

ABSTRACT
Streptococcus pyogenes (group A streptococcus, GAS) causes a wide range of clinical manifestations ranging from mild self-limiting pyoderma to invasive diseases such as sepsis. Also of concern are the post-infectious immune-mediated diseases including rheumatic heart disease. The development of a vaccine against GAS would have a large health impact on populations at risk of these diseases. However, there is a lack of suitable models for the safety evaluation of vaccines with respect to post-infectious complications. We have utilized the Lewis Rat model for cardiac valvulitis to evaluate the safety of the J8-DT vaccine formulation in parallel with a rabbit toxicology study. These studies demonstrated that the vaccine did not induce abnormal pathology. We also show that in mice the vaccine is highly immunogenic but that 3 doses are required to induce protection from a GAS skin challenge even though 2 doses are sufficient to induce a high antibody titer.
Keywords: M-protein, peptide, rheumatic fever, rheumatic heart disease, streptococcus pyogenes, vaccine
Vaccines have proven to be a very cost effective method in preventing many infectious diseases. However, vaccine development has proven to be extremely challenging for some pathogens partially due to the lack of appropriate models for testing of the vaccine candidates before clinical studies. Here we discuss an approach we have adopted by selecting optimal animal models for different aspects of vaccine development and testing safety of a vaccine candidate for Streptococcus pyogenes.
GAS infection causes death either directly due to sepsis / deep tissue infection or post-infective complications such as rheumatic heart disease (RHD). It is estimated that GAS is responsible for over 600 million cases of non-life threatening pharyngitis and over 100 million cases of pyoderma annually. If untreated, pharyngitis and pyoderma 1 can lead to Rheumatic Fever (RF) - an autoimmune process triggered by GAS antigens involving both antibodies and T cells that cross-react with host target proteins and damage host tissue, including myocardium and the heart valves (RHD). It is thus critical that any vaccine approach minimizes the possibility of targeting GAS epitopes that induce autoreactive B and T cell responses. This is an impediment to vaccine research, particularly since in an early study using a crude antigen prepared from GAS, 3 individuals developed rheumatic fever.2 In contrast, GAS antigens have been administered to volunteers with no reported serious adverse events.3,4 In 1979, the FDA regulated the exclusion of GAS from vaccine products after reviewing and considering the findings of the independent advisory panel (“Review of Bacterial Vaccines and Bacterial Antigens”).5 This exclusion was lifted in 2006.6
From an immunological perspective, GAS presents significant obstacles to vaccine development.7,8 The organism can subvert the immune system and present dominant antigens that can display an array of allelic types. In spite of these obvious decoys, the main strategy to develop vaccines for these organisms has been to focus on dominant epitopes. The obvious reason for this approach is that such antigens and epitopes are easy to define. However, our approach to vaccine development for GAS has been to avoid dominant antigens; and instead attempt to define cryptic epitopes that induce protective immune responses that do not constitute the normal responses and repertoire that follow natural infection. Such epitopes are much more likely to be conserved between strains.
As a suitable vaccine candidate, we identified and defined a cryptic epitope in the C3-repeat of the GAS M protein. We used a series of overlapping peptides from the highly conserved C3 repeat region and generated antibodies to each peptide and screened the antibodies for their capacity to kill GAS in vitro in an opsonophagocytosis assay. We identified p145, a 20-mer peptide that induced functional antibodies in mice.9,10 We further observed that most children living in streptococcal-endemic areas did not develop antibodies to p145 (demonstrating that it was ‘cryptic’), whereas most adults did have antibodies against this peptide,11 presumably as a result of prolonged exposure. We found that affinity-purified human antibodies could also kill multiple strains of GAS in vitro.12 Thus, we concluded that p145 is a poor immunogen in its natural state, and was thus not under immune pressure, explaining why it may be highly conserved across all GAS isolates. However, synthetic peptide, p145, was highly immunogenic and antibodies induced following immunization were able to recognize the epitope on the bacterial cell surface.
To develop this as a vaccine candidate, we next defined the minimal protective epitope within p145.10 To achieve this it was necessary to maintain the natural α helical structure of the p145 epitope. This was accomplished by developing a folding technology based on the GCN4 DNA binding protein from yeast. Peptide sequences from p145 were flanked by GCN4 sequences.10 The minimal vaccine epitope that we thus defined and folded correctly, J8, was able to induce antigen-specific serum IgG in outbred mice when conjugated to diphtheria toxoid (DT) and administered intramuscularly with Alum 13; it induced IgG and IgA antibodies when administered intranasally in either a proteosome vesicle 14 or when conjugated to certain lipids.15-17
Following the development of a candidate vaccine, further experiments were performed to exclude the possibility that the epitope would trigger autoimmunity. Here we report the preclinical immunogenicity testing and safety evaluation of the vaccine formulation using 3 different but complementary models.
Intramuscular immunogenicity of the vaccine formulation
To investigate the potential of the vaccine formulation to induce an immune response, cohorts of male and female outbred SWISS mice were immunized intramuscularly (IM) with the J8-DT/alum; specific cohorts received one or 2 boosts, 21 d apart. Blood was collected from animals at specific time points and antigen-specific serum IgG titers were determined (Fig. 1).
Figure 1.
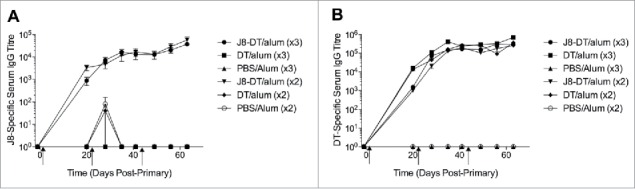
(A) J8-specific or (B) DT-specific serum IgG in SWISS mice (n = 5 female and n = 5 male mice) administered J8-DT/alum, DT/alum or PBS/alum with 2 or 3 intramuscular immunizations on days 0, 21 and 42 (x3 immunisations only). Mice were immunized as previously described30 with minor modifications. Briefly, 4–6 week old SWISS mice (n = 5 female and n = 5 male/group housed separately) were immunized intramuscularly on day 0 with J8-DT (50 ug), DT (50 ug) or PBS formulated with alum (Alhydrogel 2%). Selected cohorts were boosted on day 21 or on days 21 and 42. Sera from the cohorts were collected on selected days pre and post-immunisation. SWISS mice were purchased from The Animal Resource Centre, Western Australia. Statistical significance was calculated using GraphPad Prism version 6. One-way Analysis of Variance (ANOVA) was applied to evaluate significant differences in absorbance levels. The Dunnett's test was performed as a post-hoc test. A P value of < 0.05 was considered significant. Data was expressed as mean ± SEM.
ELISAs were performed as previously described.18 In brief, Nunc Maxisorp F96 plates (Thermo Fisher, Scoresby, Australia) were coated with 100 ul of antigen (rM5 at 1 ug/ml or J8 at 5 ug/ml) in bicarbonate coating buffer (pH 9.6) overnight at 4°C. The wells were washed 4 times with PBS-Tween 20 buffer and blocked with 200 µl of postcoating buffer (TropBio) for 1 h at 37°C. After the wells were washed as described above, 2-fold serial dilutions of sera, starting at 1:100, were added to duplicate wells at 100 µl per well and incubated for 1 h at 37°C. Followed by a third wash, horseradish peroxidase-conjugated goat anti-IgG (Jackson ImmunoResearch, West Grove, PA, USA) at 1:5,000 dilution was added and incubated for 1 h at 37°C. Pooled sera from rM5- or J8-DT-immunized animals were used as positive control for rM5 ELISA and J8 ELISA respectively, and pooled normal sera were used as negative control per plate. After a final wash, 2,2′-Azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) substrate solution (KPL, Gaithersburg, USA) was added and incubated at room temperature for 20 min. The absorbance was measured at 414 nm with a reference wavelength of 492 nm on a Multiskan microplate reader (Titertek). The reaction was defined as positive when its OD value was 3SD above the mean OD of control wells containing serum from naïve animals. Titres were expressed as the highest dilution giving a positive reaction.
Interestingly, the titer of J8-specific serum IgG was not significantly different between cohorts of mice immunized twice compared to cohorts administered the vaccine 3 times (p > 0.05). J8-specific immune responses were monitored until day 168 post-primary immunization. During this period cohorts administered J8-DT/alum (2 or 3 immunizations) maintained strong J8-specific serum IgG titers, that were significantly higher than the control groups. As expected cohorts of animals administered 2 or 3 immunizations of either J8-DT/alum or DT/alum also developed significant DT-specific serum IgG titers. Expanding on this we subcutaneously administered cohorts of the inbred BALB/c mice with 3 doses at days 0, 21 and 28. Immunogenicity was determined by ELISA (Fig. 2A). Similar to that observed with the Swiss mice, the inbred mice also developed a significant antigen-specific IgG response (p < 0.05).
Figure 2.
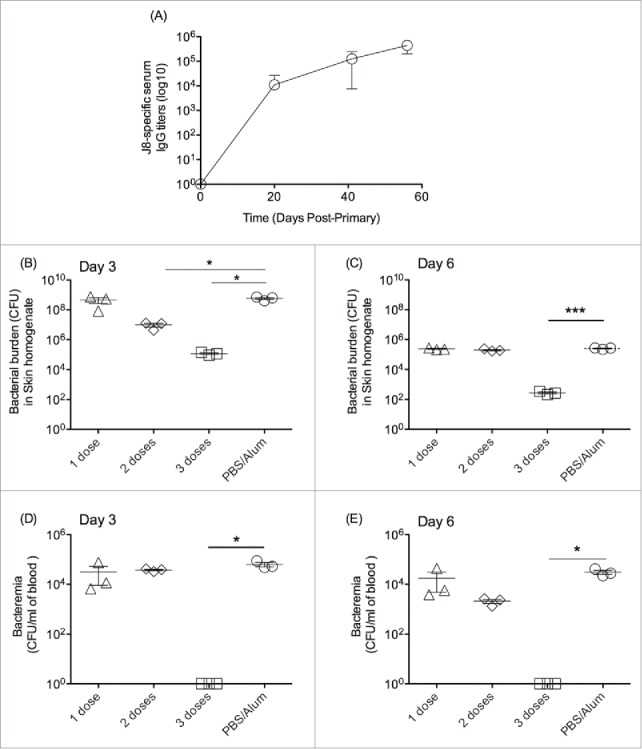
Immunogenicity and protective efficacy of J8-DT/alum in BALB/c mice. (A) Cohorts of BALB/c mice (4–6 weeks old) were subcutaneously immunised with 50 ug of J8-DT vaccine formulation on days 0, 21 and 28. The J8-specific IgG titers on days 20, 27 and 35 are shown. Efficacy of the vaccine formulation following bacterial skin challenge with 88/30 GAS strain for cohorts of mice that received either 1, 2 or 3 doses of vaccine and the control cohort administered PBS plus adjuvant. Skin (B and C) and blood (D and E) bioburden was determined for days 3 and 6 post-exposure respectively. Mean (+/− SEM) shown. A P value of <0.05 was considered significant. Statistical significance was calculated using GraphPad Prism version 6.
Sera collected from the cohorts of BALB/c mice was used an in vitro direct killing assay using the GAS strain 88/30. Sera from immunised mice was pre-incubated with GAS for 1 hour before being mixed with fresh whole mouse blood (1:2 ratio) and incubated at 37°C for 3 hours. The number of CFU was determined for each cohort at zero and 3 hours. We observed that the percentage (%) of opsonised bacteria (when compared to the control group) was significantly reduced for the cohort administered 2 doses compared to the cohort administered 3 doses (Table 1).
Table 1.
In vitro opsonisation (mean %+/−SEM) of the GAS strain 88/30.
| Control | 2 x Dose Cohort | 3 x Dose Cohort |
|---|---|---|
| 0+/−7.13% | 40.2+/−2.7% | 66.9+/−4.5% |
p < 0.05 for Immunised vs control cohorts.
p < 0.05 for 2x doses vs 3x doses cohorts.
The cohorts of BALB/c mice administered 1, 2, or 3 doses of vaccine were then challenged via the skin with 88/30 GAS as previously described.19 On days 3 and 6 post-challenge animals were culled and bacterial burden determined in the skin homogenate and blood (Fig. 2B-E). Mice administered 3 doses of vaccine had significantly reduced bacterial burden in the skin and blood on days 3 and 6 post-challenge compared to cohorts that received only 1 or 2 doses of vaccine. While mice administered 2 or 3 doses of vaccine developed comparable antibody titres, only cohorts administered the third dose were protected from GAS challenge and significantly opsonised bacteria in vitro suggesting that affinity maturation of the antigen-specific IgG following the third vaccine dose was required for protection.
Similar to that observed for mice, we demonstrated that J8-DT/alum also induced J8-specific serum IgG titers in New Zealand White Rabbits when administered intramuscularly (Fig. 3). This is similar to what we had previously observed using different J8-containing vaccine formulations.20,21
Figure 3.
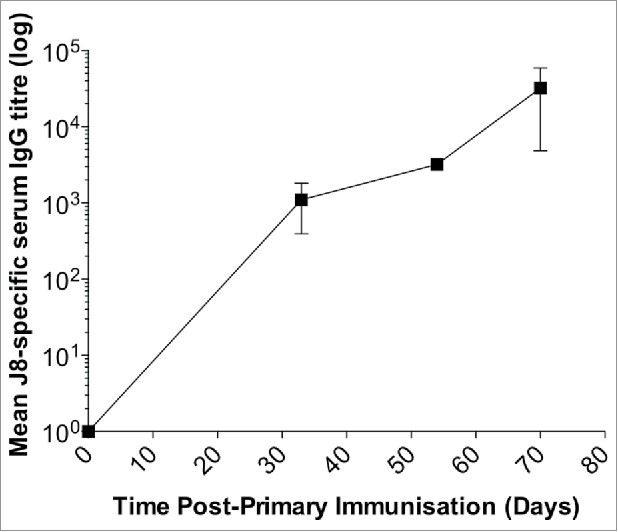
Immunogenicity of the vaccine formulation (J8-DT/alum) in New Zealand White rabbits. Rabbits were administered intramuscularly 50 ug of J8-DT/alum vaccine formulation on days 0, 21 and 42. J8-specific serum IgG titers were determined on the days indicated using a similar protocol used for the mice studies. New Zealand White Rabbits were sourced by Veterinary Institute, South Australia.
Dose escalating toxicology assessment of the vaccine formulations in rabbits
To assess the safety of the vaccine formulation, a toxicology study was undertaken in New Zealand White Rabbits. Initially, we demonstrated that J8-DT/alum induced J8-specific serum IgG titers in rabbits when administered intramuscularly (Fig. 3).
A toxicology study was then conducted according to Good Laboratory Practice (GLP) at the lIT Research Institute (IITRI), Chicago, USA. Clinical and histological findings were compiled by IITRI and reported to the sponsor. J8-DT with/without alum adjuvant (Alhydrogel 2%) was administered to male and female New Zealand white rabbits (n = 10/sex/group) in a repeat-dose toxicity study. The vaccine with/without adjuvant, adjuvant only or the control (phosphate-buffered saline; PBS) was administered on study days 1, 29 and 57 via intramuscular (i.m.) injection as follows: Group 1: PBS; Group 2: alum (Alhydrogel, 2%); Group 3: 50 ug J8-DT/alum; Group 4: 150 ug J8-DT/alum; and Group 5: 150 ug J8-DT. All study animals continued to be observed throughout treatment until half of the study animals were euthanized and necropsied on study day 59; the remaining rabbits were euthanized and necropsied on study day 71.
Experimental endpoints included moribundity/mortality, clinical signs/physical examinations, inoculation site scoring for reactogenicity (erythema and edema), inoculation site pain evaluation/reflexive responses, body weight change, body temperatures, food consumption, ophthalmology, clinical pathology (clinical chemistry, hematology, coagulation), serum protein electrophoresis, organ weights, immunogenicity, gross necropsy observations, histopathology analyses).
All study rabbits survived to the scheduled necropsy on days 59 or 71 during which 58 different tissues (including heat tissue) were collected for microscopic evaluation from each animal. No treatment-related or toxicologically significant effects were observed including inoculation site reactogenicity, inoculation site pain evaluation/reflexive responses, body weight change, food consumption, body temperatures, ophthalmology, clinical pathology, and serum protein electrophoresis.
Heart tissue from only one rabbit from cohort 2 (adjuvant only) on Day 71 was found to have abnormal pathology. On dissection of the heart a 1mm trackable gross lesion was observed on a valve and this correlated with minimal muscular ectopia. Since this cohort was only administered adjuvant alone it was not considered to be related to the J8-DT vaccine.
Microscopic findings were observed at the injection site (skeletal muscle and/or skin) and consisted of macrophage infiltration. These infiltrates, however, were associated with the adjuvant (alum, Alhydrogel) component and not the J8-DT component since the infiltrates were also noted in adjuvant-treated groups (Groups 2, 3 and 4). No other treatment-related microscopic findings were observed in any of the vaccine with adjuvant groups (Groups 3, 4 and 5). Therefore, co-administration of the adjuvant (Alhydrogel) with the J8-DT vaccine may be contributing to the observed increase in macrophage infiltrates at the injection site. However, this is considered a common response following administration of an immunogenic substance or vaccine.
Antigen-specific serum IgG titers were determined by EISA for the serum samples collected and mean half-maximal effective concentration (EC50) reported for each cohort. Administration of the vaccine alone (150 ug of J8-DT, no adjuvant) induced antibody titer following repeated administration (Mean EC50: 1063 +/−336 at day 31), whereas administration of the vaccine plus adjuvant (150 ug of J8-DT with alum) resulted in significantly higher antibody responses (Mean EC50: 15898 +/−5027 at day 31). The highest antibody levels were detected on study day 71 (Mean EC50: 25166 +/− 7958) in the high dose vaccine plus adjuvant group (Group 4). High levels of antigen-specific serum IgG were also observed in the low dose vaccine plus adjuvant group (Group 3) peaking with a mean EC50 of 15730 +/− 4974 on day 71. In general, female rabbits (Mean EC50 17951 +/− 8027 for Group 3 at day 71) appeared to mount a stronger immune response than males (Mean EC50 13509 +/− 6041 for Group 3 at day 71), which is in contrast to the mouse studies where no significant difference was observed in J8-specific antibody titers.
Despite there being no recognized serological markers for rheumatic fever/rheumatic heart disease, we undertook immunohistological assays as previously described with sera from the rabbits immunised with 150ug J8-DT with/without alum adjuvant, 50ug J8-DT/alum, alum alone or PBS used in conjunction with goat anti-rabbit secondary antibody conjugated to FITC on a panel of human tissues including heart, brain cortex, basal ganglia, cartilage and kidney, specifically examining cross-reactivity. Slides were analyzed in a blinded fashion by 2 veterinary pathologists. Transient positive and equivocal responses were seen on tissue sections with both negative control and vaccine sera; no significant or consistent differences in staining were seen between control and vaccine sera for any tissues.
Thus, repeated administration of J8-DT with and without adjuvant by IM injection was well tolerated and there were no significant toxicological findings noted throughout the study.
Vaccine formulation does not induce pathology in a Lewis rat model for rheumatic heart disease
A rat model was previously developed to mimic rheumatic heart disease.22-26 Histological examination of cardiac tissue in rats immunised with recombinant M5 (rM5) revealed the presence of inflammatory lesions in both the myocardium and valve tissue 22 which were akin to the histological changes observed in RF/RHD in humans. In this investigation the rat autoimmune valvulitis (RAV) model has been used to evaluate the safety of our streptococcal vaccine candidates.
Briefly, recombinant streptococcal M5 protein was prepared as previously described.18 Proteins purified from Escherichia coli contain endotoxins that need to be removed for in vivo applications. Bacterial endotoxin concentration was quantified by the Protein Expression Facility (PEF), University of Queensland, Australia, using an Endosafe-PTS Limulus Amebocyte Lysate (LAL) test kit (Charles River Laboratory, Charleston, SC, USA). The endotoxin levels were reduced using a HiTrap Q FF anion-exchange column (GE Healthcare, Rydalmere, Australia) to achieve a suitable concentration of endotoxin for animal injection.
Protein quantification was performed by measuring absorbance at 280 nm using a NanoDrop spectrophometer (Thermo Fisher Scientific, Scoresby, Australia). The vaccine candidate J8-DT, and DT alone, were supplied by Institute for Glycomics, Griffith University (Gold Coast, Australia), and prepared as described elsewhere.27,28 Porcine cardiac myosin and human collagen IV were purchased from Sigma (Australia).
Immunogenicity of the vaccine formulations was evaluated and confirmed in Lewis rats (Fig. 4). The rats were immunized as described previously.18,22,26 Briefly, 8-week-old female Lewis rats (n = 5/group) were immunized subcutaneously on day 0 into the hock with either rM5 (500 ug), J8-DT (150 ug) or DT (150 ug) emulsified 1:1 with Complete Freund's Adjuvant (Sigma, Australia) in a total volume of 200 ul following anesthesia with isofluorane (5%) in 100% oxygen. On day 1 and 3, the rats received an intraperitoneal injection of 1 × 1010 whole killed Bordetella pertussis cells as an additional adjuvant.22 Seven days after initial immunization (Day 7), rats received subcutaneous flank boost injections of antigen emulsified 1:1 with incomplete Freund's adjuvant in a total volume of 200 ul. Control rats were immunized with PBS and adjuvant only. All rats were euthanized 21 d after the initial immunization.
Figure 4.
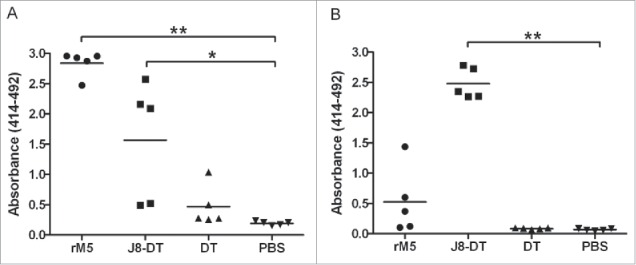
Antibody responses to rM5 and J8-peptide in immunized Lewis rats. IgG antibody reactivity of serum from Lewis rats immunized with rM5 (•), J8-DT (▪), DT (▴) or PBS (negative control) (▾), to rM5 (A) or J8-peptide (B). All rats received a primary immunization on day 0, followed by an intraperitoneal injection of B. pertussis on day 1 and 3. A booster was administered on day 7. Absorbance values of rat sera at 1:100 dilution are shown, with mean absorbance value presented as horizontal lines. Significance was determined by ANOVA with the post hoc Dunnet's test (*, P < 0.01; **, P < 0.001). Lewis rats were purchased from The Animal Resource Centre, Western Australia.
Histological evaluation of tissues collected post-euthanasia indicated that the vaccine formulation did not induce significant histological changes (Fig. 5) in cardiac tissue of rats immunized with the different formulations of the vaccine and alum. Histology of cardiac tissue from groups of rats immunized with vaccine peptide J8, J8-DT or J8-DT/alum (n = 15) were indistinguishable from the PBS-immunized controls (n = 5; negative control). In contrast, inflammatory changes in the myocardium and valvular tissue with fibrosis were observed in groups of rats (n = 10) immunized with recombinant M5 protein (positive control). A veterinary pathologist blinded to the groupings analyzed the histology for all groups in the study. This study confirmed the validity of the model and demonstrated that J8-DT did not cause rheumatic carditis in Lewis rats.
Figure 5.
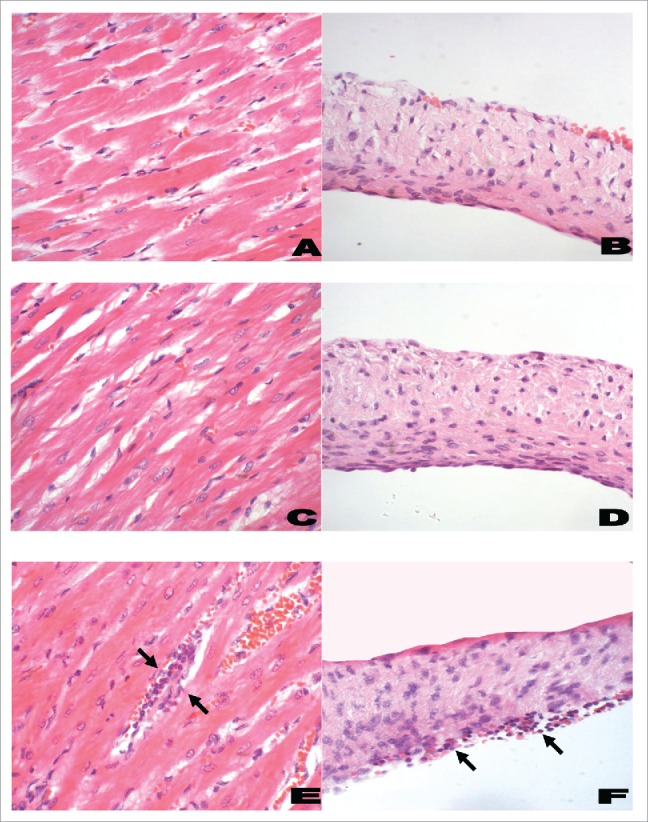
Histological features of myocardium (left panel) and heart valves (right panel) from Lewis rats. A representative histological section from rats immunized with PBS and adjuvant (negative control; A and B) had little or no infiltration of inflammatory cells into the myocardium or valvular tissue. Rats immunized with J8-DT (C and D) had minimal changes comparable to the negative control rats. However, in rM5-immunized rats (positive control; E and F) there was evidence of mononuclear cell infiltration (Arrows) in the myocardial and valvular tissue (H& E; original magnifications: A, C and E, x200; B, D and F, x400). The rat hearts were excised and fixed in 10% neutral buffered formalin for 48 h prior to being embedded in paraffin. Tissue samples were cut in 5 µm-thick sections on a microtome and stained with hematoxylin and eosin (H& E) using standard procedures. Sections were examined by a pathologist who was blinded to the treatment groups, using a light microscope fitted with a QImaging camera. Evidence of inflammatory changes and cellular infiltration in the myocardium and mitral, aortic and tricuspid valve leaflets were assessed.
Discussion
A significant challenge to development and evaluation of candidate vaccines for GAS has been the lack of a suitable animal model for the evaluation of efficacy and safety. While nonhuman primates are susceptible to GAS colonization, the cost and animal welfare issues are barriers to use. Here we report on the immunogenicity of the vaccine formulation in mice that has been the traditional model used in many research programs, and in rabbits, which are routinely used as part of toxicology studies.
We observed that 2 or 3 doses of vaccine were required to induce an optimal antibody titer; however, the third dose of vaccine was required to protect mice from a skin challenge with GAS. We evaluated immunogenicity of several vaccine formulations in a toxicology study using rabbits. The route of administration, amount of vaccine and administration schedule were designed according to industry standard recommendations for evaluation of vaccine safety including an additional dose in the group administered the highest amount of vaccine beyond the planned human dosing schedule.
No morbidity or mortality occurred in any animals, and only minor skin reactions (e.g. erythema or edema) were reported at the site of injection for several animals including animals that received only adjuvant and therefore they were considered to be due to the Alhydrogel adjuvant. The injection site lesions were localized, transient, and consistent with alum adjuvant-induced lesions reported by other investigators.29 The clinical chemistry and hematology parameters were also reported to be within the normal range for New Zealand White rabbits across all cohorts.
A major impediment in developing GAS vaccines based on the M-protein is the concern of inducing autoimmune sequelae in vaccine recipients. Here we demonstrate that our vaccine formulations did not induce pathology in the RAV model. The immunological and functional characterization of the RAV model has enabled us to evaluate the safety of vaccine formulations prior to use in Phase I clinical studies. This model complements existing industry standard practices for toxicological and safety assessments. We also observed that serum from vaccinated rabbits did not recognize human tissues at levels above background staining of negative control sera. However, in our opinion, this assay is of limited predictive value, particularly as there are no serological assays of tissue cross-reactivity that are useful for diagnosis of rheumatic heart disease. To the best of our knowledge, the RAV model is the only pre-clinical model that could be used to test the possibility of a candidate vaccine to induce an immune response that may lead to cardiac tissue damage similar to pathology observed in rheumatic heart disease.
The results of the RAV model are also consistent with the rabbit toxicology study in which no abnormal heart tissue pathology was observed during the necropsies except for a rabbit in the cohort administered adjuvant alone. A gross lesion was observed on a heart valve from this rabbit but was not associated with the vaccine J8-DT. Since the post-infectious sequelae, rheumatic fever is induced by Streptococcus pyogenes and that subsequent GAS infections can reactivate rheumatic fever symptoms an important future direction of this research would be to evaluate safety in the RAV model for animals immunised with the vaccine formulations and then subsequently exposed to the bacteria.
If the vaccine is ultimately successful, the crux will be that the p145 peptide is antigenic in its native configuration but not immunogenic. For this reason alone, the organism has not had to evolve multiple allelic forms; however, vaccine-induced antibodies can target the epitope and kill the organism. If successful, this strategy of defining non-dominant immune responses and epitopes will be more seriously considered for other organisms that readily evade natural immunity.
Disclosure of potential conflicts of interest
No authors have declared conflicts of interest or financial disclosures relating to this work.
Acknowledgments
All experimental procedures were approved by one of the following committees: the Queensland Institute of Medical Research (QIMR) Animal Ethics Committee; Griffith University Animal Ethics Committee or James Cook University Animal Ethics Committee in accordance with National Health and Medical Research Council (NHMRC) of Australia guidelines.
We would like to thank Drs F. Rubin and R. Mason at NIH for their guidance in aspects of this project.
Funding
This work was supported by the National Heart Foundation of Australia, National Health and Medical Research Council (Australia), the National Institute of Health (USA) and the Cooperative Research Center for Aboriginal Health. Part of this work was supported by a National Institutes of Health Grant (U01-AI060579-01) and conducted at IIT Research Institute (IITRI), Chicago, USA and the Queensland Institute of Medical Research (QIMR, Queensland, Australia).
References
- [1].McDonald M, Currie BJ, Carapetis JR. Acute rheumatic fever: a chink in the chain that links the heart to the throat? Lancet Infect Dis 2004; 4:240-5; PMID:15050943; http://dx.doi.org/ 10.1016/S1473-3099(04)00975-2 [DOI] [PubMed] [Google Scholar]
- [2].Massell BF, Honikman LH, Amezcua J. Rheumatic fever following streptococcal vaccination. Report of three cases. JAMA 1969; 207:1115-9; PMID:5818242; http://dx.doi.org/ 10.1001/jama.1969.03150190037007 [DOI] [PubMed] [Google Scholar]
- [3].D'Alessandri R, Plotkin G, Kluge RM, Wittner MK, Fox EN, Dorfman A, Waldman RH. Protective studies with group A streptococcal M protein vaccine. III. Challenge of volunteers after systemic or intranasal immunization with Type 3 or Type 12 group A Streptococcus. J Infect Dis 1978; 138:712-8; PMID:368261; http://dx.doi.org/ 10.1093/infdis/138.6.712 [DOI] [PubMed] [Google Scholar]
- [4].Fox EN, Waldman RH, Wittner MK, Mauceri AA, Dorfman A. Protective study with a group A streptococcal M protein vaccine. Infectivity challenge of human volunteers. J Clin Invest 1973; 52:1885-92; PMID:4719668; http://dx.doi.org/ 10.1172/JCI107372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].FDA Status of specific products; Group A Streptococcus. Federal Register January 5, 1979; 44(4):1544-9. [PubMed] [Google Scholar]
- [6].FDA . Status of specific products; Group A Streptococcus. Federal Register December 2, 2005; 70:72197-9; PMID:16323338 [PubMed] [Google Scholar]
- [7].Hu MC, Walls MA, Stroop SD, Reddish MA, Beall B, Dale JB. Immunogenicity of a 26-valent group A streptococcal vaccine. Infect Immun 2002; 70:2171-7; PMID:11895984; http://dx.doi.org/ 10.1128/IAI.70.4.2171-2177.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dale JB, Beachey EH. Multiple, heart-cross-reactive epitopes of streptococcal M proteins. J Exp Med 1985; 161:113-22; PMID:2578539; http://dx.doi.org/ 10.1084/jem.161.1.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pruksakorn S, Currie B, Brandt E, Martin D, Galbraith A, Phornphutkul C, Hunsakunachai S, Manmontri A, Good MF. Towards a vaccine for Rheumatic-Fever - Identification of a Conserved Target Epitope on M-Protein of Group-a Streptococci. Lancet 1994; 344:639-42; PMID:7520963; http://dx.doi.org/ 10.1016/S0140-6736(94)92083-4 [DOI] [PubMed] [Google Scholar]
- [10].Hayman WA, Brandt ER, Relf WA, Cooper J, Saul A, Good MF. Mapping the minimal murine T cell and B cell epitopes within a peptide vaccine candidate from the conserved region of the M protein of group A streptococcus. Int Immunol 1997; 9:1723-33; PMID:9418133; http://dx.doi.org/ 10.1093/intimm/9.11.1723 [DOI] [PubMed] [Google Scholar]
- [11].Brandt ER, Hayman WA, Currie B, Pruksakorn S, Good MF. Human antibodies to the conserved region of the M protein: opsonization of heterologous strains of group A streptococci. Vaccine 1997; 15:1805-12; PMID:9364687; http://dx.doi.org/ 10.1016/S0264-410X(97)00178-3 [DOI] [PubMed] [Google Scholar]
- [12].Brandt ER, Hayman WA, Currie B, Carapetis J, Wood Y, Jackson DC, Cooper J, Melrose WD, Saul AJ, Good MF. Opsonic human antibodies from an endemic population specific for a conserved epitope on the M protein of group A streptococci. Immunology 1996; 89:331-7; PMID:8958044; http://dx.doi.org/ 10.1046/j.1365-2567.1996.d01-754.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Batzloff MR, Hayman WA, Davies MR, Zeng M, Pruksakorn S, Brandt ER, Good MF. Protection against Group A Streptococcus by immunization with J8-Diphtheria toxoid: Contribution of J8- and Diphtheria Toxoid-specific antibodies to protection. J Infect Dis 2003; 187:1598-608; PMID:12721940; http://dx.doi.org/ 10.1086/374800 [DOI] [PubMed] [Google Scholar]
- [14].Batzloff MR, Yan H, Davies MR, Hartas J, Lowell GH, White G, Burt DS, Leanderson T, Good MF. Toward the development of an antidisease, transmission-blocking intranasal vaccine for group a streptococcus. J Infect Dis 2005; 192:1450-5; PMID:16170764; http://dx.doi.org/ 10.1086/466528 [DOI] [PubMed] [Google Scholar]
- [15].Batzloff MR, Hartas J, Zeng W, Jackson DC, Good MF. Intranasal vaccination with a lipopeptide containing a conformationally constrained conserved minimal peptide, a universal T cell epitope, and a self-adjuvanting lipid protects mice from group A streptococcus challenge and reduces throat colonization. J Infect Dis 2006; 194:325-30; PMID:16826480; http://dx.doi.org/ 10.1086/505146 [DOI] [PubMed] [Google Scholar]
- [16].Olive C, Batzloff M, Horvath A, Clair T, Yarwood P, Toth I, Good MF. Potential of lipid core peptide technology as a novel self-adjuvanting vaccine delivery system for multiple different synthetic peptide immunogens. Infect Immun 2003; 71:2373-83; PMID:12704107; http://dx.doi.org/ 10.1128/IAI.71.5.2373-2383.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Olive C, Ho MF, Dyer J, Lincoln D, Barozzi N, Toth I, Good MF. Immunization with a tetraepitopic lipid core peptide vaccine construct induces broadly protective immune responses against group A streptococcus. J Infect Dis 2006; 193:1666-76; PMID:16703510; http://dx.doi.org/ 10.1086/504266 [DOI] [PubMed] [Google Scholar]
- [18].Gorton D, Govan B, Olive C, Ketheesan N. B- and T-cell responses in group a streptococcus M-protein- or Peptide-induced experimental carditis. Infect Immun 2009; 77:2177-83; PMID:19273562; http://dx.doi.org/ 10.1128/IAI.01514-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pandey M, Langshaw E, Hartas J, Lam A, Batzloff MR, Good MF. A synthetic M protein peptide synergizes with a CXC chemokine protease to induce vaccine-mediated protection against virulent streptococcal pyoderma and bacteremia. J Immunol 2015; 194:5915-25; PMID:25980008; http://dx.doi.org/ 10.4049/jimmunol.1500157 [DOI] [PubMed] [Google Scholar]
- [20].Sheel M, Pandey M, Good MF, Batzloff MR. Correlation between bioluminescence and bacterial burden in passively protected mice challenged with a recombinant bioluminescent M49 group A streptococcus Strain. Clin Vaccine Immunol 2010; 17:127-33; PMID:19889937; http://dx.doi.org/ 10.1128/CVI.00256-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pandey M, Batzloff MR, Good MF. Mechanism of protection induced by group A Streptococcus vaccine candidate J8-DT: contribution of B and T-cells towards protection. PLoS One 2009; 4:e5147; PMID:19340309; http://dx.doi.org/ 10.1371/journal.pone.0005147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lymbury RS, Olive C, Powell KA, Good MF, Hirst RG, LaBrooy JT, Ketheesan N. Induction of autoimmune valvulitis in Lewis rats following immunization with peptides from the conserved region of group A streptococcal M protein. J Autoimmun 2003; 20:211-7; PMID:12753806; http://dx.doi.org/ 10.1016/S0896-8411(03)00026-X [DOI] [PubMed] [Google Scholar]
- [23].Li Y, Heuser JS, Kosanke SD, Hemric M, Cunningham MW. Cryptic epitope identified in rat and human cardiac myosin S2 region induces myocarditis in the Lewis rat. J Immunol 2004; 172:3225-34; PMID:14978130; http://dx.doi.org/ 10.4049/jimmunol.172.5.3225 [DOI] [PubMed] [Google Scholar]
- [24].Cunningham MW. T cell mimicry in inflammatory heart disease. Mol Immunol 2004; 40:1121-7; PMID:15036918; http://dx.doi.org/ 10.1016/j.molimm.2003.11.023 [DOI] [PubMed] [Google Scholar]
- [25].Wegmann KW, Zhao W, Griffin AC, Hickey WF. Identification of myocarditogenic peptides derived from cardiac myosin capable of inducing experimental allergic myocarditis in the Lewis rat. The utility of a class II binding motif in selecting self-reactive peptides. J Immunol 1994; 153:892-900; PMID:8021520 [PubMed] [Google Scholar]
- [26].Rush CM, Govan BL, Sikder S, Williams NL, Ketheesan N. Animal models to investigate the pathogenesis of rheumatic heart disease. Front Pediatr 2014; 2:1-6; PMID:25414841; http://dx.doi.org/ 10.3389/fped.2014.00116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Houghten RA. General method for the rapid solid-phase synthesis of large numbers of peptides: specificity of antigen-antibody interaction at the level of individual amino acids. Proc Natl Acad Sci U S A 1985; 82:5131-5; PMID:2410914; http://dx.doi.org/ 10.1073/pnas.82.15.5131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Coligan JKA, Margulie D, Shevach E, Strober W. Current protocols in immunology. New York: John Wiley, 1991 [Google Scholar]
- [29].Verdier F, Burnett R, Michelet-Habchi C, Moretto P, Fievet-Groyne F, Sauzeat E. Aluminium assay and evaluation of the local reaction at several time points after intramuscular administration of aluminium containing vaccines in the Cynomolgus monkey. Vaccine 2005; 23:1359-67; PMID:15661384; http://dx.doi.org/ 10.1016/j.vaccine.2004.09.012 [DOI] [PubMed] [Google Scholar]
- [30].Batzloff MR, Hayman WA, Davies MR, Zeng M, Pruksakorn S, Brandt ER, Good MF. Protection against group A streptococcus by immunization with J8-diphtheria toxoid: Contribution of J8-and diphtheria toxoid-specific antibodies to protection. J Infect Dis 2003; 187:1598-608; PMID:12721940; http://dx.doi.org/ 10.1086/374800 [DOI] [PubMed] [Google Scholar]