

Abstract
Non‐alcoholic fatty liver disease (NAFLD) is a continuous spectrum of diseases characterized by excessive lipid accumulation in hepatocytes. NAFLD progresses from simple liver steatosis to non‐alcoholic steatohepatitis and, in more severe cases, to liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). Because of its growing worldwide prevalence, various animal models that mirror both the histopathology and the pathophysiology of each stage of human NAFLD have been developed. The selection of appropriate animal models continues to be one of the key questions faced in this field. This review presents a critical analysis of the histopathology and pathogenesis of NAFLD, the most frequently used and recently developed animal models for each stage of NAFLD and NAFLD‐induced HCC, the main mechanisms involved in the experimental pathogenesis of NAFLD in different animal models, and a brief summary of recent therapeutic targets found by the use of animal models. Integrating the data from human disease with those from animal studies indicates that, although current animal models provide critical guidance in understanding specific stages of NAFLD pathogenesis and progression, further research is necessary to develop more accurate models that better mimic the disease spectrum, in order to provide both increased mechanistic understanding and identification/testing of novel therapeutic approaches. © 2016 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: non‐alcoholic fatty liver disease (NAFLD), non‐alcoholic steatohepatitis, hepatocellular carcinoma, animal model, disease histopathology
Introduction
Non‐alcoholic fatty liver disease (NAFLD) is the hepatic manifestation of the metabolic syndrome. NAFLD has a strong association with metabolic abnormalities such as obesity 1, 2, insulin resistance (IR) 3, 4, fasting hyperglycaemia, dyslipidaemia, and altered adipokine profiles 4. Its worldwide prevalence continues to increase with the growing epidemic of obesity and insulin resistance, and it is becoming the most common cause of chronic liver disease 5. NAFLD is a continuous spectrum of diseases characterized by excessive lipid accumulation in hepatocytes. It progresses from simple liver steatosis to non‐alcoholic steatohepatitis (NASH) and, in more severe cases, to liver fibrosis and cirrhosis 6. NASH with fibrosis or cirrhosis increases the risk of developing hepatocellular carcinoma (HCC) 7. Each stage of the disease spectrum has distinctive histopathological characteristics. Simple hepatic steatosis encompasses fat droplet accumulation in hepatocytes 8. As the disease progresses to NASH, hepatocellular injury, ballooning and inflammation develop. Further worsening of NASH leads to liver fibrosis and ultimately to cirrhosis 6.
Hepatic steatosis is caused by excessive import or diminished export or oxidation of free fatty acids (FFAs). NASH is the resultant inflammatory response that is stimulated by various additional hits 6. However, the exact pathogenetic mechanism(s) of NAFLD remain unclear. Further research on pathogenic pathways and potential drug treatments is crucial, given the rapid growth in NAFLD prevalence. Animal models that mirror both the histopathology and pathophysiology of each stage of human NAFLD provide critical guidance for understanding disease pathogenesis and progression. This review will summarize the current and most frequently used animal models for each stage of NAFLD: (1) non‐alcoholic fatty liver (simple steatosis); (2) NASH; and (3) NASH‐associated HCC. We will also outline possible therapeutic targets that have been found recently by the use of animal models.
Table 1.
Animal models of non‐alcoholic fatty liver diseases
| Model | Summary of diet composition | Obese | Steatosis | NASH | Fibrosis | HCC |
|---|---|---|---|---|---|---|
| High‐fat diet | 45–75% of the animals' total calorie intake is derived from fat. The classic reported HFD model comprised 71% fat, 11% carbohydrates, and 18% protein | Yes | Yes | Yes (mild) | Yes | No |
| ob/ob mice | NA | Yes | Yes | No (does not develop spontaneously) | No (resistant to fibrosis) | No |
| db/db mice | NA | Yes | Yes | No (does not develop spontaneously) | No (does not develop spontaneously) | No |
| Methionine and choline‐deficient diet | Diet usually consists of sucrose (40% of energy) and fat (10%); however, it is deficient in methionine and choline | No | Yes | Yes | Yes | No |
| High‐cholesterol diet | Approximately 1% of animals' total calorie intake is from cholesterol. Often fed in conjunction with high fat (15%) or high cholate (0.5%) | Yes | Yes | Yes | Yes | No |
| foz/foz mice | NA | Yes | Yes | Yes | Yes | No |
| Choline‐deficient high‐fat diet | 20% protein, 35% carbohydrate, and 45% fat, without choline added | Yes | Yes | Yes | Yes | Yes |
| Choline‐deficient l‐amino acid‐defined diet | 28.9 kcal/g l‐glutamic acid, 15.8 kcal/g l‐aspartic acid, 12.7 kcal/g l‐arginine, and 10.5 kcal/g l‐leucine, without choline bitartrate | Yes | Yes | Yes | Yes | Yes |
| Choline‐deficient l‐amino acid‐defined diet + carbon tetrachloride | 28.9 kcal/g l‐glutamic acid, 15.8 kcal/g l‐aspartic acid, 12.7 kcal/g l‐arginine, and 10.5 kcal/g l‐leucine, without choline bitartrate, with CCl4 injection | No | Yes | Yes | Yes | Yes |
| High‐fat diet + streptozotocin | 24.8% protein, 46.7% nitrogen‐free extract, and 14.4% fat, with 200‐µg streptozotocin injection | Yes | Yes | Yes | Yes | Yes |
| Hepatocyte‐specific PTEN‐deficient mice | NA | Yes | Yes | Yes | Yes | |
| Db/db mice + DEN | NA | Yes | Yes | Yes | ? | Yes |
DEN, diethylnitrosamine; HCC, hepatocellular carcinoma; NA, not available; NASH, non‐alcoholic steatohepatitis; PTEN, phosphatase and tensin homologue.
Histopathology and pathogenesis of NAFLD
Hepatic steatosis is the hallmark feature of NAFLD, whereby fat droplets accumulate in the form of triglycerides in hepatocytes. NAFLD is histologically diagnosed when accumulation occurs in >5% of hepatocytes 9. The extent of steatosis can be graded according to the percentage of steatotic hepatocytes: mild, 0–33%; moderate, 33–66%; and severe, >66%. In severe cases, steatosis can occupy the entire acinus 10.
Triglycerides in the livers of patients with NAFLD derive from esterification of glycerol and FFAs 6. Triglyceride accumulation occurs when the rate of import or synthesis of FFAs by hepatocytes exceeds the rate of export or catabolism 11, 12. Obesity, and particularly IR, are tightly associated with the genesis of NAFLD 11, 13. Overexpression of tumour necrosis factor (TNF)‐α in obese patients activates IκB kinase β, which plays an important role in IR development by inhibiting the phosphorylation of insulin receptor substrate (IRS)‐1 and IRS‐2 14, 15. IR leads to an increase in the liver triglyceride level and ultimately liver steatosis through various mechanisms 14. First, insulin fails to suppress adipose tissue lipolysis via hormone‐sensitive lipase, resulting in increased efflux of FFAs into the circulation and consequent uptake by the liver 14. Second, IR‐associated hyperinsulinaemia and hyperglycaemia promote hepatic de novo lipid synthesis via upregulation of the membrane‐bound transcription factor sterol regulatory element‐binding protein‐1c (SREBP‐1c) and carbohydrate response element‐binding protein (ChREBP), respectively (Figure 1) 16, 17. Third, hyperinsulinaemia directly inhibits β‐oxidation of FFAs 18. Together, these phenomena promote hepatic FFA accumulation and, through esterification, hepatic triglyceride accumulation and steatosis.
Figure 1.
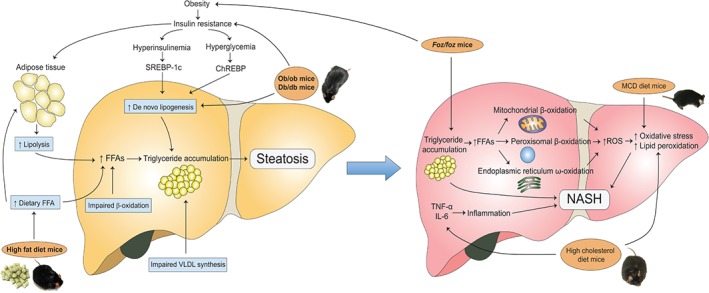
The main mechanisms involved in the experimental pathogenesis of NAFLD and NASH in different animal models. In NAFLD, the mechanisms include increased de novo lipogenesis, increased adipose tissue lipolysis, increased dietary FFA levels, impaired β‐oxidation, and impaired VLDL synthesis. These all lead to hepatic triglyceride accumulation and ultimately NAFLD. db/db mice and ob/ob mice develop NAFLD because of both increased de novo lipogenesis and IR, whereas mice fed an HFD develop NAFLD because of increased dietary FFA levels. In NASH, the two main mechanisms for progression of steatosis to steatohepatitis are increased oxidative stress and proinflammatory cytokines. Mice fed an MCD diet develop NASH because of increased oxidative stress; mice fed a high‐cholesterol diet develop NASH because of both increased oxidative stress and proinflammatory cytokines; foz/foz mice develop NASH because of obesity‐induced IR.
Animal models of NAFLD
High‐fat diet (HFD)
The association between NAFLD and obesity led to the development of an HFD that matches modern Western diets. In HFD animal models, 45–75% of the animals' total calorie intake is derived from fat, and animals are fed predominantly ad libitum, although sometimes forcibly. The classic HFD model used rats fed a diet composed of 71% fat, 11% carbohydrates and 18% protein for 3 weeks, as compared with control rats fed a standard Lieber–DeCarli diet of 35% fat, 47% carbohydrates and 18% protein. The HFD caused hepatic steatosis, and lipid concentrations were almost two‐fold of those in control rats, owing to the increased dietary load of FFAs. Similarly to human NAFLD patients, rats developed IR, as shown by elevated plasma insulin levels. Weight change, however, was the same in both HFD and control rats 19.
A recent study reported similar results in male C57BL/6 mice fed the same HFD for up to 16 weeks. Body weights increased in the HFD and control diet groups. HFD mice showed hepatic steatosis, as verified by the presence of increased liver triglyceride levels, hepatocyte ballooning, Mallory bodies, higher fasting serum glucose levels, and decreased adiponectin levels, suggesting hyperglycaemia and IR 20. Similarly, our group found that male C57BL/6 mice fed an HFD (45% fat, 35% carbohydrates, and 20% protein) for 12 weeks develop steatosis, as shown by increased lipid accumulation (Figure 2A). An HFD has been reported to result in a higher percentage of cells enriched in fat than other diets. For example, Wistar male rats were fed diets with the same quantity (15 g/rat per day) for 16 weeks but with different compositions, including high‐fat, moderate‐fat, high‐sucrose and high‐fructose groups. The high‐fat group had the highest body and liver weights, and the highest percentage of liver steatosis (40%) 21.
Figure 2.
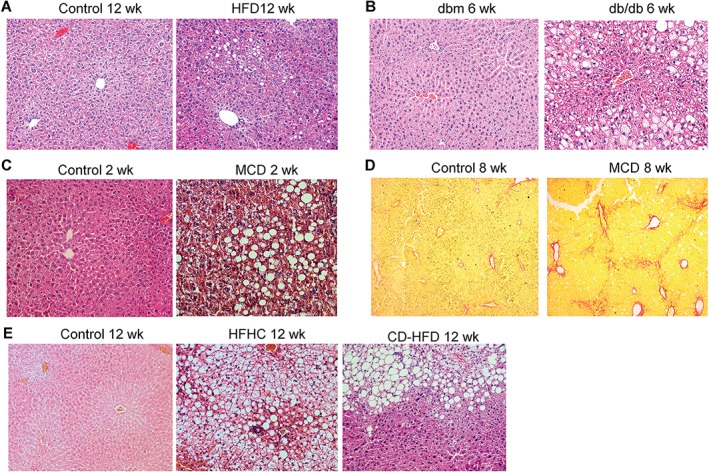
Histopathological features of NAFLD in different animal models. (A–C) Representative haematoxylin and eosin (H&E) staining of liver sections of: (A) C57BL/6 mice fed a control diet or an HFD for 12 weeks; (B) db/db and dbm control mice fed normal chow for 6 weeks; and (C) C57BL/6 mice fed a control diet or an MCD diet for 2 weeks. (D) Representative Sirius Red staining of liver sections of C57BL/6 mice fed a control diet or an MCD diet for 8 weeks. (E) Representative H&E staining of liver sections of C57BL/6 mice fed a control diet, an HFHC diet or a CD‐HFD for 12 weeks.
Unlike various other animal models, animals fed an HFD mimic both the histopathology and pathogenesis of human NAFLD, as they have the hallmark features observed in human NAFLD patients, including obesity and IR. The degree of hepatic steatosis, however, seems to depend on various factors, including rodent strain.
db/db and ob/ob mice
db/db mice are homozygous for the autosomal recessive diabetic gene (db). The db gene encodes a point mutation of the leptin receptor (Ob‐Rb), which leads to defective leptin signalling 22. Therefore, db/db mice have normal or elevated levels of leptin, but are resistant to its effects. Leptin is responsible for regulating feeding behaviour by promoting satiety. These mice have persistent hyperphagia, and are obese and diabetic 23. They show severe hyperglycaemia, hyperinsulinaemia, and elevated serum leptin levels, and develop macrovesicular hepatic steatosis 11, 24, 25 (Figure 2B). db/db mice do not spontaneously develop inflammation when fed a normal control diet. Prolonged calorie overconsumption (>1 month) may lead to slightly aggravated hepatic inflammation 22. Nevertheless, db/db mice rarely show features of NASH when fed a control diet. Thus, db/db mice alone are good models of NAFLD but not of NASH. Despite this, NASH can be induced if db/db mice are given a second hit with a methionine and choline‐deficient (MCD) diet or trans‐fat intake.
ob/ob mice carry an autosomal recessive mutation in the leptin gene. Unlike db/db mice, ob/ob mice have functional leptin receptors, but have truncated and non‐functional leptin. Similarly, these mice are grossly overweight, hyperphagic, hyperinsulinaemic, hyperglycaemic, and resistant to insulin, and develop spontaneous liver steatosis 22 but not steatohepatitis. Secondary insults are also required to trigger steatohepatitis, such as an MCD diet, an HFD, small doses of lipopolysaccharide endotoxin 23, ethanol, or hepatic ischaemia–reperfusion challenge 11. However, unlike db/db mice, ob/ob mice are resistant to hepatic fibrosis, owing to hepatic fibrosis requiring leptin 24.
The advantage of db/db and ob/ob mouse models is that they show characteristics of human metabolic syndrome, unlike various diet models, such as an MCD diet. When fed a standard diet without an additional hit, these mice are useful models of NAFLD, as they develop pronounced hepatic steatosis. With the addition of a second hit such as an MCD diet, db/db mice can also be used to study the progression of steatosis to NASH. However, congenital leptin deficiency and leptin resistance caused by gene mutations in obese humans are extremely rare 26, so db/db and ob/ob mouse models are limited in their ability to reflect the aetiology of human obesity, IR, and hepatic steatosis.
Histopathology and pathogenesis of NASH
The development of steatosis is followed by progression to NASH in one‐third of patients with NAFLD 27. NASH is diagnosed when hepatocellular steatosis occurs with concurrent necroinflammatory reactions of the liver and hepatocellular ballooning with or without fibrosis and/or cirrhosis. Lobular inflammation (usually in acinar zone 3) and portal inflammation are both present in NASH. Lobular inflammation is followed by infiltration of affected areas by innate immune cells 28, 29. Portal inflammation is common and usually mild. Increased portal inflammation may be a marker of severe and advanced NAFLD 30. Other histological lesions present in NASH include hepatocellular ballooning, fibrosis, apoptotic bodies, sinusoidal collagen formation, Mallory–Denk bodies (MDBs), megamitochondria, glycogenated nuclei, and iron deposition 29, 31.
The progressive transition from steatosis to NASH was initially explained by a two‐hit hypothesis 32, although recent studies have proposed a modified ‘multiple‐hit’ model. In this case, the first hit is IR and metabolic disturbance, which leads to liver steatosis. This is followed by a series of hits, including oxidative stress, proinflammatory cytokine‐mediated hepatocyte injury, altered lipid partitioning and hepatoxicity mediated by FFAs, abnormal intrahepatic cholesterol loading, hyperinsulinaemia, hyperleptinaemia, and hypoadiponectinaemia 33, 34. Additionally, genetic predisposition may be involved 15. Of all these factors, two mechanisms are considered to be pivotal: oxidative stress and inflammatory cytokines (Figure 1).
Oxidative stress
Studies have found a strong association between the degree of oxidative stress and the severity of NASH 35, and also the presence of biological markers of oxidative stress, in both patients and animal models of steatohepatitis 36, 37. A major source of oxidative stress in NASH is the excess FFA load resulting from obesity and IR. FFA oxidation occurs in three subcellular organelles: β‐oxidation in mitochondria and peroxisomes, and CYP4A‐catalysed ω‐oxidation in the endoplasmic reticulum 38. In the context of FFA load, mitochondrial β‐oxidation can become overwhelmed, reesulting in an increase in reactive oxygen species (ROS) production 6 (Figure 1). Under continuous oxidative stress, an imbalance between ROS and the antioxidant capacity of the cell leads to lipid peroxidation and ultimately cellular damage 39. Lipid peroxidation of polyunsaturated fatty acids generates toxic aldehyde byproducts, which, together with ROS, cause damage to intracellular organelles, cell death, and activation of fibrogenic hepatic stellate cells 37 (Figure 1).
Proinflammatory cytokines
NASH is tightly associated with chronic hepatic inflammation and abnormal cytokine production. An increase in the synthesis of proinflammatory cytokines, including TNF‐α and interleukin (IL)‐6, has been reported in NASH patients 40. Both TNF‐α and IL‐6 affect adipokine levels, as they: (1) decrease the levels of adiponectin, which has anti‐inflammatory, anti‐atherogenic and anti‐diabetic properties; and (2) increase leptin levels, resulting in perpetuation of the loop of chronic inflammation in obesity (Figure 1) 40, 41.
Animal models of NASH
MCD dietary model
Feeding mice a lipogenic MCD diet is a frequently used and reproducible nutritional model of NASH. The diet usually consists of considerable amounts of sucrose (40% of energy) and is only moderately enriched with fat (10%), but is deficient in methionine and choline. Choline is an essential nutrient that is stored and metabolized chiefly in the liver. Depriving animals of choline alone impairs hepatic very‐low‐density lipoprotein (VLDL) secretion and results in hepatosteatosis, oxidative stress, liver cell death, and changes in cytokines and adipocytokines 42, but causes only slight hepatic inflammation and fibrosis. However, mice fed a diet lacking both choline and methionine develop extensive hepatic inflammation as early as 2 weeks of feeding, and significant fibrosis after 6 weeks 11, 43 (Figure 2C, D). Serum alanine aminotransferase (ALT) levels also increase alongside with ballooning degeneration of hepatocytes 44. Recent literature suggests that the progression of steatosis to steatohepatitis in MCD mouse models involves downregulation of proteins affecting methionine metabolism and oxidative stress, especially peroxiredoxin, which may participate in cellular defence against the development of hepatitis 45. An MCD diet better mimicked the pathological findings of severe human NASH than did other dietary models. Inflammation, fibrosis and hepatocyte apoptosis developed much more quickly and severely than in mice fed an HFD or Western diets. The diet also better models the mechanisms implicated in the pathogenesis of human NASH. Endoplasmic reticulum stress, oxidative stress and autophagocytic stress are all more active in MCD models than in other dietary models 46. Thus, this model is appropriate for studying histologically advanced NASH and the mechanisms of inflammation and fibrosis in NASH.
The MCD model is limited, because it has known disparities with the metabolic profile of human NASH. Instead of being obese, mice fed an MCD diet show significant weight loss, cachexia, no IR, and low serum insulin, fasting glucose, leptin and triglyceride levels 47. Thus, MCD diets are often fed to db/db or ob/ob mice to better replicate human NASH. db/db mice fed an MCD diet show marked hepatic inflammation and fibrosis 25. In addition, the responsiveness of different mouse strains to an MCD diet varies considerably. The release of transaminases differs between mouse strains, and can be ranked as follows: A/J > C57BL/6 > C3H/HeN = Balb/c = DBA/2 J. Long‐term feeding with a methionine‐deficient diet caused more pronounced liver injury in DBA/2 J mice than in C57BL/6 mice, and caused hepatocarcinogenesis in DBA/2 J mice but not in C57BL/6 mice 48.
High‐cholesterol diet (HCD)
Many foods consumed by humans contain high levels of cholesterol. Recent reports have suggested that dietary cholesterol is a critical factor in the progression of steatohepatitis and hepatic inflammation in both animal models 49, 50, 51 and humans 52. Mice fed an HCD (1%) alone show striking increases in serum insulin levels but only slight increases in liver weight, triglyceride levels, FFA levels, and serum ALT levels 52. However, features of NASH are more pronounced when a high amount of cholesterol is given in conjunction with a high amount of fat or a high amount of cholate. Mice fed a high‐fat (15%), high‐cholesterol (1%) diet (HFHC) showed greater weight gain, greater hepatic lipid accumulation, 10‐fold elevations in serum ALT levels, decreased adiponectin levels, adipose tissue inflammation (high gene expression for TNF‐α), and fibrosis. All of these features were more pronounced in HFHC mice than in HFD or HCD mice 52. Similarly, mice fed a high‐cholesterol (1.25%), high‐cholate (0.5%) diet also showed greater steatosis, inflammation, hepatocellular ballooning, and fibrosis 31, 49. Mice fed with a high‐fat (23%), high‐sucrose (424 g/kg) and high‐cholesterol (1.9 g/kg) diet or a choline‐deficient high‐fat diet (CD‐HFD) for 3 months developed pronounced steatohepatitis (Figure 2E). Several studies have suggested that dietary cholesterol reduces VLDL synthesis and β‐oxidation of fatty acids, and increases apoptosis and hepatic oxidative stress 51, 52.
High‐fructose diet
Humans consume a significant number of calories from fructose‐rich foods, and this has been linked with the development of obesity and NASH 53. Findings obtained with C57BL/6 mice fed an HFD or high‐fat, high‐fructose (HFHF) diet suggested that fructose consumption is necessary for the progression of liver fat deposition to fibrogenesis, because, although weight gain, body fat, insulin resistance and liver steatosis were similar between the two groups, hepatic oxidative stress, liver CD11b+F4/80+Gr1+ macrophage numbers, transforming growth factor (TGF)‐β1‐driven fibrogenesis and collagen deposition were increased in mice fed the HFHF diet 53. In a more recent study, Cxcr3‐knockout and C57BL/6 wild‐type mice were fed a similar HFHF diet consisting of an HFHC diet supplemented with drinking water containing 23 g/l fructose. Cxcr3‐knockout mice showed improved liver histology, a lower level of necroinflammation and reduced lipid peroxidation, suggesting that CXCR3 plays a pivotal role in NASH development in HFHF mouse models 54.
foz/foz mice
foz/foz mice have a mutated Alms1 gene, which encodes a protein found in the basal body of the primary cilium. Although its function has not been fully elucidated, ALMS1 may have a role in intracellular transport and appetite regulation 55. foz/foz mice are morbidly obese and hyperphagic, and they show IR, significantly reduced adiponectin levels, increased cholesterol levels, and steatosis. An HFD promotes the transition of steatosis to NASH with severe fibrosis by aggravating metabolic complications, resulting in further decreases in adiponectin levels and increases in cholesterol levels. However, the severity of diet‐induced NASH in foz/foz mice depends on the strain. When foz/foz BALB/c and C57BL6/J mice were fed an HFD, weight gain was equivalent, suggesting that the appetite defect in foz/foz mice is independent of strain, but NAFLD was more severe in foz/foz C57BL6/J mice than in foz/foz BALB/c mice. IR, hyperinsulinaemia, obesity and substantial NAFLD‐related liver fibrosis were seen in foz/foz C57BL6/J mice but not in foz/foz BALB/c mice. These findings suggest that, although the levels of obesity are equal, the responses to obesity, including features of NASH, are strain‐dependent 56.
db/db mice supplemented with iron
In addition to an MCD diet, a recent study found that iron overload in db/db mice can also cause progression of NAFLD to NASH and fibrosis. Unlike db/db mice fed a normal chow diet, db/db mice fed a chow diet supplemented with high iron showed hepatocellular ballooning, fibrogenesis, increased hepatic oxidative stress, inflammasome activation, hepatic inflammatory immune cell activation, and impaired hepatic mitochondrial fatty acid β‐oxidation 57.
NAFLD‐induced HCC
HCC is the third most common cause of cancer‐related death worldwide. Liver cirrhosis is the most important risk factor for HCC, and HCC occurring in patients with non‐cirrhotic NASH is very rare 29. Increased fat uptake, hepatic steatosis and NASH are all incremental risk factors for HCC. Approximately 4–27% of patients with NASH‐related cirrhosis ultimately develop HCC 7. Long‐term follow‐up studies have revealed that HCC in NAFLD patients is less common than HCC in NASH patients, with prevalence rates of 0–0.5% and 0–2.8%, respectively 7, 58, 59. Current mouse models of NAFLD and NASH do not replicate the pathological progression from fatty liver, NASH and fibrosis to HCC. Various experimental mouse models for HCC are available, but only a few represent NAFLD‐induced HCC 60. Thus, more recent studies have focused on establishing novel NASH‐associated HCC mouse models.
Dietary NAFLD‐induced HCC
Models fed only one type of diet have distinctive limitations. C57BL/6 mice fed an HFD do not show NASH‐like pathology, whereas mice fed an MCD diet or a choline‐deficient diet do. However, MCD or choline‐deficient diets could not induce features of metabolic syndrome or obesity. Wolf et al proposed a mixed diet model combining a choline‐deficient diet and an HFD. Liver steatosis, features of metabolic syndrome and liver damage, as reflected by elevated serum ALT and aspartate aminotransferase levels, were present concurrently in this novel model. Features of liver damage were reminiscent of human NASH, including oxidative stress, hepatocyte ballooning, immune cell infiltration, glycogenated nuclei, and MDBs. The tumour incidence in HFD mice was only 2.5%, as compared with 25% in CD‐HFD mice 61. In another combination dietary model, C57BL/6 mice fed a choline‐deficient l‐amino acid‐defined diet (CDAA) developed liver injury that mimicked NASH features and led to HCC. Feeding with CDAA induced IR and increased hepatic steatosis, altered carbohydrate and lipid metabolism enzymes, and caused liver damage and fibrosis. HCC developed after 9 months of feeding 62. Asgharpour et al recently reported a diet‐induced animal model of NAFLD that recapitulates the key human NASH‐associated HCC features. They generated an isogenic strain from C57Bl/6 J and 129S1/SvlmK mice. B6/129 mice fed a high‐fat, high‐carbohydrate diet sequentially developed steatosis in 4–8 weeks, NASH in 16–24 weeks, and HCC at week 52, which may be an ideal preclinical model of NASH‐associated HCC 63.
Combined chemical and dietary model
C57BL/6 mice fed a CDAA diet and subjected to low‐dose intraperitoneal injections of carbon tetrachloride (CCl4) have more marked features of NASH and HCC. Mice had greater steatosis, lobular inflammation and fibrogenesis than mice fed a CDAA diet alone. Additionally, although only 35% of CDAA C57BL/6 mice developed HCC, all of the CDAA + CCl4 mice developed HCC, with a higher average tumour diameter 62. In another combined model, C57BL/6 mice were fed an HFD and treated with streptozotocin (STZ). STZ, a glucosamine‐nitrosourea compound, is toxic towards pancreatic β‐cells, and induces hypoinsulinaemia, hyperglycaemia and diabetes in mice. STZ‐primed mice stimulated with an HFD induced histological changes, including steatosis, lobular inflammation, fibrosis and, at 20 weeks, tumour protrusion. Other observed features resembling human NASH included body weight gain, increases in fasting blood sugar levels, and increases in serum ALT levels. Male STZ HFD mice had increased proliferation of hepatocytes at 16 weeks, and eventually developed HCC. The model provides insights into the mechanisms linking metabolic disorder, NASH, and HCC 64.
Genetic NAFLD‐induced HCC
Phosphatase and tensin homolog (PTEN) is a tumour suppressor because of its lipid phosphatase activity, and is mutated in many human cancers 65. PTEN is important for preventing oncogenesis in the liver, and deficiency results in proliferation, antiapoptosis, and oncogenesis. Hepatocyte‐specific PTEN‐deficient mice develop features similar to those of human NASH and NASH‐associated HCC 66. Tumours were present in the livers of 66% of male and 30% of female PTEN‐deficient mice at 40–44 weeks, and HCC was present in 83% of male and 50% of females at 74–78 weeks 66. Thus, this model is useful for understanding not only the pathogenesis of NASH, but also the progression of NASH to HCC.
Combined genetic and chemical NAFLD‐induced HCC
Genetic obesity in db/db mice is a direct promoter of NASH‐associated HCC development. db/db mice treated with the carcinogen diethylnitrosamine when aged 13–15 days had higher body weights, higher liver weights, hepatic steatosis, a higher HCC incidence, and higher numbers of and larger tumour nodules. Findings from this mouse model suggest that obesity and NASH increase susceptibility to HCC development 67.
Using animal models for pathogenesis and treatment
Animal models are important in elucidating the mechanisms and pathways involved in the pathogenesis of the NAFLD spectrum, and studies using the aforementioned animal models may provide promising results for possible future treatments for NAFLD and NASH. A recent study using HFD mouse models found that activation of cyclin D3/cyclin‐dependent kinase 4 is a key event in the development of NAFLD 68. In db/db mice, carboxylesterase 2 was demonstrated to be a novel triglyceride hydrolase involved in triglyceride homeostasis and NAFLD 69. In db/db mice fed an MCD diet, administration of exendin‐4 (a glucagon‐like peptide‐1 analogue) improved MCD diet‐induced steatohepatitis and reduced hepatic triglyceride and FFA contents, suggesting that exendin‐4 could be used for the treatment of non‐obese patients with NASH 70.
Conclusion
The reviewed animal models all have individual limitations in representing the full disease spectrum of NAFLD. Some replicate the histopathology of NAFLD but not the physiological properties, and others replicate the physiological properties but not the histopathology. Despite their shortcomings, they are useful tools for studying the pathogenesis and progression of NAFLD, and uncovering potential treatment targets, as indicated by those mentioned in this review. Nevertheless, more accurate animal models that better mimic the disease spectrum are necessary, and require further research.
Author contributions statement
All authors contributed to writing the manuscript and approved the final version.
Acknowledgements
The project was supported by research funds from RGC‐GRF Hong Kong (1406415), the National Basic Research Programme of China (973 Programme, 2013CB531401), and a Dr and Madam Tzu‐leung Ho Full Scholarship for Medical Students 2014/15 to JKCL.
No conflicts of interest were declared.
References
- 1. Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology 1990; 12: 1106–1110. [DOI] [PubMed] [Google Scholar]
- 2. Ratziu V, Giral P, Charlotte F, et al. Liver fibrosis in overweight patients. Gastroenterology 2000; 118: 1117–1123. [DOI] [PubMed] [Google Scholar]
- 3. Marchesini G, Brizi M, Morselli‐Labate AM, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med 1999; 107: 450–455. [DOI] [PubMed] [Google Scholar]
- 4. Gaggini M, Morelli M, Buzzigoli E, et al. Non‐alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013; 5: 1544–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wree A, Broderick L, Canbay A, et al. From NAFLD to NASH to cirrhosis – new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol 2013; 10: 627–636. [DOI] [PubMed] [Google Scholar]
- 6. Dowman JK, Tomlinson JW, Newsome PN. Pathogenesis of non‐alcoholic fatty liver disease. QJM 2010; 103: 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 2010; 51: 1820–1832. [DOI] [PubMed] [Google Scholar]
- 8. Hubscher SG. Histological assessment of non‐alcoholic fatty liver disease. Histopathology 2006; 49: 450–465. [DOI] [PubMed] [Google Scholar]
- 9. Tandra S, Yeh MM, Brunt EM, et al. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J Hepatol 2011; 55: 654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brunt EM, Janney CG, Di Bisceglie AM, et al. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999; 94: 2467–2474. [DOI] [PubMed] [Google Scholar]
- 11. Anstee QM, Goldin RD. Mouse models in non‐alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol 2006; 87: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bradbury MW, Berk PD. Lipid metabolism in hepatic steatosis. Clin Liver Dis 2004; 8: 639–671, xi. [DOI] [PubMed] [Google Scholar]
- 13. Day CP. Pathogenesis of steatohepatitis. Best Pract Res Clin Gastroenterol 2002; 16: 663–678. [DOI] [PubMed] [Google Scholar]
- 14. Nieto‐Vazquez I, Fernandez‐Veledo S, Kramer DK, et al. Insulin resistance associated to obesity: the link TNF‐alpha. Arch Physiol Biochem 2008; 114: 183–194. [DOI] [PubMed] [Google Scholar]
- 15. Medina J, Fernandez‐Salazar LI, Garcia‐Buey L, et al. Approach to the pathogenesis and treatment of nonalcoholic steatohepatitis. Diabetes Care 2004; 27: 2057–2066. [DOI] [PubMed] [Google Scholar]
- 16. Araya J, Rodrigo R, Videla LA, et al. Increase in long‐chain polyunsaturated fatty acid n ‐ 6/n ‐ 3 ratio in relation to hepatic steatosis in patients with non‐alcoholic fatty liver disease. Clin Sci (Lond) 2004; 106: 635–643. [DOI] [PubMed] [Google Scholar]
- 17. Dentin R, Girard J, Postic C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein‐1c (SREBP‐1c): two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005; 87: 81–86. [DOI] [PubMed] [Google Scholar]
- 18. Hamel FG, Bennett RG, Upward JL, et al. Insulin inhibits peroxisomal fatty acid oxidation in isolated rat hepatocytes. Endocrinology 2001; 142: 2702–2706. [DOI] [PubMed] [Google Scholar]
- 19. Lieber CS, Leo MA, Mak KM, et al. Model of nonalcoholic steatohepatitis. Am J Clin Nutr 2004; 79: 502–509. [DOI] [PubMed] [Google Scholar]
- 20. Eccleston HB, Andringa KK, Betancourt AM, et al. Chronic exposure to a high‐fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxid Redox Signal 2011; 15: 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fakhoury‐Sayegh N, Trak‐Smayra V, Khazzaka A, et al. Characteristics of nonalcoholic fatty liver disease induced in wistar rats following four different diets. Nutr Res Pract 2015; 9: 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trak‐Smayra V, Paradis V, Massart J, et al. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high‐calorie diet. Int J Exp Pathol 2011; 92: 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang SQ, Lin HZ, Lane MD, et al. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA 1997; 94: 2557–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol 2002; 37: 206–213. [DOI] [PubMed] [Google Scholar]
- 25. Sahai A, Malladi P, Pan X, et al. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short‐form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol 2004; 287: G1035–G1043. [DOI] [PubMed] [Google Scholar]
- 26. Paz‐Filho G, Mastronardi C, Delibasi T, et al. Congenital leptin deficiency: diagnosis and effects of leptin replacement therapy. Arq Bras Endocrinol Metabol 2010; 54: 690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 2006; 43: S99–S112. [DOI] [PubMed] [Google Scholar]
- 28. Ganz M, Szabo G. Immune and inflammatory pathways in NASH. Hepatol Int 2013; 7(suppl 2): 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brunt EM, Tiniakos DG. Histopathology of nonalcoholic fatty liver disease. World J Gastroenterol 2010; 16: 5286–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brunt EM, Kleiner DE, Wilson LA, et al. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): a histologic marker of advanced NAFLD – clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology 2009; 49: 809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol 2012; 18: 2300–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Day CP, James OF. Steatohepatitis: a tale of two ‘hits’? Gastroenterology 1998; 114: 842–845. [DOI] [PubMed] [Google Scholar]
- 33. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010; 52: 1836–1846. [DOI] [PubMed] [Google Scholar]
- 34. Yilmaz Y. Is non‐alcoholic fatty liver disease a spectrum, or are steatosis and non‐alcoholic steatohepatitis distinct conditions? Aliment Pharmacol Ther 2012; 36: 815–823. [DOI] [PubMed] [Google Scholar]
- 35. Petta S, Muratore C, Craxi A. Non‐alcoholic fatty liver disease pathogenesis: the present and the future. Dig Liver Dis 2009; 41: 615–625. [DOI] [PubMed] [Google Scholar]
- 36. Sanyal AJ, Campbell‐Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001; 120: 1183–1192. [DOI] [PubMed] [Google Scholar]
- 37. George J, Pera N, Phung N, et al. Lipid peroxidation, stellate cell activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. J Hepatol 2003; 39: 756–764. [DOI] [PubMed] [Google Scholar]
- 38. Reddy JK. Nonalcoholic steatosis and steatohepatitis. III. Peroxisomal beta‐oxidation, PPAR alpha, and steatohepatitis. Am J Physiol Gastrointest Liver Physiol 2001; 281: G1333–G1339. [DOI] [PubMed] [Google Scholar]
- 39. Pessayre D, Berson A, Fromenty B, et al. Mitochondria in steatohepatitis. Semin Liver Dis 2001; 21: 57–69. [DOI] [PubMed] [Google Scholar]
- 40. Tilg H. The role of cytokines in non‐alcoholic fatty liver disease. Dig Dis 2010; 28: 179–185. [DOI] [PubMed] [Google Scholar]
- 41. Stojsavljevic S, Gomercic Palcic M, Virovic Jukic L, et al. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol 2014; 20: 18070–18091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Corbin KD, Zeisel SH. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol 2012; 28: 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamada T, Obata A, Kashiwagi Y, et al. Gd‐EOB‐DTPA‐enhanced‐MR imaging in the inflammation stage of nonalcoholic steatohepatitis (NASH) in mice. Magn Reson Imaging 2016; 34: 724–729. [DOI] [PubMed] [Google Scholar]
- 44. Larter CZ, Yeh MM, Williams J, et al. MCD‐induced steatohepatitis is associated with hepatic adiponectin resistance and adipogenic transformation of hepatocytes. J Hepatol 2008; 49: 407–416. [DOI] [PubMed] [Google Scholar]
- 45. Lee SJ, Kang JH, Iqbal W, et al. Proteomic analysis of mice fed methionine and choline deficient diet reveals marker proteins associated with steatohepatitis. PLoS One 2015; 10: e0120577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Machado MV, Michelotti GA, Xie G, et al. Mouse models of diet‐induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One 2015; 10: e0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rinella ME, Green RM. The methionine‐choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol 2004; 40: 47–51. [DOI] [PubMed] [Google Scholar]
- 48. Kanuri G, Bergheim I. In vitro and in vivo models of non‐alcoholic fatty liver disease (NAFLD). Int J Mol Sci 2013; 14: 11963–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matsuzawa N, Takamura T, Kurita S, et al. Lipid‐induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007; 46: 1392–1403. [DOI] [PubMed] [Google Scholar]
- 50. Zheng S, Hoos L, Cook J, et al. Ezetimibe improves high fat and cholesterol diet‐induced non‐alcoholic fatty liver disease in mice. Eur J Pharmacol 2008; 584: 118–124. [DOI] [PubMed] [Google Scholar]
- 51. Subramanian S, Goodspeed L, Wang S, et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor‐deficient mice. J Lipid Res 2011; 52: 1626–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Savard C, Tartaglione EV, Kuver R, et al. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013; 57: 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kohli R, Kirby M, Xanthakos SA, et al. High‐fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010; 52: 934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang X, Han J, Man K, et al. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J Hepatol 2016; 64: 160–170. [DOI] [PubMed] [Google Scholar]
- 55. Bell‐Anderson KS, Aouad L, Williams H, et al. Coordinated improvement in glucose tolerance, liver steatosis and obesity‐associated inflammation by cannabinoid 1 receptor antagonism in fat Aussie mice. Int J Obes (Lond) 2011; 35: 1539–1548. [DOI] [PubMed] [Google Scholar]
- 56. Farrell GC, Mridha AR, Yeh MM, et al. Strain dependence of diet‐induced NASH and liver fibrosis in obese mice is linked to diabetes and inflammatory phenotype. Liver Int 2014; 34: 1084–1093. [DOI] [PubMed] [Google Scholar]
- 57. Handa P, Morgan‐Stevenson V, Maliken BD, et al. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am J Physiol Gastrointest Liver Physiol 2016; 310: G117–G127. [DOI] [PubMed] [Google Scholar]
- 58. Rafiq N, Bai C, Fang Y, et al. Long‐term follow‐up of patients with nonalcoholic fatty liver. Clin Gastroenterol Hepatol 2009; 7: 234–238. [DOI] [PubMed] [Google Scholar]
- 59. Ong JP, Pitts A, Younossi ZM. Increased overall mortality and liver‐related mortality in non‐alcoholic fatty liver disease. J Hepatol 2008; 49: 608–612. [DOI] [PubMed] [Google Scholar]
- 60. Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol 2009; 90: 367–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wolf MJ, Adili A, Piotrowitz K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross‐talk with hepatocytes. Cancer Cell 2014; 26: 549–564. [DOI] [PubMed] [Google Scholar]
- 62. De Minicis S, Agostinelli L, Rychlicki C, et al. HCC development is associated to peripheral insulin resistance in a mouse model of NASH. PLoS One 2014; 9: e97136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Asgharpour A, Cazanave SC, Pacana T, et al. A diet‐induced animal model of non‐alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 2016; 65: 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fujii M, Shibazaki Y, Wakamatsu K, et al. A murine model for non‐alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol 2013; 46: 141–152. [DOI] [PubMed] [Google Scholar]
- 65. Horie Y, Suzuki A, Kataoka E, et al. Hepatocyte‐specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest 2004; 113: 1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Watanabe S, Horie Y, Kataoka E, et al. Non‐alcoholic steatohepatitis and hepatocellular carcinoma: lessons from hepatocyte‐specific phosphatase and tensin homolog (PTEN)‐deficient mice. J Gastroenterol Hepatol 2007; 22(suppl 1): S96–S100. [DOI] [PubMed] [Google Scholar]
- 67. Shen J, Tsoi H, Liang Q, et al. Oncogenic mutations and dysregulated pathways in obesity‐associated hepatocellular carcinoma. Oncogene 2016. DOI: 10.1038/onc.2016.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jin J, Valanejad L, Nguyen TP, et al. Activation of CDK4 triggers development of non‐alcoholic fatty liver disease. Cell Rep 2016; 16: 744–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li Y, Zalzala M, Jadhav K, et al. Carboxylesterase 2 prevents liver steatosis by modulating lipolysis, endoplasmic reticulum stress, and lipogenesis and is regulated by hepatocyte nuclear factor 4 alpha in mice. Hepatology 2016; 63: 1860–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yamamoto T, Nakade Y, Yamauchi T, et al. Glucagon‐like peptide‐1 analogue prevents nonalcoholic steatohepatitis in non‐obese mice. World J Gastroenterol 2016; 22: 2512–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]