

Abstract
Two divergent synthetic routes are reported offering access to four quinolone natural products from Pseudonocardia sp. CL38489. Key steps to the natural products involved a regioselective epoxidation, an intramolecular Buchwald–Hartwig amination and a final acid‐catalysed 1,3‐allylic‐alcohol rearrangement to give two of the natural products in one step. This study completes the synthesis of all eight antibacterial quinolone natural products reported in the family. In addition, this modular strategy enables an improved synthesis towards two natural products previously reported.
Keywords: Quinolones, Natural products, Antibacterial agents, Cross‐coupling, Michael addition
Introduction
Quinolones, both natural and synthetic, have repeatedly distinguished themselves as molecules of high biological relevance, which display a plethora of activities. The synthetic fluoroquinolone antibiotics have seen widespread use, with over 800 million patients treated worldwide.1 These antibiotic properties have also been utilised by nature: Pseudomonas aeruginosa produces a number of quinolone N‐oxides, termed Pyo compounds, which are strongly active against gram‐positive bacteria.2, 3 It is believed that this enables P. aeruginosa to outcompete Staphylococcus aureus in cystic‐fibrosis lung infections.4
An area of increasing interest, in which quinolones have been implicated, is that of quorum sensing, whereby bacteria use signaling molecules to modulate their activity in a population‐density‐dependent manner. P. aeruginosa is known to use quinolones as such signaling molecules,5 and it has recently emerged that quinolones are also produced by Buckerholdia and Altermonas spp., which raises the intriguing possibility of interspecies signaling.6, 7, 8
Given this wealth of activities, we became interested in a group of quinolone natural products produced by the actinomycete Pseudonocardia sp. CL38489, which was originally isolated by Dekker et al. from an Indian soil sample.9 (Figure 1). The molecules were originally noted for their antibacterial activity against Helicobacter pylori, which is implicated in the pathogenesis of chronic gastritis, peptic ulcers and gastric cancers.10, 11
Figure 1.
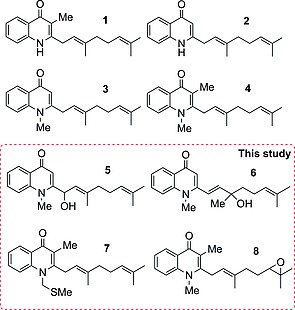
Eight quinolone natural products isolated from Pseudonocardia sp. CL38489.
We recently described the synthesis of compounds 1–4.12 The key step was an sp2–sp3 Suzuki–Miyaura reaction, whereby the various requisite quinolone cores were coupled with a common boronate ester (Scheme 1). However, one drawback of this modular strategy was the necessity to separately synthesise each quinolone coupling partner, with up to seven steps required for each quinolone core.
Scheme 1.
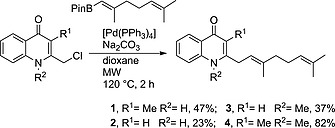
Previously employed sp2–sp3 Suzuki–Miyaura coupling to give natural products 1–4.12
In this study, our attention turned towards the synthesis of the remaining four natural products. While devising the strategy towards these, the similarity between them was noted. It was envisaged that this would enable a divergent approach towards the natural products, with decoration of mutual late‐stage intermediates leading to multiple products through the same synthetic scheme. This approach would offer a much improved efficiency over the original routes.13 In particular, the isomeric relationship between 5 and 6 was noted. It was anticipated that under acidic conditions, the allylic alcohol moiety in 5 could potentially undergo a 1,3‐rearrangement to give 6, which allows interconversion between the two molecules (Scheme 2A). We hypothesised that the additional conjugated system in 6 would be the driving force for the rearrangement. However, due to the differences in the lateral chain of 5 and 6 compared to the rest of the natural products, a retrosynthetic strategy alternative to the sp2–sp3 Suzuki–Miyaura route was required. For natural products 7 and 8 it was noted that the natural products contained previously synthesised 1 as the core structure, which could act as an intermediate in the synthesis of both (Scheme 2B). The synthesis of 8 would also require natural product 4 as an intermediate. The proposed strategy would therefore enable the synthesis of a total of six natural products from just two synthetic routes.
Scheme 2.
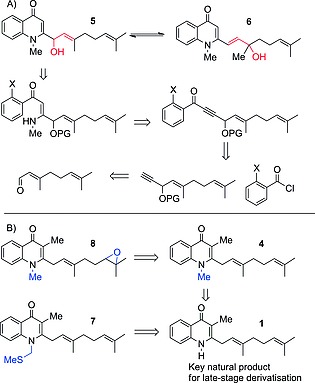
(A) Anticipated equilibration between 5 and 6 could enable access to both products from one synthetic scheme; retrosynthesis of natural product 5. X = halogen, PG = protecting group. (B) Retrosynthesis of natural‐product quinolones 8, 4 and 7 from key natural product 1.
Results and Discussion
The synthesis of 5 took its inspiration from the method of Bernini et al., in which the 4‐quinolone core is assembled by means of a copper‐catalysed heterocyclisation of 1‐(2‐halophenyl)‐2‐en‐3‐amin‐1‐ones, which are in turn synthesised from α,β‐ynones and primary amines.14 Several similar methodologies exist;15, 16 however, the use of these to attain 1,2‐dialkyl‐4‐quinolones has been limited thus far.
The requisite alkyne commenced with the already reported oxidation of geraniol 9 to geranial 10,17 followed by the reaction with ethynyl magnesium bromide to give propargylic alcohol 11 (Scheme 3). It was then necessary to protect the hydroxyl group to preclude the competing esterification in the subsequent Sonogashira‐coupling step; triisopropylsilyl (TIPS) and methoxymethyl ether (MOM) protection proceeded smoothly with good yields to give 12 and 13.
Scheme 3.
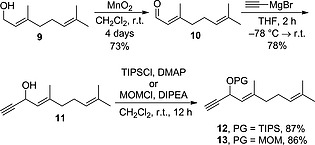
Synthesis of protected propargylic alcohol coupling partners 12 and 13 (DMAP = 4‐dimethylaminopyridine; DIPEA = N,N‐diisopropylethylamine).
Compounds 12 and 13 were then subjected to Bernini's Sonogashira conditions (Scheme 4).14 Compound 12 gave no reaction, presumably due to the presence of the bulky silyl group; however, 13 gave α,β‐ynone 14 in a moderate yield. This ynone was then subjected to a Michael addition with methylamine, which proceeded quantitatively to give 15. A number of conditions were then trialled to elicit the key heterocyclisation. Notably, the copper‐catalysed conditions by Bernini et al. gave no product, as monitored by liquid chromatography–mass spectrometry (LCMS), but instead led to the formation of a dimer. However, Buchwald–Hartwig conditions18 brought about the required transformation to give 16 in a quantitative yield, which then merely required deprotection to give 5. To our delight, when this was attempted by using pyridinium tosylate, 6 was also produced, which indicated that the predicted 1,3‐allylic‐alcohol rearrangement was taking place. The two compounds could be separated by preparative HPLC, which gave access to both natural products. We speculate that the reaction proceeds through elimination of water to give an allylic cation, which reacts with water to give either 5 or 6 depending on the position of attack. The fact that both products were originally isolated in optically enriched form may perhaps imply the existence of an analogous enzymatic process, because our proposed mechanism would result in racemisation.
Scheme 4.
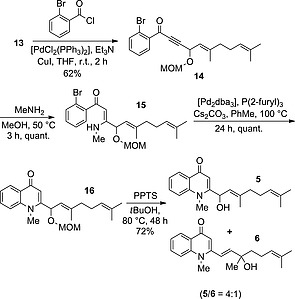
Synthesis of natural products 5 and 6. Attempted deprotection of 16 also resulted in a partial 1,3‐allylic‐alcohol rearrangement, which allowed both to be isolated (PPTS = pyridinium p‐toluenesulfonate).
With these natural products in hand, attention was then turned to 7 and 8. The proposed strategy required natural product 1 as point of divergence; it was prepared according to our previously published procedure.12 Natural product 1 was then subjected to methylation conditions by using LiOtBu as a base (Scheme 5); the choice of base is important to ensure selectivity for N‐ versus O‐methylation.19 This gave natural product 4 in a moderate yield, which then underwent regioselective epoxidation to give 8. The production of three natural products in direct succession demonstrates the high efficiency of this divergent strategy.
Scheme 5.
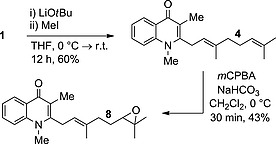
Synthesis of natural products 4 and 8 directly from 1 (mCPBA = meta‐chloroperoxybenzoic acid).
The methylthiomethylenation of 1 to give 7 was then attempted. Analogous conditions to those used in the methylation of 1 were trialled to give 7 with a very poor yield (6 %); starting material was also recovered (37 %; Scheme 6). Optimisation was attempted with a variety of bases (NaH, LiH, nBuLi), solvents (DMF, THF, DMSO), temperatures and concentrations, none of which offered any improvement. TLC analysis of these reactions revealed that a non‐polar species was present, which appeared to gradually decompose to starting material. This prompts us to postulate that a competing elimination reaction leads induces the formation of a dimer, which hydrolyses to starting material upon workup and hence precludes high reaction conversion (Scheme 7; similar dimers have been isolated in the literature, but this was not possible in our case).20 Additionally, a pure sample of 7 was observed to be stable with respect to the work‐up conditions, which implied that the low yield is not due to product decomposition following the quench. Nonetheless, a sufficient amount of 7 was isolated to facilitate future biological testing.
Scheme 6.
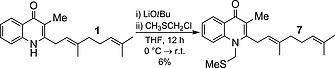
Low‐yielding conversion of 1 to 7.
Scheme 7.
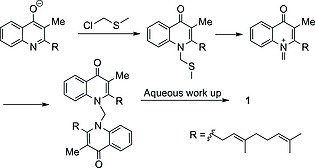
Proposed competing dimerisation accounting for low conversion of 1 to 7.
Conclusions
Two synthetic routes were developed, offering divergent access to six natural products, four of which (5–8) have never before been synthesised. The yields were moderate to excellent, with one exception; however, the main benefit of the divergent strategy is the low number of steps required to yield multiple targets. Considering the essential need for new antibacterial agents, such compounds could play a crucial role in discovering novel modes of action and act as new leads. Following biological screening, the routes will also be suitable for rapid generation of analogues, which will enable the investigation of structure–activity relationships of any activities detected.
Experimental Section
Supporting Information (see footnote on the first page of this article): Full experimental protocols, characterisation data and 1H and 13C NMR spectra.
Supporting information
Supporting Information
Acknowledgements
The research leading to these results has received funding from the European Reserach Council (ERC) under the European Union's Seventh Framework Programme (FP7/2007‐2013) and ERC grant agreement number 279337/DOS. Research in the D. R. S. lab is also supported by the Engineering and Physical Sciences Research Council (EPSRC), Biotechnology and BiologicalSciences Research Council (BBSRC), Medical Research Council (MRC), Cancer Research UK and the Wellcome Trust. Work in the M. W. lab is supported by the BBSRC and MRC. J. T. H. was supported by Trinity College Cambridge. Data accessibility: all data supporting this study are included in the paper and provided as Supporting Information accompanying this paper.
References
- 1. Van Bambeke F., Michot J.‐M., Van Eldere J. and Tulkens P. M., Clin. Microbiol. Infect., 2005, 11, 256–280. [DOI] [PubMed] [Google Scholar]
- 2. Cornforth J. W. and James A. T., Biochem. J., 1956, 63, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hays E. E. and Wells I. C., J. Biol. Chem., 1945, 159, 725–750. [Google Scholar]
- 4. Machan Z. A., Taylor G. W., Pitt T. L., Cole P. J. and Wilson R., J. Antimicrob. Chemother., 1992, 30, 615–623. [DOI] [PubMed] [Google Scholar]
- 5. Heeb S., Fletcher M. P., Chhabra S. R., Diggle S. P., Williams P. and Cámara M., FEMS Microbiol. Rev., 2011, 35, 247–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vial L., Lépine F., Milot S., Groleau M.‐C., Dekimpe V., Woods D. E. and Déziel E., J. Bacteriol., 2008, 190, 5339–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Diggle S. P., Lumjiaktase P., Dipilato F., Winzer K., Kunakorn M., Barrett D. A., Chhabra S. R., Cámara M. and Williams P., Chem. Biol., 2006, 13, 701–710. [DOI] [PubMed] [Google Scholar]
- 8. Reen F. J., Mooij M. J., Holcombe L. J., McSweeney C. M., McGlacken G. P., Morrissey J. P. and O'Gara F., FEMS Microbiol. Ecol., 2011, 77, 413–428. [DOI] [PubMed] [Google Scholar]
- 9. Dekker K. A., Inagaki T., Gootz T. D., Huang L. H., Kojima Y., Kohlbrenner W. E., Matsunaga Y., McGuirk P. R., Nomura E., Sakakibara T., Sakemi S., Suzuki Y., Yamauchi Y. and Kojima N., J. Antibiot., 1998, 51, 145–152. [DOI] [PubMed] [Google Scholar]
- 10. Rho T. C., Bae E. A., Kim D. H., Oh W. K., Kim B. Y., Ahn J. S. and Lee H. S., Biol. Pharm. Bull., 1999, 22, 1141–1143. [DOI] [PubMed] [Google Scholar]
- 11. Hamasaki N., Ishii E., Tominaga K., Tezuka Y., Nagaoka T., Kadota S., Kuroki T. and Yano I., Microbiol. Immunol., 2000, 44, 9–15. [DOI] [PubMed] [Google Scholar]
- 12. Salvaggio F., Hodgkinson J. T., Carro L., Geddis S. M., Galloway W. R. J. D., Welch M. and Spring D. R., Eur. J. Org. Chem., 2016, 434–437. [Google Scholar]
- 13. Shimokawa J., Tetrahedron Lett., 2014, 55, 6156–6162. [Google Scholar]
- 14. Bernini R., Cacchi S., Fabrizi G. and Sferrazza A., Synthesis, 2009, 1209–1219. [DOI] [PubMed] [Google Scholar]
- 15. Vinayaka A. C., Swaroop T. R., Chikkade P. K., Rangappa K. S. and Sadashiva M. P., RSC Adv., 2016, 6, 11528–11535. [Google Scholar]
- 16. Miliutina M., Ivanov A., Ejaz S. A., Iqbal J., Villinger A., Iaroshenko V. O. and Langer P., RSC Adv., 2015, 5, 60054–60078. [Google Scholar]
- 17. Lautens M. and Maddess M. L., Org. Lett., 2004, 6, 1883–1886. [DOI] [PubMed] [Google Scholar]
- 18. Wolfe J. P., Rennels R. A. and Buchwald S. L., Tetrahedron, 1996, 52, 7525–7546. [Google Scholar]
- 19. Adams M., Wube A. A., Bucar F., Bauer R., Kunert O. and Haslinger E., Int. J. Antimicrob. Agents, 2005, 26, 262–264. [DOI] [PubMed] [Google Scholar]
- 20. Gaul C., Schärer K. and Seebach D., J. Org. Chem., 2001, 66, 3059–3073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information