV latency and raise exciting questions that will direct future research. For example, are these “lytic” mRNAs functional, and do they influence latency, neuronal survival, or some aspect of reactivation? IE protein ICP0, for instance, stimulates LAT expression during latency and alters the chromatin composition of the latent genome (38). Lytic gene activity may also be sensed by the neuron, reinforcing the expression of host antiviral factors that help maintain latency.
CONCLUSIONS AND FUTURE DIRECTIONS
These exciting advances highlight the value of modeling HSV latency and reactivation in both cultured neurons and infected animals. The cultured-neuron models have shown that continuous neuronal signaling is necessary to sustain the repressive chromatin state of the viral episome and expression of latency-associated RNAs. Importantly, this provides new ideas for how transcription might be initiated from epigenetically silenced chromatin in the absence of viral factors. Release from epigenetic silencing through a bivalent histone mark (the methyl/phospho switch) is appealing because it directly links viral transcription to neuronal stress response pathways known to induce reactivation. Host transcription factors activated by the same stress pathways will also benefit from the altered chromatin state through increased access to binding sites and greater RNA polymerase processivity. Although these studies have focused on histone H3 serine 10 (H3S10) phosphorylation, stress-associated kinases can also phosphorylate histone H3 serine 28 (H3pS28), which is immediately adjacent to H3K27me3 and represents a second methyl/phospho switch, triggering gene activation from cellular promoters through polycomb displacement (28). Whether this contributes to the initiation of phase I remains to be determined.
Clearly, there has been much progress, but important questions remain. For instance, what limits the progression from phase I/animation to phase II and full reactivation? What are the specific chromatin-remodeling events that need to occur in each step, and what factors are required? How do changes in the higher-order structure of the episome (chromatin looping) observed during reactivation (39) correlate with these events? How do levels of reactivation and chromatin silencing vary between different types of neurons and between HSV-1 and HSV-2? Lastly, we should not forget that neurons are highly specialized cells and often use nuclear functions differently from other cell types. We have just begun to explore epigenetic processes in a neuronal context, so there will doubtless be more surprises along the way.
REFERENCES
- 1.Bloom DC. 2016. Alphaherpesvirus latency: a dynamic state of transcription and reactivation. Adv Virus Res 94:53–80. doi: 10.1016/bs.aivir.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 3.Thompson RL, Sawtell NM. 2001. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J Virol 75:6660–6675. doi: 10.1128/JVI.75.14.6660-6675.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63:2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q-Y, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A 102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwiatkowski DL, Thompson HW, Bloom DC. 2009. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83:8173–8181. doi: 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicoll MP, Hann W, Shivkumar M, Harman LER, Connor V, Coleman HM, Proença JT, Efstathiou S. 2016. The HSV-1 latency-associated transcript functions to repress latent phase lytic gene expression and suppress virus reactivation from latently infected neurons. PLoS Pathog 12:e1005539. doi: 10.1371/journal.ppat.1005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan D, Flores O, Umbach JL, Pesola JM, Bentley P, Rosato PC, Leib DA, Cullen BR, Coen DM. 2014. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe 15:446–456. doi: 10.1016/j.chom.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wysocka J, Herr W. 2003. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem Sci 28:294–304. doi: 10.1016/S0968-0004(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 12.Kristie TM. 2015. Dynamic modulation of HSV chromatin drives initiation of infection and provides targets for epigenetic therapies. Virology 479-480:555–561. doi: 10.1016/j.virol.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh HS, Bryant KF, Nieland TJF, Mazumder A, Bagul M, Bathe M, Root DE, Knipe DM. 2014. A targeted RNA interference screen reveals novel epigenetic factors that regulate herpesviral gene expression. mBio 5:e01086-13. doi: 10.1128/mBio.01086-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M. 2015. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18:649–658. doi: 10.1016/j.chom.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JS, Raja P, Knipe DM. 2016. Herpesviral ICP0 protein promotes two waves of heterochromatin removal on an early viral promoter during lytic infection. mBio 7:e02007-15. doi: 10.1128/mBio.02007-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deshmane SL, Fraser NW. 1989. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol 63:943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog 8:e1002540. doi: 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilcox CL, Johnson EM. 1987. Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J Virol 61:2311–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson AC, Mohr I. 2012. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol 20:604–611. doi: 10.1016/j.tim.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camarena V, Kobayashi M, Kim JY, Roehm PC, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi M, Wilson AC, Chao MV, Mohr I. 2012. Control of viral latency in neurons by axonal mTOR signaling and the 4E-BP translation repressor. Genes Dev 26:1527–1532. doi: 10.1101/gad.190157.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du T, Zhou G, Roizman B. 2011. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc Natl Acad Sci U S A 108:18820–18824. doi: 10.1073/pnas.1117203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson RL, Preston CM, Sawtell NM. 2009. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog 5:e1000352. doi: 10.1371/journal.ppat.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coffey ET. 2014. Nuclear and cytosolic JNK signalling in neurons. Nat Rev Neurosci 15:285–299. doi: 10.1038/nrn3729. [DOI] [PubMed] [Google Scholar]
- 25.Hargett D, McLean T, Bachenheimer SL. 2005. Herpes simplex virus ICP27 activation of stress kinases JNK and p38. J Virol 79:8348–8360. doi: 10.1128/JVI.79.13.8348-8360.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soshnev AA, Josefowicz SZ, Allis CD. 2016. Greater than the sum of parts: complexity of the dynamic epigenome. Mol Cell 62:681–694. doi: 10.1016/j.molcel.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischle W, Wang Y, Allis CD. 2003. Binary switches and modification cassettes in histone biology and beyond. Nature 425:475–479. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 28.Gehani SS, Agrawal-Singh S, Dietrich N, Christophersen NS, Helin K, Hansen K. 2010. Polycomb group protein displacement and gene activation through MSK-dependent H3K27me3S28 phosphorylation. Mol Cell 39:886–900. doi: 10.1016/j.molcel.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 29.Hill JM, Quenelle DC, Cardin RD, Vogel JL, Clement C, Bravo FJ, Foster TP, Bosch-Marce M, Raja P, Lee JS, Bernstein DI, Krause PR, Knipe DM, Kristie TM. 2014. Inhibition of LSD1 reduces herpesvirus infection, shedding, and recurrence by promoting epigenetic suppression of viral genomes. Sci Transl Med 6:265ra169–265ra169. doi: 10.1126/scitranslmed.3010643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Messer HGP, Jacobs D, Dhummakupt A, Bloom DC. 2015. Inhibition of H3K27me3-specific histone demethylases jMJD3 and UTX blocks reactivation of herpes simplex virus 1 in trigeminal ganglion neurons. J Virol 89:3417–3420. doi: 10.1128/JVI.03052-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. 2009. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med 15:1312–1317. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pesola JM, Zhu J, Knipe DM, Coen DM. 2005. Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J Virol 79:14516–14525. doi: 10.1128/JVI.79.23.14516-14525.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fletcher GC, Xue L, Passingham SK, Tolkovsky AM. 2000. Death commitment point is advanced by axotomy in sympathetic neurons. J Cell Biol 150:741–754. doi: 10.1083/jcb.150.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margolis TP, Sedarati F, Dobson AT, Feldman LT, Stevens JG. 1992. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virology 189:150–160. doi: 10.1016/0042-6822(92)90690-Q. [DOI] [PubMed] [Google Scholar]
- 35.Speck PG, Simmons A. 1991. Divergent molecular pathways of productive and latent infection with a virulent strain of herpes simplex virus type 1. J Virol 65:4001–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Proença JT, Coleman HM, Connor V, Winton DJ, Efstathiou S. 2008. A historical analysis of herpes simplex virus promoter activation in vivo reveals distinct populations of latently infected neurones. J Gen Virol 89:2965–2974. doi: 10.1099/vir.0.2008/005066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC. 2014. Lytic gene expression is frequent in HSV-1 latent infection and correlates with the engagement of a cell-intrinsic transcriptional response. PLoS Pathog 10:e1004237. doi: 10.1371/journal.ppat.1004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raja P, Lee JS, Pan D, Pesola JM, Coen DM, Knipe DM. 2016. A herpesviral lytic protein regulates the structure of latent viral chromatin. mBio 7:e00633–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ertel MK, Cammarata AL, Hron RJ, Neumann DM. 2012. CTCF occupation of the HSV-1 genome is disrupted at early times post-reactivation in a transcription-dependent manner. J Virol 86:12741–12759. doi: 10.1128/JVI.01655-12. [DOI] [PMC free article] [PubMed] [Google Scholar]