

Abstract
Deposits comprised of amyloid‐β (Aβ) are one of the pathological hallmarks of Alzheimer's disease (AD) and small hydrophobic ligands targeting these aggregated species are used clinically for the diagnosis of AD. Herein, we observed that anionic oligothiophenes efficiently displaced X‐34, a Congo Red analogue, but not Pittsburgh compound B (PIB) from recombinant Aβ amyloid fibrils and Alzheimer's disease brain‐derived Aβ. Overall, we foresee that the oligothiophene scaffold offers the possibility to develop novel high‐affinity ligands for Aβ pathology only found in human AD brain, targeting a different site than PIB.
Keywords: Alzheimer's disease, amyloid ligands, fluorescence, luminescent conjugated oligothiophenes, proteins
Protein aggregates are the pathological hallmarks of a wide range of neurodegenerative diseases, including Alzheimer's disease (AD), and reagents for visualizing these proteinaceous species are essential for diagnosis.1, 2 In this regard, small hydrophobic ligands that are selective for protein aggregates that have an extensive cross β‐pleated sheet conformation and sufficient structural regularity have been developed.3, 4, 5, 6, 7, 8 The most common ligands are derivatives of Congo Red or Thioflavins. Some of these molecular scaffolds targeting amyloid‐β (Aβ) deposits have also been modified for diagnosis of AD pathology in living subjects by positron emission tomography (PET) imaging.9 Recent studies have shown that different morphotypes of Aβ deposits exist.10, 11, 12, 13 Thus, in order to evaluate the contribution of these morphotypes to the complex Aβ pathology in AD brain, it is imperative to understand which aggregated morphotypes and which specific binding sites are recognized by individual ligands.
Recently, luminescent conjugated oligothiophenes (LCOs) have been employed as novel tools for fluorescence imaging of protein aggregates. Compared to conventional ligands, LCOs have been shown to detect a wider range of disease‐associated protein aggregates.14, 15, 16, 17, 18 However, the binding mode of LCOs in comparison to other ligands has never been reported. Herein, we initially explored the binding of three LCOs (Figure 1 A) to Aβ aggregates in comparison to the conventional ligands, 3H‐PIB and 3H‐X‐34 (Figure 1 A) which bind to distinct sites on Aβ fibrils. PIB is a benzothiazole derivative which binds to a different site than Thioflavin T or Congo Red, whereas X‐34 is a bis‐styrylbenzene and an analogue of Congo Red. First, we investigated the binding of nonradioactive q‐FTAA, p‐FTAA, and h‐FTAA to recombinant Aβ 1–42 fibrils in competition with radiolabelled PIB and X‐34. Displacement studies with 1 μm compounds showed that all three LCOs predominantly competed with 3H‐X‐34, and not 3H‐PIB (Figure 1 B). p‐FTAA displayed an EC50 value around 15 nm, whereas q‐FTAA and h‐FTAA showed higher EC50 values, 630 nm and 250 nm, respectively (Table 1). By contrast, for 3H‐PIB the displacement was less than 50 % at 1 μm h‐FTAA and less for the other LCOs, suggesting that the effects of the compounds on 3H‐PIB binding may be due to LCO binding to secondary low affinity sites that affect PIB binding. Thus, we conclude that on recombinant Aβ 1–42 fibrils, LCOs predominantly competed with the Congo Red derivative, 3H‐X‐34. This finding is consistent with earlier studies showing that p‐FTAA binds in a similar fashion as Congo Red to HET‐s amyloid fibrils.19, 20 p‐FTAA displaced 3H‐X‐34 much more efficiently than did Congo Red, whereas q‐FTAA and h‐FTAA produced similar EC50 values as Congo Red (Table 1).
Figure 1.
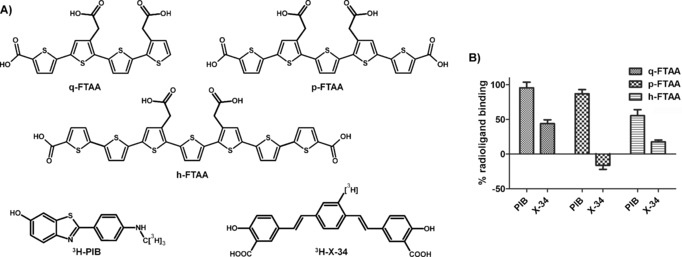
A) Chemical structures of q‐FTAA, p‐FTAA, h‐FTAA, 3H‐PIB and 3H‐X‐34. B) Displacement of 3H‐PIB or 3H‐X‐34 from recombinant Aβ1‐42 fibrils by the three different LCOs. Percent radioligand binding=(binding in absence of competitor–binding in the presence of 1 μm LCO). Mean of two assays on separate days ± SD.
Table 1.
EC50 Values (nm) of LCO competition for 3H‐X‐34 binding to Aβ preparations.
| Ligand | Synthetic Aβ1‐42 fibrils | ADPBC |
|---|---|---|
| q‐FTAA | 330–630 | 300–500 |
| p‐FTAA | 15 | 0.7 |
| h‐FTAA | 250 | 57 |
| Congo Red | 400 | N.D.* |
*Not determined.
PIB binds with high affinity to an isolatable insoluble fraction of the total AD brain Aβ pathology that is only observed in humans and is negligible in primates, canine, and transgenic mouse animal models.21, 22, 23 Therefore, we tested the LCOs against the Alzheimer's disease PIB binding complex (ADPBC) purified from AD brain.5 As shown in Table 1, the displacement of 3H‐X‐34 by the LCOs was also observed with the ADPBC. q‐FTAA displayed a similar EC50 value, 300–500 nm, to that with recombinant Aβ 1–42 fibrils. In contrast, the EC50 values for p‐FTAA and h‐FTAA, 0.7 nm and 57 nm, respectively, were lower (higher affinity) than those compounds for recombinant Aβ 1–42 fibrils. Thus, both p‐FTAA and h‐FTAA were even more efficient competitors of 3H‐X‐34 when using an isolated fraction of the Aβ pathology from human brain.
After establishing that the LCOs competed for binding to Aβ with 3H‐X‐34, we next investigated the effect of minor chemical alterations of the LCOs on the displacement of 3H‐X‐34. Previous studies have shown that chemical modifications of the α‐terminal positions can markedly improve the binding to protein aggregates, as well as increase the therapeutic effect of LCOs in mice infected with prions.15, 16, 20 In this regard, the q‐FTAA scaffold was selected, since it was rather straightforward to replace the α‐terminal hydrogen with other chemical moieties. Furthermore, improvements in affinity due to such minor chemical modifications would be readily apparent, as q‐FTAA displayed a higher EC50‐value than the other two LCOs. To produce a palette of ligands, we selected the previously reported tetrameric building block 1 14 (Scheme 1). By applying various electrophilic aromatic substitution, Ullman type coupling, carbonylation, and hydrolysis protocols, seven tetrameric analogues, q‐FTAA‐NO2, q‐FTAA‐Br, q‐FTAA‐I, q‐FTAA‐OMe, q‐FTAA‐CN, q‐FTAA‐CONH2 and q‐FTAA‐CO2H with different moieties in one of the α‐terminal positions along the thiophene backbone were synthesized (Scheme 1).
Scheme 1.
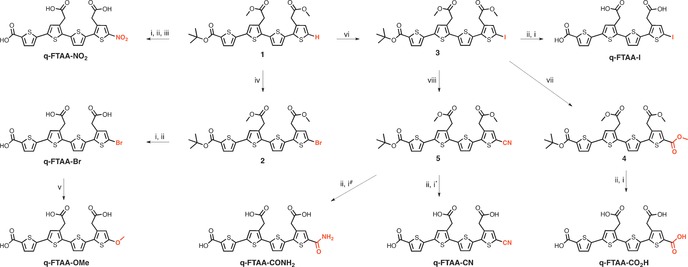
Reagents and conditions: (i) NaOH (1 m), dioxane, H2O; (ii) TFA, DCM; (iii) NaNO2, TFA, DCM; (iv) NBS, DMF; (v) NaOMe, CuBr, MeOH, DMF; (vi) NIS, TFA, DMF; (vii) Pd(PPh3)4, Mo(CO)6, TEA, DBU, MeOH, dioxane; (viii) CuCN, DMF. * Reaction temperature 0 °C. # Reaction temperature 50 °C.
From the binding competition with the q‐FTAA derivatives, it was evident that the nature of the α‐terminal chemical moiety had a major influence on the LCO efficiency to displace 3H‐X‐34 from recombinant Aβ 1–42 fibrils (Table 2). Introducing a nitro (q‐FTAA‐NO2) or an amide (q‐FTAA‐CONH2) group at the α‐terminal slightly decreased the EC50 values, whereas introduction of a bromo (q‐FTAA‐Br), iodo (q‐FTAA‐I), methoxy (q‐FTAA‐OMe) or nitrile (q‐FTAA‐CN) group at the α‐terminal position had a major impact on the displacement of 3H‐X‐34.
Table 2.
EC50 Values (nm) of q‐FTAA analogues competition for 3H‐X‐34 binding to Aβ preparations.
| LCO | Synthetic Aβ1‐42 fibrils | ADPBC |
|---|---|---|
| q‐FTAA | 330–630 | 300–500 |
| q‐FTAA‐Br | 20 | 2.2 |
| q‐FTAA‐I | 90 | 50 |
| q‐FTAA‐NO2 | 120 | 18 |
| q‐FTAA‐CONH2 | 220 | 55 |
| q‐FTAA‐OMe | 64 | 40 |
| q‐FTAA‐CO2H | 830 | 100 |
| q‐FTAA‐CN | 15 | <0.1 |
The lowest EC50 value for recombinant Aβ 1–42 fibrils, 15 nm, was obtained for q‐FTAA‐CN, suggesting that having a linear polar moiety, such as the nitrile group, in one of the α‐terminal positions of the tetrameric backbone is favorable for having an efficient binding to recombinant Aβ 1–42 fibrils. In contrast, attaching an additional α‐terminal carboxyl group (q‐FTAA‐CO2H) rendered an LCO less efficient in displacing 3H‐X‐34 (Table 2). Thus, compared to the pentameric‐ and heptameric analogues, p‐FTAA and h‐FTAA, which have bi‐terminal carboxyl groups, a tetrameric oligothiophene with carboxyl group functionalities at both α‐terminal positions was a strikingly inefficient competitor for 3H‐X‐34. Hence, the spacing of the terminal carboxyl groups is also a major chemical determinant for achieving an efficient competitive ligand for 3H‐X‐34 binding. Distinct spacing of the carboxyl groups along the thiophene backbone has also been shown to influence the LCOs performance for spectral assignment of different protein aggregates, as well as their therapeutic potency in prion‐infected mice.17, 20
The effectiveness of the competition of the q‐FTAA‐analogues for 3H‐X‐34 binding was even more striking for the ADPBC (Table 2). Except for q‐FTAA, all the analogues displayed lower EC50 values with ADPBC compared to the values obtained with recombinant Aβ 1–42 fibrils. In addition, on the ADPBC isolated from human AD brain, q‐FTAA‐CO2H was more efficient in displacing 3H‐X‐34 than q‐FTAA. Similar to the results obtained when using recombinant Aβ 1–42 fibrils, the most efficient competitor of 3H‐X‐34 was q‐FTAA‐CN. Overall, these experiments verified that an alteration of the chemical moiety in the α‐terminal position highly influences the binding mode of the tetrameric LCOs to Aβ species derived from human AD brain.
Finally, we employed q‐FTAA‐CN for histological staining of human brain tissue sections with AD pathology (Figure 2). When using 100 nm q‐FTAA‐CN, specificity towards Aβ plaque pathology was observed even in the presence of tau pathology. As shown in Figure 2, Aβ aggregates and tau neurofibrillary tangles were identified by antibody staining, whereas q‐FTAA‐CN fluorescence was only observed from the immunopositive Aβ deposits. Thus, even at >1000 times the EC50 of q‐FTAA‐CN for ADPBC (<0.1 nm), q‐FTAA‐CN displayed a dominant selectivity for Aβ plaque pathology in AD brain. The q‐FTAA‐CN selectivity for Aβ pathology was also confirmed by applying an LCO, h‐FTAA, previously shown to bind both Aβ deposits, NFTs and dystrophic neurites, to a section pre‐stained with 100 nm q‐FTAA‐CN (Supporting Information, Figure S1). The tau pathology, dystrophic neurites and NFTs, was only stained by h‐FTAA. In addition, when using 3 μm of q‐FTAA‐CN for staining, fluorescence was also observed from NFTs (Supporting Information, Figure S1). Hence, q‐FTAA‐CN had a strikingly higher affinity for Aβ deposits than aggregated species composed of tau and this high affinity towards Aβ pathology was achieved by introducing a nitrile group at one of the α‐terminal positions of the tetrameric thiophene backbone.
Figure 2.
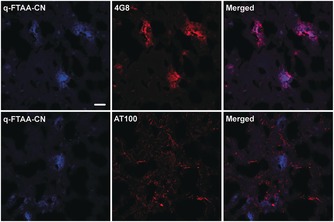
Images of q‐FTAA‐CN (100 nm) and antibody labelling in human AD brain tissue. q‐FTAA‐CN fluorescence (blue) are observed from immunopositive Aβ plaques (4G8 antibody), whereas no co‐localization are obtained from q‐FTAA‐CN and an antibody (AT100) towards neurofibrillary tangles. Scale bar=20 μm.
In conclusion, we have shown that anionic oligothiophenes compete for binding of 3H‐X‐34 but not 3H‐PIB to recombinant Aβ amyloid fibrils as well as to Aβ deposits derived from AD brain. In addition, for a tetrameric thiophene scaffold, the nature of the α‐terminal chemical moiety was demonstrated to be a key determinant for efficient tetrameric LCO displacing 3H‐X‐34 binding from Aβ fibrillar pathology. Overall, we foresee that optimized oligothiophenes might be utilized as high‐affinity ligands, targeting Aβ pathology in human AD brain in a different fashion than PIB, potentially recognizing different polymorphs of Aβ deposits.
Experimental Section
Frozen brain sections from human AD brain was purchased from Tissue Solutions Ltd, Glasgow, Scotland. Tissue Solutions Ltd confirmed that these human tissue samples have been collected with ethics committee approval and with permission to use these sections for research, and that all samples have been collected from donors followed written consent. Full experimental details, additional figures and NMR spectra of new compounds are given in the Supporting Information.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Our work is supported by the Swedish Foundation for Strategic Research (K.P.R.N, M.B.) and the Erling Persson foundation (K.P.R.N, H.A., M.B.). H.L. was supported by the Coins for Alzheimer's Research Trust (C.A.R.T.) and NIH R21 NS080576.
M. Bäck, H. Appelqvist, H. LeVine, K. P. R. Nilsson, Chem. Eur. J. 2016, 22, 18335.
References
- 1. Ross C. A., Poirier M. A., Nat. Med. 2004, 10, S10–S17. [DOI] [PubMed] [Google Scholar]
- 2. Lockhart A., Drug Discovery Today 2006, 11, 1093–1099. [DOI] [PubMed] [Google Scholar]
- 3. Styren S. D., Hamilton R. L., Styren G. C., Klunk W. E., J. Histochem. Cytochem. 2000, 48, 1223–1232. [DOI] [PubMed] [Google Scholar]
- 4. Kung H. F., Lee C. W., Zhuang Z. P., Kung M. P., Hou C., Plossl K., J. Am. Chem. Soc. 2001, 123, 12740–12741. [DOI] [PubMed] [Google Scholar]
- 5. Mathis C. A., Wang Y., Holt D. P., Huang G. F., Debnath M. L., Klunk W. E., J. Med. Chem. 2003, 46, 2740–2754. [DOI] [PubMed] [Google Scholar]
- 6. Nesterov E. E., Skoch J., Hyman B. T., Klunk W. E., Bacskai B. J., Swager T. M., Angew. Chem. Int. Ed. 2005, 44, 5452–5456; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5588–5592. [Google Scholar]
- 7. Furumoto S., Okamura N., Iwata R., Yanai K., Arai H., Kudo Y., Curr. Top. Med. Chem. 2007, 7, 1773–1789. [DOI] [PubMed] [Google Scholar]
- 8. Nilsson K. P. R., FEBS Lett. 2009, 583, 2593–2599. [DOI] [PubMed] [Google Scholar]
- 9. Klunk W. E., Engler H., Nordberg A., Wang Y., Blomqvist G., Holt D. P., Bergström M., Savitcheva I., Huang G. F., Estrada S., Ausen B., Debnath M. L., Barletta J., Price J. C., Sandell J., Lopresti B. J., Wall A., Koivisto P., Antoni G., Mathis C. A., Långström B., Ann. Neurol. 2004, 55, 306–319. [DOI] [PubMed] [Google Scholar]
- 10. Maarouf C. L., Daugs I. D., Spina S., Vidal R., Kokjohn T. A., Patton R. L., Kalback V. L., Luehrs D. C., Walker D. G., Castano E. M., Beach T. G., Ghetti B., Roher A. B., Mol. Neurodegener. 2008, 3, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.H. LeVine,3rd, L. C. Walker, Neurobiol. Aging 2016, 42, 205–212. [DOI] [PMC free article] [PubMed]
- 12. Eisenberg D., Jucker M., Cell 2012, 148, 1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu J. X., Qiang W., Yau W. M., Schwieters C. D., Meredith S. C., Tycho R., Cell 2013, 154, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Åslund A., Sigurdson C. J., Klingstedt T., Grathwohl S., Bolmont T., Dickstein D. L., Glimsdal E., Prokop S., Lindgren M., Konradsson P., Holtzman D. M., Hof P. R., Heppner F. L., Gandy S., Jucker M., Aguzzi A., Hammarström P., Nilsson K. P. R., ACS Chem. Biol. 2009, 4, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klingstedt T., Åslund A., Simon R. A., Johansson L. B. G., Mason J. J., Nyström S., Hammarström P., Nilsson K. P. R., Org. Biomol. Chem. 2011, 9, 8356–8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klingstedt T., Shirani H., Åslund K. O. A., Cairns N. J., Sigurdson C. J., Goedert M., Nilsson K. P. R., Chemistry 2013, 19, 10179–10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klingstedt T., Shirani H., Mahler J., Wegenast-Braun B. M., Nyström S., Goedert M., Jucker M., Nilsson K. P. R., Chemistry 2015, 21, 9072–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shirani H., Linares M., Sigurdson C., Lindgren M., Norman P., Nilsson K. P. R., Chemistry 2015, 21, 15133–15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schütz A. K., Soragni A., Hornemann S., Aguzzi A., Ernst M., Böckmann A., Meier B. H., Angew. Chem. Int. Ed. 2011, 50, 5956–5960; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6078–6082. [Google Scholar]
- 20. Herrmann U. S., Schütz A. K., Shirani H., Huang D., Saban D., Nuvolone M., Li B., Ballmer B., Åslund A. K., Mason J. J., Rushing E., Budka H., Nyström S., Hammarström P., Böckmann A., Caflisch A., Meier B. H., Nilsson K. P. R., Hornemann S., Aguzzi A., Sci. Transl. Med. 2015, 7, 299ra123. [DOI] [PubMed] [Google Scholar]
- 21. Klunk W. E., Lopresti B. J., Ikonomovic M. D., Lefterov I. M., Koldamova R. P., Abrahamson E. E., Debnath M. L., Holt D. P., Huang G. F., Shao L., DeKosky S. T., Price J. C., Mathis C. A., J. Neurosci. 2005, 25, 10598–10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rosen R. F., Walker L. C., LeVine 3rd H., Neurobiol. Aging 2011, 32, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matveev S. V., Spielmann H. P., Metts B. M., Chen J., Onono F., Zhu H., Scheff S. W., Walker L. C., LeVine 3rd H., J. Neurochem. 2014, 131, 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary