

Abstract
The pyrrolo[2,1‐c][1,4]benzodiazepines (PBDs) are a family of sequence‐selective DNA minor‐groove binding agents that form a covalent aminal bond between their C11‐position and the C2‐NH2 groups of guanine bases. The first example of a PBD monomer, the natural product anthramycin, was discovered in the 1960s, and the best known PBD dimer, SJG‐136 (also known as SG2000, NSC 694501 or BN2629), was synthesized in the 1990s and has recently completed Phase II clinical trials in patients with leukaemia and ovarian cancer. More recently, PBD dimer analogues are being attached to tumor‐targeting antibodies to create antibody–drug conjugates (ADCs), a number of which are now in clinical trials, with many others in pre‐clinical development. This Review maps the development from anthramycin to the first PBD dimers, and then to PBD‐containing ADCs, and explores both structure–activity relationships (SARs) and the biology of PBDs, and the strategies for their use as payloads for ADCs.
Keywords: antibody–drug conjugates, antitumor agents, DNA, interstrand cross-linking, pyrrolobenzodiazepines
1. Introduction
The pyrrolo[2,1‐c][1,4]benzodiazepines (PBDs) are sequence‐selective DNA minor‐groove binding agents based on the naturally occurring anthramycin family of antitumor antibiotics,1 the best known member of which is anthramycin itself (1, Figure 1) which was isolated from Streptomyces refuineus in 1965.2 The skeletal structure of the PBDs contains a substituted aromatic A‐ring, a diazepine B‐ring and a pyrrolidine C‐ring, with an S‐chiral center at the C11a‐position between the B and C rings. This provides a 3‐dimensional shape perfectly crafted for the molecules to fit within the DNA minor groove (Figure 2). They also possess an electrophilic imine moiety (or equivalent carbinolamine or carbinolamine methyl ether) at the N10–C11 position within their B‐ring which can form a covalent aminal linkage between their C11 carbon and the C2‐NH2 group of a guanine base (Figure 2 A), but only after the molecule is secure within the minor groove (Figure 2 B). PBD monomers such as anthramycin (1) typically span three DNA base pairs with a reported preference for 5′‐Pu‐G‐Pu‐3′ sequences,1b, 3 although more recent data suggest that they have a kinetic preference for 5′‐Py‐G‐Py‐3′ sequences (Pu=purine, Py=pyrimidine).4
Figure 1.

Structures of the naturally occurring anthramycin (1), the PBD C8‐conjugates GWL‐78 (2) and KMR‐28–39 (3), and examples of C7/C7′‐linked (4) and C8/C8′‐linked (e.g., DSB‐120, 5 a, n=3 and SJG‐136, 6 a, n=3) PBD Dimers.
Figure 2.

A) The mechanism of covalent binding of a PBD to DNA once it has located in a low‐energy position within the minor groove. B) Molecular model of the crystal structure of anthramycin (PDB ID 274D)13 covalently bound to G19 (magenta) of the sequence 5′‐CC(G)AACGTTGG‐3′ (covalently modified guanine underlined and in brackets as it on the opposing DNA strand) as an example of a PBD–DNA adduct. Due to the perfect fit of anthramycin in the DNA minor groove (as a consequence of its 3‐dimensional shape created by the C11a(S) chiral center), normal base‐pairing is maintained (cyan) with negligible distortion of the minor groove.
Since the discovery of anthramycin in the early 1960s2 and its subsequent evaluation in the clinic,5 a range of synthetic PBDs1f, 6 has been developed by various academic groups and industrial laboratories. For example, non‐covalent minor‐groove binding components have been appended to the C8‐position of the PBD A‐ring (e.g., GWL‐783a (2) and KMR‐28–397 (3), Figure 1), and monomeric PBD units have been joined together through their C7/C7′‐8 (4) and C8/C8′‐positions (e.g., 5 a–d and 6 a,b) to afford PBD dimers (e.g., DSB‐1209 (5 a) and SJG‐13610 (6 a), Figure 1). PBD units have also been joined through their C8/C2′‐ and C2/C2′‐positions but the resulting dimers do not have the appropriate shape to fit into the DNA minor groove, and so have poor DNA‐binding affinity and cytotoxicity (see Figure 6).11 C2/C8′‐linked triazolo PBD dimers, connected using a click chemistry approach, have a similarly poor DNA‐binding profile.12 The unique structure of the PBD dimers which contain two alkylating imine functionalities allows them to form interstrand or intrastrand DNA cross‐links in addition to mono‐alkylated adducts,14 thus resulting in greater DNA stabilization10, 15 compared to monomeric PBDs. Due to the additional types of DNA adducts possible and the greater adduct stability, the C8‐linked PBD dimers generally have significantly greater cytotoxicity,9 antitumor activity16 and antibacterial activity17 compared to PBD monomers, and one such agent, SJG‐136, has reached Phase II clinical trials in ovarian cancer and leukaemia.18 Furthermore, related PBD dimers are now being used as payloads for antibody–drug conjugates (ADCs) due to their significant cytotoxicity.19 The covalent binding of both monomeric and dimeric PBDs to DNA is thought to be a two‐step process, the first involving recognition of a favored low‐energy binding site by fast, reversible non‐covalent association of the molecules in the minor groove through interactions including hydrogen bonding, van der Waals and electrostatic contacts. If these non‐covalent intermolecular interactions are weak, the PBD presumably dissociates and re‐associates at another site, with this process repeating itself until an appropriate low‐energy site is found containing one or two guanine C2‐NH2 groups (for PBD monomers or dimers, respectively) suitably aligned for nucleophilic attack at the PBD C11‐position(s). In the second step, the PBD forms one or two covalent bonds with the guanine base(s), and the molecule is then locked into position. Whereas the initial non‐covalent association is a fast process, the covalent attachment step is variable in rate depending on the PBD structure and DNA sequence, and can take between 34 to 2414 hours to complete (Figure 2 A).
Figure 6.

Examples of C8/C2′‐ and C2/C2′‐linked PBD dimers synthesized by the Thurston,11a Kamal61 and Lown11b groups. Dimer 6C differs from the others in possessing only one electrophilic N10–C11 imine moiety, and so cannot cross‐link DNA.
Once covalently bound to DNA, PBD monomers and dimers have been shown to mediate a number of biological effects in cells including DNA strand breakage,20 inhibition of DNA processing enzymes (e.g., endonuclease BamH1,21 RNA polymerase3a, 22 and Ligase 123) and specific transcription factors (e.g., Sp1,24 NF‐Y25 and NF‐κB),25a, 26 and modulation of various signaling pathways (e.g., p53‐dependent and ‐independent apoptogenic,27 JNK/AP‐1,8 VEGF28 and SDF1α28 signaling). Many of these proteins and signaling pathways are upregulated in tumor cells compared to healthy cells which could, in part, explain the anticancer activity of the PBDs in animal models and humans.3c
2. Structure–Activity Relationships of PBDs
Since their discovery in the 1960s, a substantial body of structure–activity relationship (SAR) data has been generated based on both the known PBD natural products and the large number of synthetic analogues that have become available. A summary of the key SAR features of PBD monomers (Figure 3 A) and dimers (Figure 3 B), and of their structural analogues, the indolinobenzodiazepine dimers (Figure 3 C), is provided below.
Figure 3.

Summary of structure–activity relationships (SARs) for A) the PBD monomers, B) the PBD dimers, and C) the indolinobenzodiazepine analogues. Modifications which enhance activity are shown in blue, and those which reduce activity in red.
2.1. N10–C11 Imine (or Equivalent)
The presence of an imine, carbinolamine or carbinolamine methyl ether group at the N10–C11 position is essential for covalent interaction of a PBD molecule with DNA. Early studies by Kohn29 and Hurley30 demonstrated that PBDs covalently bind to guanine‐containing DNA sequences, and also established that they do not interact through an intercalation mechanism29b (Figure 2).
Crucially, replacement of the N10–C11 imine with lactam (Figure 4 A) or secondary amine (Figure 4 B) functionalities results in a dramatic reduction in DNA‐binding ability31 and cytotoxicity. For example, the loss of DNA‐binding affinity upon replacing the two N10–C11 imine functionalities of the PBD dimer SJG‐136 (6 a) with lactams has been demonstrated.10 Similarly, didehydroanhydroanthramycin32 (Figure 4 C) which has extended N10–C11/C11a–C1/C2–C3 conjugation is biologically inactive, as the N10–C11 imine is too stable to react with a guanine base. Although PBD dilactams (e.g., Figure 4 A) cannot bind covalently to DNA, they are still isohelical with the DNA minor groove due to their chiral C11a‐position, and so can possess weak DNA‐binding properties through non‐covalent hydrogen bonding and other interactions such as van der Waals. For example, a series of dilactams synthesized by Lown and co‐workers33 provided a thermal stabilization (i.e., ΔT m) of up to approximately 3 °C with calf thymus DNA, although this compares unfavorably with a simple PBD monomer such as anthramycin which provides a ΔT m of 13.1 °C under the same conditions. PBD dimers produce significantly higher ΔT m values compared to monomers due to their length (and hence greater number of non‐covalent contacts in the minor groove) and cross‐linking ability.15 Libraries of PBD dilactams synthesized by Thurston and co‐workers have shown similar ΔT m ranges (e.g., up to approximately 2.4 °C for C2‐aryl substituted dilactams)31a to those reported by Lown and co‐workers.33a
Figure 4.

Structures of PBD analogues in which the electrophilic N10–C11 imine moiety has been replaced with a lactam (i.e., dilactam, A) or a secondary amine (B), or in which the imine has been rendered non‐electrophilic by conjugation with double bonds in the C‐ring to create a fully aromatized system (i.e., didehydroanhydroanthramycin, C). All molecules of types A, B and C cannot bond covalently to DNA, and so have negligible cytotoxicity. Some examples (e.g., dilactams of type A) can still bind to DNA, albeit weakly, through non‐covalent interactions because they still have the appropriate 3‐dimensional shape to fit into the DNA minor groove.31a
2.2. C11a/C11 Stereochemistry
It has been shown through NMR34 and X‐ray crystallography13 studies that all naturally occurring PBDs exist in a single stereochemical form (i.e., S) at their C11a‐position which provides the right‐handed twist that allows the molecules to follow the curvature of the minor groove within the right‐handed B‐DNA helix. Molecules with the (R)‐stereochemistry at C11a are not isohelical with the DNA minor groove, causing them to sterically clash with the groove wall.13, 35 Examples of PBDs with the incorrect (R)‐stereochemistry at C11a have been synthesized and, as predicted, do not possess significant DNA‐binding affinity or cytotoxicity.36 Although PBDs themselves can exist as imines at their N10–C11 position, or carbinolamines or carbinolamine methyl ethers with different stereochemistry at their C11‐position, there are conflicting reports in the literature as to the stereochemistry at the C11‐position of a PBD molecule once bound to DNA. Although an anthramycin–DNA crystal structure13 and a number of NMR studies of both PBD monomers34, 35 and dimers37 bound to DNA have shown that the adduct formed is in the C11(S)‐configuration, molecular mechanics calculations38 and one NMR‐determined structure39 suggest that, in some situations, both R‐ and S‐configurations are equally preferred. It is now thought that PBD monomers can form either R‐ or S‐adducts at C11, and that the outcome may depend on the substitution pattern of a particular PBD (e.g., C8‐substitution may be partially influential) and the DNA sequence to which it is covalently bound.38c
2.3. Functionalization of the A‐Ring
The number and pattern of substituents in the A‐ring of the PBD skeleton can affect the DNA binding ability and cytotoxicity. The presence of electron‐donating substituents, particularly at the C7‐ and C8‐positions, can enhance DNA interactivity by increasing the electrophilicity (through enhanced ability of the N10‐atom to protonate) and alkylating potential of the N10–C11 imine moiety.40 Furthermore, extensive molecular modeling studies have shown that C8‐substituents can position themselves along the floor of the minor groove leading to significant additional stabilization of the adduct.7 Thus heterocyclic fragments (e.g., pyrroles, furans, imidazoles, thiophenes, indoles, benzofurans and biaryl units) have been added to this position to create PBD C8‐conjugates3a, 7, 25b, 28, 41 (e.g., GWL‐78,3a KMR‐28‐39,7 Figure 1). Similarly, two PBD units have been joined through their C8‐positions to create C8/C8′‐linked PBD dimers,10, 37a, 42 and this approach led to the design and synthesis of the C8/C8′‐linked PBD dimer SJG‐13643 (Figure 1). The C7‐position has also been used as a potential linking point for PBD dimers,8a but molecules joined in this way have significantly less DNA cross‐linking potential and cytotoxicity, as the two PBD units are not properly aligned for alkylation of guanine bases.8b Based on experiments with anthramycin in mice and rats, the presence of a hydroxy group at C9 is thought to render a PBD cardiotoxic due to free radical production which can damage cardiac tissue.5 Other studies on PBDs such as anthramycin13 have suggested that a C9‐OH group has the potential to hydrogen bond to bases within the minor groove of DNA, thus increasing binding affinity and influencing sequence selectivity.13 The presence of a larger group (e.g., a methoxy group) at C9 appears to render the PBD biologically inactive44 potentially due to steric hindrance between this substituent and functional groups on the minor groove floor, as can be demonstrated through molecular dynamics simulations.45
2.4. C‐Ring Unsaturation
The degree of unsaturation associated with the C‐ring of a PBD can also influence its DNA‐binding ability and cytotoxicity. In general, PBD monomers with C2‐exo or C2‐endo unsaturation (e.g., anthramycin,30 tomaymycin46 and sibiromycin)47 have enhanced DNA‐binding affinity relative to PBD monomers with fully saturated C‐rings (e.g., neothramycin A,48 chicamycin49 and DC‐81).1a This feature has been adopted in the rational design of PBD dimers (see Figures 3 B and 3 C), where introduction of exo‐unsaturation at the C2/C2′‐positions of the PBD dimer DSB‐12037a (5 a, Figure 1) led to SJG‐136 (6 a, Figure 1). DSB‐120 has poor activity in vivo, attributed partly to its high reactivity with cellular thiol‐containing molecules such as glutathione.50 However, introduction of C2/C2′‐exo unsaturation as in SJG‐136 led to an overall increase in DNA‐binding affinity and cytotoxicity, and a lower reactivity toward cellular nucleophiles with more of the agent potentially reaching its target DNA. Modeling studies have shown that C2/C2′‐unsaturation causes a flattening of the C‐ring which may lead to superior van der Waal contacts within the minor groove10 thus contributing to the enhanced DNA‐binding affinity.38a Interestingly, complete unsaturation of the C‐ring significantly reduces the electrophilicity of the N10–C11 imine by producing a fully aromatic system across the N10–C11/C11a–C1/C2–C3 positions which ablates DNA‐binding ability and cytotoxicity (e.g., didehydroanhydroanthramycin,32 Figure 4 C).
2.5. C‐Ring Substitution
In a similar manner to C9‐substitution, a substituent at C3 can affect the activity of a PBD. For example, methylation of the C3‐hydroxy substituent of neothramycin A reduces cytotoxicity relative to unsubstituted neothramycin.51 However, extended C2‐substituents (e.g., the conjugated acrylamide side chain of anthramycin)13 significantly enhance DNA interactivity because they locate along the minor groove and stabilize the adduct through van der Waals interactions and sequence‐specific hydrogen bonds to functional groups in the minor groove floor. For example, early studies on anthramycin suggested that removal of the C2‐acrylamide side‐chain significantly reduced its DNA‐interactivity.44, 52 This knowledge of SAR at the C2‐position has been used to design novel C2‐substituted PBD monomers and dimers with enhanced DNA‐binding ability and cytotoxicity.19, 31a For example, some C2‐substituents (e.g., p‐tolyl) can lead to cytotoxicities in the low nanomolar range.31a Studies have also shown that replacement of the C1‐atom itself with an oxygen can provide analogues (e.g., oxazolobenzodiazepines) with similar activity to the parent PBDs.53 C1‐substituted PBD monomeric conjugates have also been synthesized recently, and shown to have cytotoxicity in the nanomolar range.54 Molecular modeling studies have shown that the C1‐polyamide substituents locate in the minor groove and generate additional hydrogen bonding and other interactions, thus enhancing DNA‐binding affinity and cytotoxicity.54
2.6. Other Modifications
Many of the modifications outlined above have been incorporated into PBD dimers that are now being used as chemical payloads for ADCs (Figure 3 B,C). For example, the ADC SGN‐CD33A (Vadastuximab talirine) produced by Seattle Genetics Inc. (see Section 5) contains a C2–C3/C2′–C3′‐endo‐unsaturated C2/C2′‐bis‐aryl PBD with a C8/C8′‐propylenedioxy linker, and another PBD‐based ADC Rovalpituzumab tesirine (“Rova‐T”) produced by Stemcentrx Inc. contains a PBD dimer with C2–C3/C2′–C3′‐endo‐unsaturated C2/C2′‐bis‐methyl substitutions. In the case of both ADCs, the A‐rings contain oxygen substituents in both the C7‐ and C8‐positions which enhance interactivity with DNA, but the C9‐position remains unsubstituted, thereby minimizing any steric interactions within the minor groove. However, in addition, researchers at Sanofi S.A. and ImmunoGen Inc. have introduced a fourth D‐ring to the C‐ring of the PBD skeleton to produce indolinobenzodiazepines (Figure 3 C). They have also introduced an aromatic ring into the center of the C8/C8′‐linker which is thought to enhance DNA‐binding affinity and provides an antibody attachment point (Figure 3 C).
3. Evolution of the PBD Dimers
The first PBD dimers were synthesized by Suggs and co‐workers in 19888 with the two PBD units joined through linkers attached at the C7/C7′‐positions of the aromatic A‐rings (4 a–c, Figure 1). Although modest DNA cross‐linking activity was reported, no cytotoxicity data were disclosed.
Based on a rational approach to the way in which a PBD dimer might best fit into the DNA minor groove, in 1992 Thurston and co‐workers9 reported the PBD dimer DSB‐120 (5 a, Figure 1) which consists of two unsubstituted PBD units coupled through their C8/C8′‐positions via a 1,3‐dioxypropyl linker. This molecule, which was designed to more closely follow the curvature of the minor groove, had a very high stabilizing effect on calf thymus DNA (i.e., ΔT m >15.1 °C), significant interstrand cross‐linking activity toward linear pBR322 plasmid DNA (i.e., >90 % cross‐linking for 0.4 μm drug concentration at 1:1 DSB‐120/DNA), and sub‐micromolar cellular cytotoxicity in some tumor cell lines (e.g., lymphosarcoma ADJ/PC6).9 SAR studies55 demonstrated that changing the central methylene linker from 3 (5 a) to 4, 5 or 6 methylene units (5 b–d) still allowed DNA binding but significant differences in binding affinity were observed. Increasing the linker length to n=5 (5 c) afforded a molecule with similar cross‐linking activity to DSB‐120 (n=3) but with approximately half of the DNA stabilizing ability, while the cellular cytotoxicity was significantly enhanced. However, increasing the linker length to n=4 or 6 afforded a reduction in all three parameters, and led to the conclusion that linkers with an odd number of methylenes are preferred for maximum cytotoxicity. This was later explained through molecular modeling studies,55 as only dimers with odd numbers of methylenes in their C8/C8′‐linkers allow the two PBD units to arrange themselves in the correct orientations for their N10–C11 imine moieties to alkylate guanine C2‐NH2 groups. Although DSB‐120 had potent activity in biophysical and cellular assays, it proved to be inactive in human tumor xenograft studies in mouse models. This was thought to be due to the N10–C11 imine moieties being too electrophilic due to the fully saturated state of the C‐rings, leading to the molecule reacting with extra‐ and intra‐cellular thiol‐containing nucleophiles before it could reach its DNA target.50, 56
The addition of methylene substituents to the C2/C2′‐positions to afford SJG‐136 (6 a, Figure 1)10 appeared to reduce the electrophilicity of the N10–C11 imine moieties, increasing the potential for the molecule to reach its target DNA in the nucleus of cells. This was attributed to flattening of the C‐rings due to the presence of the C2/C2′‐exo‐methylene substituents which appear to modify the electrophilicity of the N10–C11 imines through an, as yet, unidentified mechanism, and also provide a better fit of the molecule in the minor groove. This analogue was not only more potent in biophysical and cellular assays but was also active in vivo in human tumor xenograft mouse models.16, 57 Due to the presence of two electrophilic imine moieties, SJG‐136 (6 a) can form interstrand and intrastrand DNA cross‐links in addition to monoalkylated adducts (Figure 5).10, 58 The type of adducts that form appears to depend on both the precise DNA sequence and the distance between reacting guanines.14, 59 Although earlier studies suggested that SJG‐136 (6 a) formed only Pu‐GATC‐Py interstrand cross‐linked adducts,1i, 60 more recent HPLC‐MS studies have established that it can also form intrastrand cross‐links at longer Pu‐GAATG‐Py sequences for which, in some cases, it can have a preference, and mono‐adducts.14
Figure 5.

Examples of the main types of adducts that can form between SJG‐136 (6 a) and DNA (i.e., interstrand and intrastrand cross‐links, and mono‐adducts) (X=any base).
Examples also exist in the literature of PBD dimers linked through their C8/C2′‐11a or C2/C2′‐positions11b rather than their C8/C8′‐positions (Figure 6), but these are at least 103‐fold less cytotoxic than the most potent C8/C8′‐linked dimers. For example, Thurston and co‐workers11a synthesized a C8/C2′‐linked PBD dimer (Figure 6 A) containing C2′/C3′‐endo unsaturation at the C2‐linkage, which had an IC50 of >25 μm in A2780 cells. This is less cytotoxic than the C2‐unsubstiuted PBD monomer DC‐81, suggesting that the second PBD unit may be impeding rather than enhancing DNA‐interaction. Kamal and co‐workers61 synthesized two similar C8/C2′‐linked dimers consisting of a C8′‐benzyloxy‐PBD linked via an exocyclic double bond at the C2′‐position to the C8 of a second PBD unit (Figure 6 B; n=1 or 2). The most potent dimer (n=1) had cytotoxicity in the micromolar range (e.g., 2.6 μm in the MCF‐7 cell line), and in DNA thermal denaturation studies gave a ΔT m of 5.7 °C after incubation with calf thymus DNA for 18 hours. This compared unfavorably with the ΔT m for the C8/C8′‐linked dimer DSB‐120 under similar conditions (i.e., 15.1 °C), again suggesting a poor fit in the DNA minor groove. In the same study, Kamal and co‐workers synthesized other C8/C2′‐coupled analogues in which one of the N10–C11 imine functionalities had been converted to a lactam (Figure 6 C). Interestingly, an example of this type of dimer (n=1) was still cytotoxic, but with a level of activity approximately 40‐fold lower than the bis‐imine parent molecule (i.e., Figure 6 B; n=1) suggesting that, although the molecule could no longer cross‐link DNA, some form of DNA interaction (presumably mono‐alkylation) was still occurring. A series of C2/C2′‐linked dimers have also been synthesized by Lown and co‐workers (Figure 6 D).11b Cytotoxicity studies indicated that one member of the series (where n=3) had an average LC50 of approximately 50 μm across 60 tumor cell lines. Molecular dynamics studies have since confirmed that the 3‐dimensional shape of both C8/C2′‐ and C2/C2′‐linked PBD dimers is not conducive to a good fit within the DNA minor groove.62
4. SJG‐136: The First PBD Dimer to Undergo Clinical Evaluation
As SJG‐136 is the best known and most characterized PBD dimer, the only example to reach clinical trials as a single agent, and is similar in structure to those currently being used as ADC payloads, its biophysical and biological properties, sequence‐selectivity, and pre‐clinical and clinical evaluation are described below.
4.1. Biophysical and Biological Properties of SJG‐136
The DNA binding affinity of SJG‐136 was initially investigated in DNA thermal denaturation studies using calf thymus DNA.10 The ΔT m values obtained (Table 1) demonstrated an increase in DNA melting temperature for each ratio after various incubation times at 37 °C. A gradual increase in helix melting temperature to 33.6 °C and 34.4 °C after 18 h and 72 h incubation, respectively, for a 1:5 ratio (SJG‐136/DNA) was obtained with a significant stabilization effect already evident at t=0, suggesting a strong kinetic component to the interaction. It is noteworthy that the ΔT m values obtained were significantly higher than for the PBD monomers tomaymycin and sibiromycin under identical conditions, with sibiromycin providing the highest ΔT m value at 18 hours for all of the known naturally occuring PBD monomers.10, 21 The extent of interstrand cross‐link formation produced by SJG‐136 was also investigated using a gel electrophoresis assay developed by Hartley and co‐workers.10, 63 It was evaluated at concentrations between 0.001 to 10 μm with pBR322 DNA (0.2 nm), producing highly efficient DNA cross‐linking (Figure 7 a). After 2 h incubation of DNA with SJG‐136, cross‐links were visible starting from a concentration as low as 0.01 μm, with single‐stranded DNA disappearing completely at 0.3 μm. The degree of cross‐linking was quantitated using laser densitometry as shown in Figure 7 b. Overall, these results demonstrated the high potency of SJG‐136 as a DNA interstrand cross‐linking agent.
Table 1.
Thermal denaturation data for the interaction of SJG‐136 with calf thymus DNA after incubation at different ratios for 0, 4 and 18 hours at 37 °C.[10, 21] [a,b]
| Compound | [PBD]:[DNA] molar ratio | Induced ΔTm (°C) after incubation at 37 °C for: | ||
|---|---|---|---|---|
| 0 h | 4 h | 18 h | ||
| SJG‐136 | 1:100 | 7.1 | 8.0 | 9.1 |
| SJG‐136 | 1:50 | 11.3 | 12.3 | 15.0 |
| SJG‐136 | 1:5 | 25.7 | 31.9 | 33.6 |
| Tomaymycin | 1:5 | 0.97 | 2.38 | 2.56 |
| Sibiromycin | 1:5 | – | – | 16.3 |
[a] Values for tomaymycin and sibiromycin under identical conditions are provided for comparison. [b] The induced ΔTm for a 1:5 [SJG‐136]:[DNA] molar ratio after 72 hours incubation at 37 °C was 34.4°C. 
Figure 7.

A) Autoradiograph of a denaturing agarose gel showing DNA interstrand cross‐linking of linear 32P‐end‐labeled pBR322 DNA (at 0.2 nm) by SJG‐136 following a 2 h incubation at 37 °C. The lanes are: C, double‐stranded DNA control; 0, single‐stranded DNA control; and 0.001, 0.003, 0.01, 0.03, 0.1, 0.3, 1.0, 3.0, 10.0 μm SJG‐136. DS and SS are double‐ and single‐stranded DNA, respectively. B) Quantification of the gel in (A) to show the concentration dependence of DNA cross‐linking for SJG‐136 in linear 32P‐end‐labeled pBR322 DNA.10 Reprinted with permission from S. J. Gregson, et al. (2001), J. Med. Chem. 44, 737–748. Copyright 2001 American Chemical Society.
Molecular modeling studies of cross‐link formation were carried out to support the observations made in the thermal denaturation and DNA cross‐linking studies.10 The results suggested that either no or insignificant disruption to DNA secondary structure occurred upon adduct formation and cross‐linking, and that no induced distortion of base pairs was evident within the helical structure.10 Overall, energy‐minimized models demonstrated excellent accommodation of SJG‐136 within the minor groove, with no significant part of the molecule exposed beyond the periphery of the duplex. DNA repair is usually initiated after repair enzymes recognize distortion or helical perturbations of the DNA helix (which usually occurs after interaction with the majority of DNA alkyating agents64), and so the excellent accommodation of SJG‐136 in the minor groove may explain the observed resistance of its adducts to DNA repair. These computational studies allowed models to be created for the interaction of SJG‐136 with DNA (Figure 8) consistent with the high ΔT m values observed (Table 1).
Figure 8.

A) Schematic diagram of the PBD dimer SJG‐136 forming an interstrand cross‐link with the DNA sequence 5′‐AGATCT‐3′. Its sequence‐selectivity is due in part to a) covalent bonds formed between the C11/C11′‐positions of the PBD units and the exocyclic C2‐amino groups of guanines on both strands, and b) hydrogen bonds (shown as ‐ ‐ ‐ ‐) formed between the N10‐H/N10′‐H protons of the PBD units and the ring nitrogen N3 acceptors of the 3′‐adenines adjacent to the covalently modified guanines.37a B) Molecular model of SJG‐136 forming an interstrand cross‐link within the sequence 5′‐TATAGATC(G)TATA‐3′ (covalently modified guanines underlined, and the guanine on the opposing strand in brackets) between the guanines on each strand (both magenta).
Initial evaluation of the in vitro cytotoxicity of SJG‐136 was carried out in the human ovarian carcinoma cell lines SKOV‐3, A2780 and CH1, together with their cisplatin‐resistant counterparts, A2780cis R and CH1cis R, using 96 hour continuous exposure in the sulforhodamine B (SRB) growth delay assay.65 The IC50 (half maximal inhibitory concentration) values obtained demonstrated a significant cytotoxic effect, with one of the lowest IC50 values obtained at the time for any synthetic PBD monomer or dimer, and significantly lower (i.e., 1500‐fold lower in CH1) than for cisplatin. Furthermore, in the A2780/A2780cisR pair of cell lines, SJG‐136 produced extremely low IC50 values of approximately 0.00002 μm with little cross‐resistance (i.e., resistance factor=1.1). Similar observations were made in the intrinsically cisplatin‐resistant cell line SKOV‐3, in which SJG‐136 had an IC50 value of 0.0091 μm compared to 10.5 μm for cisplatin.
The effect of SJG‐136 on DNA processing was initially studied in parallel DNA footprinting and in vitro transcription stop (T‐Stop) assays.66 The sequence selectivity of SJG‐136 toward isolated DNA was initially studied using a 262‐bp duplex DNA sequence from the MS2 plasmid which contained the predicted interstrand cross‐linking site for SJG‐136 (i.e., 5′‐Pu‐GATC‐Py‐3′) in addition to less‐favored sites (i.e., 5′‐GXXC‐3′; X=any base)10 (Figure 9 A). The sequence included two preferred binding sites at the two positions 5′‐48GGATCC53‐3′ and 5′‐208GGATCC213‐3′, together with 17 other less‐favored potential binding sites containing the sequence 5′‐GXXC‐3′. As anticipated, the molecule was observed to bind with high affinity to the 5′‐48GGATCC53‐3′ and 5′‐208GGATCC213‐3′ sites, but also bound to a number of other 5′‐GXXC‐3′ sites (where XX was TA, GC, CT, TT, GG or GT) with lower affinity. This result was surprising at the time of publication, given that a guanine residue should not be well‐tolerated between two cross‐linked guanines due to the relatively bulky nature of guanine C2‐exocyclic amino groups which point into the minor groove. However, based on more recent studies demonstrating that SJG‐136 can form mono‐adducts and intrastrand cross‐links in addition to interstrand cross‐links,14 this observation can now be explained by the formation of these other adduct types.
Figure 9.

A) Footprinting gels showing the interaction of SJG‐136 with the MS2 DNA sequence (10 nm) at concentrations of 0.1, 1, 3, 10, 30, and 100 μm (left panel=top strand [MS2‐F]; right panel=bottom strand [MS2‐R]). The labels to the right of each gel correspond to the potential binding sites.66 B) T‐Stop assay based on the same DNA sequence, showing the effect of incubation time on the ability of SJG‐136 (at 1.0 μm) to inhibit transcription. The right‐hand side of the panel shows that most stop sites were already visible after only 15 min incubation of SJG‐136 and the DNA together prior to addition of transcription buffer, and that there was little significant change up to 60 min incubation. The left‐hand side of the panel shows that, once transcription had started (t=0), there was little effect upon adding SJG‐136 at either 30 or 60 min time points (labeled as −30 and −60 min, respectively). The marker numbers on the right of Panel (B) represent DNA length in base‐pairs.66
To investigate whether the in vitro and in vivo biological activity57 of SJG‐136 could be due to sequence‐selective inhibition of transcription, a T‐Stop assay was carried out on a 282‐bp DNA sequence to investigate its effect on T7 RNA polymerase and transcription termination (Figure 9 B).22, 66 Significant transcriptional stop sites (T‐stops) were observed which appeared to be mainly associated with 5′‐GXXC‐3′ sequences. Although this assay could not distinguish between mono‐alkylated and cross‐linked adducts, at the time of publication the T‐stops were assumed to be mainly associated with interstrand cross‐links.22 In accord with the DNA footprinting experiments (Figure 9 A), one of the most favored cross‐linking sites (i.e., 5′‐48GGATCC53‐3′) gave the strongest T‐stop. The kinetic behavior of SJG‐136 was also studied by time‐dependent experiments which showed that incubation with DNA for 15 minutes was sufficient to inhibit transcription with negligible changes after this up to 1 hour incubation. Intriguingly, once transcription had been initiated, addition of SJG‐136 had negligible effect, suggesting that adducts needed to be fully formed before they could block transcription (Figure 9 B). Additional temperature‐dependent experiments suggested that the activation energy for covalent SJG‐136/DNA interaction must be very low at 5′‐Pu‐GATC‐Py‐3′ sites, since footprints were clearly detectable at these sites on the gels at 4 °C. In contrast, the activation energy for the less preferred 5′‐39GGTACC44‐3′ site must be higher, as the strongest footprint was only observed at 37 °C. Further evidence for the effect of SJG‐136 on DNA processing was derived from a restriction endonuclease inhibition assay (the RED50 assay) which showed that it can inhibit cleavage of the pGL3‐C plasmid at the BglII sequence 5′‐AGATCT‐3′ (a preferred SJG‐136 cross‐linking site) at low concentrations.
Next, cross‐link formation in tumor cell lines was studied using the single cell electrophoresis (Comet) assay to correlate the extent of DNA cross‐linking with cytotoxicity.67 SJG‐136 was initially evaluated in eight different cell lines using the MTT growth inhibition assay, and an average GI50 (concentration for 50 % maximal inhibition of cell proliferation) of 212 pm was determined (range: 2.1 pm in ovarian A2780 to 2.3 nm in colon HCT‐15).67 The Comet assay was then used to detect DNA interstrand cross‐links in some of these cell lines following a one hour exposure to SJG‐136 at 0.1 nm to 10 μm. The Comet assay involves exposure of cells to X‐rays which cause the nuclear DNA to fragment and electrophorese with a long (i.e., Comet‐like) tail. However, if the cells have been pre‐treated with a DNA cross‐linking agent such as SJG‐136, the DNA fragments are held together and electrophorese as a more‐singular entity. Using this methodology, cross‐linking was detected at very low SJG‐136 concentrations (e.g., 0.5 nm in the K562 cell line), with cross‐links persisting over a 48 hours period.
Similar studies were carried out using the γ‐H2AX assay68 which is more sensitive than the Comet assay69 in its ability to detect DNA damage caused by DNA cross‐linking agents. γ‐H2AX is a phosphorylated histone protein which is rapidly recruited in the form of “foci” to sites of replication fork collapse or DNA double‐strand breaks induced by cross‐linking agents.70 This provides a signal to recruit DNA repair and cell cycle checkpoint enzymes to initiate repair. These foci can be visualized using immunostaining techniques, and the number of foci present is proportional to the extent of DNA damage. Therefore, the γ‐H2AX assay has become an important and useful biomarker assay for measuring the DNA damage caused by DNA cross‐linking agents.71 An initial study was carried out to establish whether the assay could correlate the appearance of γ‐H2AX foci in HCT‐116 cells with the rate of interstrand cross‐link formation by SJG‐136.72 The cells were exposed to either 50 nm SJG‐136 for 1 hour, or 1 nm for 24 hours, and significant foci formation was detected under both conditions at 4 hours or 8 hours post treatment, respectively (Figure 10). Despite these exposure time differences, the levels of γ‐H2AX foci formed in both cases were comparable after 24 hours. Parallel experiments using the Comet assay showed that interstrand cross‐links were formed earlier with the higher dose (i.e., maximum reached after 1 hour for the 1 hour/50 nm treatment), but maximum cross‐link formation was reached only after 24 hours for the lower 1 nm treatment. These results were consistent with the need for DNA adducts to form prior to the appearance of γ‐H2AX foci.
Figure 10.

The results of a γ‐H2AX Foci assay used to study the adducts formed by SJG‐136 in HCT‐116 cells. The panels show the concentration‐ and time‐dependent phosphorylation of histone γ‐H2AX in response to treatment with SJG‐136. HCT‐116 cells were exposed for either 1 h at 50 nm or 24 h at 1 nm, fixed at 1, 4, 8 and 24 h after the start of the time course, and then immunostained with an anti‐phospho‐histone γ‐H2AX antibody.72
Studies were also carried out to investigate whether the cytotoxicity of SJG‐136 in colon cancer cell lines is dependent on the expression of P‐glycoprotein (P‐gp),73 as over‐expression is responsible for the resistance of many tumor cell types to DNA‐interactive agents.74 SJG‐136 was initially evaluated in a range of colon and ovarian tumor cell types known to over‐express P‐gp73 The results revealed a decrease in IC50 of up to 10‐fold after 24 hours treatment with SJG‐136 in the presence of verapamil, suggesting that P‐gp could be causing SJG‐136 efflux in these cells. Similar studies were undertaken in the adriamycin‐resistant A2780AD ovarian cell line known to over‐express P‐gp due to up‐regulation of the mdr‐1 gene, and in its non‐resistant parent cell line A2780. As anticipated, after 24 hours incubation, the parent cell line was more sensitive to SJG‐136 (i.e., IC50=0.27 pm) compared to the resistant A2780AD line (i.e., IC50=13 nm). Pre‐treatment with verapamil resulted in a greater increase in cytotoxicity in the doxorubicin‐resistant cell line (i.e., A2780, IC50=0.13 pM; A2780AD, IC50=0.7 nm), further supporting the possibility that SJG‐136 is a substrate for P‐gp. In vivo human tumor xenograft mouse models based on these cell lines were also investigated. In A2780 xenograft models at doses of 300 μg kg−1 as a single IV injection or 120 μg kg−1 daily for 5 days (i.v), a significant decrease in tumor volume was observed along with growth delay, whereas no significant response was observed in the adriamycin‐resistant A2780AD xenografts regardless of the dose or schedule.
Finally, the influence of SJG‐136 on cell cycle was studied in HCT‐116 cells (colon carcinoma) at high concentration (50 nm) for 1 hour and low concentration (1 nm) for 24 hours using flow cytometry. After the shorter exposure to the higher concentration of SJG‐136, a more prominent S‐phase arrest followed by transition to G0/G1 phase after 48 hours was observed, consistent with rapid interstrand cross‐link formation. In contrast, longer exposure to the lower concentration of SJG‐136 induced limited S‐phase arrest and re‐entry into G0/G1 phase after 72 hours.72
4.2. Sequence‐Selectivity of SJG‐136
The traditional methods used to evaluate the sequence‐selectivity and interstrand cross‐linking ability of small molecules are mostly based on gel electrophoresis methods involving drug‐treated radiolabeled duplex DNA (e.g., Figure 7 A).63 If the duplex DNA is cross‐linked in an interstrand manner, then it runs as the double‐stranded form on the gel despite the denaturing conditions. The resulting band on the gel is then quantitated by densitometry (e.g., Figure 7 B). However, HPLC/MS methodology has also been developed75 that requires only small quantities of inexpensive short non‐radiolabeled oligonucleotides. More importantly, the adducts formed can be rapidly visualized (i.e., in <30 min), and so both kinetic and thermodynamic data can be obtained, along with sequence selectivity information if different oligonucleotides are used. Initial studies of SJG‐136 using this methodology involved the self‐complementary 12‐mer oligonucleotides containing Pu‐GATC‐Py (Seq‐1), Pu‐GTAC‐Py (Seq‐2), Py‐GATC‐Pu (Seq‐3) and Pu‐IATC‐Py (Seq‐4) sequences (I=Inosine) (Figure 11 A). The results clearly established the kinetic and thermodynamic preferences of SJG‐136 for these sequences (Figures 11 B and C), with a rank order of Seq‐1 > Seq‐2 > Seq‐3 > Seq‐4).
Figure 11.

A) Structures of the double‐stranded (DS) oligonucleotides used to study interaction with SJG‐136 in initial HPLC/MS studies.75 B) Graph of % cross‐linking (% CL) versus time for duplexes Seq‐1 to Seq‐4 for a molar ratio of 4:1 (SJG‐136/DS‐DNA). C) Same data as in Panel B but plotted against log time to provide rate data from the gradients (Units=% cross‐linking, log h−1). For Panels B and C, 0 h corresponds to approximately 5 min after initial mixing of the duplex DNA and SJG‐136. Seq‐4 had all guanines replaced with inosines, and so was unreactive. All data points are the means of triplicate measurements from independent experiments, with error bars showing ± standard errors. Reproduced with permission from: M. Narayanaswamy, et al., Anal. Biochem. 374, 173–181.75
Based on gel assays, until the late 2000s the biological activity of SJG‐136 was thought to be entirely due to Pu‐GATC‐Py interstrand cross‐links.10, 66, 76 However, through HPLC/MS studies reported since then, it has become clear that SJG‐136 is capable of forming longer interstrand cross‐links at Pu‐GAATC‐Py sequences, intrastrand cross‐links at standard‐length Pu‐GATG‐Py and longer Pu‐GAATG‐Py sequences, and mono‐alkylated adducts at guanine residues where cross‐linking is not possible because of the distance between potentially reacting guanines.14 Similar studies have shown that SJG‐136 can also form interstrand and intrastrand cross‐links with shorter Pu‐GAC‐Py and Pu‐GAG‐Py sequences, respectively.58a In these experiments (Figure 12), interstrand cross‐link formation at the usual Pu‐GATC‐Py66 sequence was initially confirmed using a 12‐mer double‐stranded oligonucleotide containing this preferred sequence. The possibility of intrastrand cross‐link formation was then investigated using a similar 12‐mer DNA duplex mutated to contain a 5′‐AGATGT‐3′ sequence. HPLC and MALDI‐TOF‐MS analysis confirmed the formation of an intrastrand adduct, although the rate of formation was much slower (i.e., incomplete after 24 hours) compared to the standard interstrand cross‐link of similar length. In addition, mono‐alkylated adducts were observed in this study, confirmed by their HPLC retention times and MALDI‐TOF‐MS data. Previous modeling studies15 had suggested that insertion of an additional base pair between reactive guanines would prevent cross‐link formation due to loss of the preferred Pu‐GATC‐Py motif. To investigate this, reaction with oligonucleotides containing the extended Pu‐GAATC‐Py and Pu‐GAATG‐Py sequences designed for interstrand and intrastrand cross‐link formation, respectively, was studied. Surprisingly, SJG‐136 was able to form cross‐linked adducts with both sequences, although rates of reaction were slower compared to interstrand cross‐link formation with the standard‐length Pu‐GATC‐Py. Finally, a direct competition experiment was carried out between the interstrand Pu‐GATC‐Py and Pu‐GAATC‐Py, and intrastrand Pu‐GATG‐Py and Pu‐GAATG‐Py cross‐link sequences. A rank order of reactivity of Pu‐GAATG‐Py > Pu‐GATC‐Py ≥ Pu‐GATG‐Py > Pu‐GAATC‐Py was observed (Figure 12).
Figure 12.

Comparison of the reaction rates of SJG‐136 with 12‐mer duplex oligonucleotides containing Pu‐GATC‐Py, Pu‐GATG‐Py, Pu‐GAATC‐Py and Pu‐GAATG‐Py sequences. Reactions were monitored by HPLC in separate experiments with a 4:1 molar ratio of SJG‐136/DS‐DNA, and with adduct molecular weights and stoichiometries confirmed by MS.[14] The x‐axis is time in hours (t/h).
Molecular dynamics simulations were undertaken to gain insight into these observations.14 Energy‐minimized models indicated that the two electrophilic N10–C11 imine moieties of SJG‐136 are well‐positioned to react covalently with the two guanines in the Pu‐GATC‐Py (i.e., the favored interstrand cross‐link) and Pu‐GATG‐Py (i.e., the shorter intrastrand cross‐link) sequences, with little distortion of the DNA helix. However, for the extended Pu‐GAATC‐Py and Pu‐GAATG‐Py sequences, the model predicted that bis‐alkylation should only occur at the expense of some distortion of the DNA helix at the points of covalent attachment, and with SJG‐136 adopting a slightly lower position in the DNA minor groove in order to span the necessary distances between reactive guanines. Interestingly, more recent HPLC/MS studies77 have established that SJG‐136 can also bind to a terminal guanine within duplex and hairpin oligonucleotides, with the bulk of one of the PBD units positioned over two rather than three base pairs. Previously, this was not thought to be possible as, according to the literature,60 PBDs prefer to bind to a Pu‐G‐Pu triplet with a covalent bond forming between the central guanine and the C11‐position of the PBD.
Other related HPLC studies have uncovered previously unknown properties of PBD–DNA adducts. For example, it has been demonstrated that a loss of minor‐groove structure through denaturation of the DNA helix results in dissociation of the PBD/DNA adduct, although it can re‐form upon cooling.78 Similar studies have shown that although PBD molecules cannot form covalent adducts with single‐stranded DNA, they can stay covalently attached to DNA even if a covalently‐bonded duplex DNA adduct undergoes a complete and permanent loss of minor‐groove structure.58b Together, these studies have demonstrated that a minor‐groove environment is required for covalent attachment, although single‐stranded adducts can exist if the duplex is subsequently denatured. In addition, a dynamic equilibrium has been observed between DNA hairpin and duplex forms of covalent PBD/DNA adducts.79
Overall, these HPLC and modeling studies have demonstrated that SJG‐136 can form a wider variety of covalent DNA adducts than originally thought, which has led to a re‐evaluation of the previously proposed mechanism of action involving only interstrand cross‐linking at Pu‐GATC‐Py sites. In particular, the discovery that PBDs including SJG‐136 can bind to terminal guanines80 suggests that, in cells, they may be able to bind to the ends of double‐strand DNA breaks, a previously unconsidered mechanism of action.
4.3. Preclinical Evaluation of SJG‐136
In the late 1990s, SJG‐136 was evaluated in the National Cancer Institute's (NCI's) 60‐cell line panel81, 82 in which it showed promise in terms of potency and selectivity (Figure 13), with an average LC50 (i.e., concentration killing 50 % of cells) of 7.4 nm and a range of 0.14 to 320 nm. The significant differences between the LC50, TGI (i.e., total growth inhibition) and GI50 (i.e., concentration causing inhibition of proliferation of 50 % of cells) values across the different cell lines suggested that it had selective cytotoxicity toward certain panels of cell lines and individual cell lines. For example, the leukaemia and melanoma cell panels were particularly sensitive. Furthermore, the overall cytotoxicity profile of SJG‐136 was compared to the approximately 60 000 compounds in the NCI's database using the COMPARE molecular target analysis program.82 This suggested an activity pattern for SJG‐136 similar to some known DNA‐interactive agents (e.g., melphalan, cyclophosphamide, chlorambucil), although it did not match cluster patterns associated with any specific chemotherapeutic agents, thus reflecting its unique mechanism of action.
Figure 13.

Averaged mean graphs (n=3) for the evaluation of SJG‐136 in the NCI's “In Vitro 60 Cell Line Screen”, showing graphic and tabular listings of GI50, TGI and LC50 values (log units) for each cell line. The response of each cell line relative to the mean of all cell line responses is depicted by a horizontal bar extending either to the right (more sensitive) or left (less sensitive) of the mean (vertical line) for each index of activity. Reproduced with permission from: J. A. Hartley, et al., Cancer Res. 2004, 64, 6693–6699.76
Based on these encouraging results from the NCI's 60‐cell line screen, SJG‐136 was next evaluated in the Hollow Fibre assay76 to guide the selection of tumor cell types for xenograft studies. This involved cultivation of selected tumor cells in porous hollow fibre tubes which were implanted into the intraperitoneal (i.p.) and subcutaneous (s.c.) compartments of host immunocompromised mice. These cells were then exposed to SJG‐136 by systemic administration (once daily for four days, i.p.) followed by retrieval of the hollow fibres after 4 days, and quantitation of the viability of the cells inside using the MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazol) assay.83 In the NCI's standard Hollow Fibre Assay,76, 83 SJG‐136 was evaluated against 12 different tumor cell lines, and achieved >50 % growth inhibition in 83 % of the cell lines in the i.p. cavity, and 29 % in the s.c. location. Overall, it produced cell kill in 5 of the 12 cell lines used in the study (i.e., NCI‐H522 [lung adenocarcinoma], LOX IMVI and UACC‐62 [melanoma], MDA‐MB‐435 [breast carcinoma] and OVCAR‐3 [ovarian adenocarcinoma]). During the course of the experiments, the average body weight loss in mice treated systemically with SJG‐136 was ≤9 % with no drug‐related deaths, indicating that the agent was well tolerated.
Based on these results, the NCI evaluated the in vivo efficacy of SJG‐136 in a number of human tumor xenograft mouse and rat models57, 65b using tumor cell lines selected from the 60 cell line screen and Hollow Fibre Assay data.76, 81, 83 SF‐295 and LOX IMVI were selected for assessment of maximum tolerated dose (MTD) and minimum effective dose (MED) in the mouse model, which were established as 120 μg kg−1 day−1 and 16 μg kg−1 day−1, respectively. Additionally, HL‐60 TB and NCI‐H522 were selected for evaluation of different dosing schedules. SJG‐136 was well‐tolerated and highly efficacious in both models, with HL‐60 TB found to be the most sensitive tumor type, and with a once daily for five days schedule providing the best antitumor activity in both models. In addition, a single intravenous (i.v.) bolus regimen was compared with multiple daily i.v. dosing in the SF‐295 and LOX IMVI models. Interestingly, an immediate tumor mass reduction was observed in both models after the single dose administration (MTD for single bolus=400 μg kg−1 day−1), although tumor re‐growth occurred within the next four days. In contrast, daily administration of SJG‐136 appeared to be more effective in maintaining growth suppression and/or tumor regression. Continuous infusion of SJG‐136 was also explored in the SF‐295 and LOX IMVI xenograft mouse models but did not provide any greater efficacy compared to bolus administration.
Overall, significant tumor growth delays were observed in seven of the eight models studied after i.v. administration at doses between 25–100 μg kg−1, with dose dependency after multiple daily i.v. administration, and with greater efficacy following drug accumulation (i.e., daily for five days more efficacious than every fourth day for three treatments, or a single dose), and with multilog cell kill in four of the eight models. SJG‐136 failed to show activity in only one model (based on LOX IMVI) which was thought to be due to the mode of administration (i.e., s.c. rather than i.v.),57 although the reasons for this are not fully understood.
Efficacy of SJG‐136 in a xenograft model based on the cisplatin‐sensitive human ovarian cancer parental cell line CH1 and its cisplatin‐resistant equivalent CH1cisR was also studied.76 It was found to have significant antitumor activity in the CH1 model, providing tumor growth delays at a dose of 0.2 mg kg−1 administered i.v. on days 0, 4 and 8, comparable to 4 mg kg−1 of cisplatin using the same schedule. It also produced significant growth delay in the CH1cisR model at 0.2 mg kg−1 on the same schedule whereas cisplatin was inactive at 4 mg kg−1,76 supporting the hypothesis that SJG‐136 produces non‐distortive DNA adducts that do not trigger DNA repair.
In order to evaluate the pharmacokinetics of SJG‐136 in the animal models, and to develop methodology that could be used in clinical trials, a reversed‐phase LC/MS assay was initially developed.84 Using this assay, SJG‐136 was detected in plasma in the mouse models up to 4 hours after a single intraperitoneal administration of 0.2 mg kg−1, with a peak plasma concentration (Cmax) of 336 nm after 30 minutes, a half‐life (t 1/2) of 0.98 h, and a total clearance of 17.72 mL min−1 kg−1. Furthermore, it was shown to be stable in mouse blood and plasma in vitro over a 6 hour period at 37 °C with no significant loss due to degradation, and the plasma protein binding capacity was found to be 65±11 % and 76±5 % for initial concentrations of 100 nm and 1000 nm, respectively. Comparable pharmacokinetic studies were carried out in a rat model following single‐dose (15 and 50 μg kg−1) and multiple‐dose (25 μg kg−1 day−1 for 5 days) administrations using a similar LC/MS/MS method,20 and no plasma accumulation was observed after administration of 25 μg kg−1 day−1 for 5 days. An alternative HPLC‐based assay was developed to quantify the reactive imine form of SJG‐136 which proved to be accurate, reproducible and linear.85 The metabolism of SJG‐136 was also evaluated in a rat liver microsome assay, and it was found to be metabolized by CYP3A isoforms20 resulting in oxidative cleavage of the C8/C8′‐propyl linker and the release of two PBD monomer units.
To study the pharmacodynamic (PD) endpoint of the mechanism of action of SJG‐136 in the in vivo xenograft models, the Comet assay was used to evaluate tumor biopsies taken from a subcutaneous‐growing LS174T human colon cancer in MF1 nude mice.86 SJG‐136 caused a significant growth delay in this tumor model when delivered i.v. at 0.30 or 0.45 mg kg−1, and so tumor samples were removed at 1, 3 and 24 hours, and then subjected to Comet analysis. A small but significant level of DNA cross‐linking was observed for the 0.3 mg kg−1 dose after 1 hour which remained constant over 24 hours. The extent of cross‐linking increased with the higher dose of 0.45 mg kg−1, and this also remained constant over 24 hours.86 Finally, an ex vivo bone marrow colony formation (CFU‐GM) assay was used to assess the toxicity of SJG‐136 toward the hematopoietic system.76, 87 The results demonstrated that the concentrations of SJG‐136 required to achieve cytostatic and/or cytocidal effects in HL‐60 TB and Molt‐4 human leukaemia tumor cells in culture or in a soft agar colony formation assay (i.e., 0.14 to 70.8 pmol L−1) were significantly lower than the concentrations required for similar effects in the ex vivo bone marrow assay76 (i.e., 16.2 to 536 pmol L−1), suggesting a selective cytotoxicity toward the tumor cells and a potential therapeutic window.
In summary, SJG‐136 showed significant antitumor activity in most of the in vivo models examined based on a wide variety of tumor types. Activity was observed after a single bolus administration, although it gave the best efficacy after multiple administrations over a five‐day period.57 These encouraging results, coupled with data demonstrating a linear pharmacokinetic profile and differential cytotoxicity between leukaemia and bone marrow cells, led to the decision to evaluate SJG‐136 in Phase I clinical trials.
4.4. Clinical Evaluation of SJG‐136
The first clinical evaluation of SJG‐136 as a single agent in a Phase I setting was carried out at University College London and Edinburgh University (UK) between 2004 and 2006 in patients with advanced and/or metastatic solid tumors.18b Based on data from pre‐clinical toxicology studies in mice, rats and dogs,57, 88 it was initially administered as a 10 minute i.v. infusion every 21 days at a starting dose of 15 μg m−2 (i.e., 1/16 of the total dose given to dogs) which was doubled until drug‐related adverse events were observed. This 21‐day schedule was adopted due to the myelosuppression often associated with DNA‐interactive agents after bolus administration. A MTD of 45 μg m−2 was established for this schedule. Pharmacokinetic (PK) analysis18b, 84 of urine and blood samples showed a linear relationship between AUC0–8h or Cmax and dose over the dose range studied (i.e., 15–240 μg m−2), and results from the Comet69 and γ‐H2AX foci71 assays on both peripheral blood lymphocytes and tumor tissue confirmed that SJG‐136 induced DNA cross‐links with a maximum induction period of 24 hours post‐dose.
Out of twelve evaluable patients, stable disease (SD) was observed in ten patients, but no partial (PRs) or complete responses (CRs) occurred. Although bone marrow toxicity had been anticipated, surprisingly the major dose‐limiting toxicities (DLTs) observed were reversible liver toxicity (i.e., transaminitis), fatigue and, in some patients, a delayed lower‐limb oedema which could usually be resolved with diuretics. At this time, the oedema was not classified as vascular leak syndrome, as only the lower limbs were involved with most affected patients presenting only with swollen ankles, although in a small number of patients the entire legs were involved.
A second Phase I clinical trial with a different dosing schedule was carried out between 2004 and 2007 at the Memorial Sloan‐Kettering Cancer Center and the University of Kentucky (USA) in patients with refractory solid tumors using a starting dose of 10 μg m−2 per day administered as a bolus infusion over 20 minutes on days 1, 8 and 15 of a 28‐day cycle.18a DLTs became evident at a dose level of 60 μg m−2 per day as fatigue (42 %), thrombocytopenia (14 %) and delayed transaminitis (33 %), all at the Grade 3 level. Immediate transaminitis, oedema and hypoalbuminemia were also experienced by some patients. On this basis, the MTD was determined to be 40 μg m−2 per day on this schedule, and linear pharmacokinetics were demonstrated in the evaluated dose range of 10–60 μg m−2 per day, with dose‐proportional increases of SJG‐136 following systemic exposure.89 Although no PRs or CRs were observed, of the 13 patients in the trial, one had stable disease (SD) lasting for 8 cycles.
A third Phase I clinical trial was carried out between 2005 and 2008 at the Vanderbilt University Medical Center (USA) in patients with advanced solid tumors.90 Based on the outcomes from one of the previous clinical trials,18b this study evaluated two different dose schedules. In Schedule A, patients were given doses of 6, 12, 24, or 48 μg m−2 per day for 5 consecutive days of a 21 day cycle with the aim of studying DLTs and PK after multiple dosing. In this case, the major DLTs observed included delayed oedema, reversible liver toxicity (i.e., elevations in liver transaminases and alkaline phosphatase), dyspnoea, fatigue and hypoalbuminemia. Again, no myelosuppression was experienced by any of the patients. Based on these DLTs, patients in Schedule B were given doses of 20, 25, 30 or 35 μg m−2 per day for 3 consecutive days of a 21 day cycle, a shorter and more conservative schedule. It also included dexamethasone pre‐treatment and early diuretic (i.e., spironolactone) administration if oedema occurred. For this schedule, a MTD of 30 μg m−2 was determined, with DLTs similar to those previously documented. However, in contrast to the DLTs observed in Schedule A, they were manageable with the use of steroids and diuretics. PK analysis suggested a dose‐dependent increase in systemic exposure to SJG‐136 in Cycle 1 in both Schedules. Comet assay analysis of peripheral blood mononuclear cells (PBMCs) taken from patients in Schedule B showed detectable DNA interstrand cross‐link formation prior to dose 3 of Cycle 1, which was in broad agreement with the systemic exposure calculated from the AUC and Cmax values determined in the PK studies. Further analysis indicated persistent DNA cross‐link formation still detectable prior to the first dose of Cycle 2 for Schedule B. These data confirmed the persistence of cross‐links formed between SJG‐136 and DNA, and the ability of the adducts to avoid recognition and repair by DNA repair enzymes.10, 76 In this trial, of two patients with ovarian carcinoma, one experienced a confirmed PR, and the other an unconfirmed PR. In addition, one patient with small cell lung carcinoma, and one with bladder carcinoma, achieved SDs lasting more than 12 weeks.
A final multicenter Phase I clinical evaluation of SJG‐136 was carried out at the Sloane Kettering, M.D. Anderson and Ohio State University clinical centers in the US, but the results of these trials have not yet been published.
On the basis of these encouraging Phase I results, SJG‐136 progressed to a multicenter Phase II clinical evaluation in patients with epithelial ovarian cancer not responding to cisplatin or carboplatin. This study, carried out by the NCI between 2010 and 2013, enrolled nineteen patients on a schedule of 30 μg m−2 day−1 for 3 consecutive days of a 21 day cycle. For each patient, in addition to PK analysis, the extent of DNA adduct formation was determined in PBMCs and tumor biopsies using the Comet and γ‐H2AX assays. However, patient recruitment to this trial was poor, and no clinical results have been reported to date.
A second Phase II clinical evaluation of SJG‐136 in patients with advanced chronic lymphocytic leukaemia (CLL) and acute myeloid leukaemia (AML) was started by the NCI in 2014 and closed the following year. In this trial, patients received SJG‐136 at dose levels of 15 μg m−2 day−1 (Cohort I) or 30 μg m−2 day−1 (Cohort II) on days 1, 2 and 3 every 21 days for six cycles. In addition to establishing the MTD and pharmacokinetic parameters, the aim was to obtain data from bone marrow aspirates, pulse oximetry and electrocardiogram studies, although no clinical results have been reported to date.
In summary, the main DLTs observed in the Phase I and Phase II clinical trials were surprising in that bone marrow suppression, neutropenia, GI disturbances and alopecia, frequently observed for DNA‐interactive agents (and cross‐linking agents in particular), were absent. Instead, the most frequently observed side effects of lower limb oedema, fatigue and transaminitis were all reversible, and could be limited by pre‐treatment of patients with the steroid dexamethasone. Similarly, the oedema, if problematic, could be treated with a self‐administered diuretic (i.e., spironolactone). It is noteworthy that, overall, clinical signals of stable disease and partial responses were observed.
5. PBD‐Based ADCs in Clinical Development
During the last 30 years the use of monoclonal antibodies in cancer therapy has gained increasing importance.91 This tumor‐selective approach is based on the specific interaction of antibodies with antigenic biomarkers (e.g., proteins, glycoproteins or carbohydrates) on the surface of tumor cells, or intracellular signalling proteins, essential for their proliferation. One therapeutic approach is based on the use of antibodies alone to bind to these tumor antigens which exert an antitumor effect by attracting T‐cells to the area (i.e., the antibody‐dependent cell‐mediated cytotoxicity or “ADCC” mechanism).92 For example, the CD20‐targeting antibody Rituximab (Rituxan®, Mabthera®) is an example of this approach, and is used as a single agent for the treatment of non‐Hodgkin's lymphoma. However, an alternative approach is to attach a cytotoxic chemical payload to the antibody through a suitable linker to produce an antibody–drug conjugate (ADC) (Figure 14).91, 93 The chemical payload is chosen to be a highly cytotoxic small‐molecule, protein or glycoprotein that can be released at the tumor site through cleavage of the linker, thus killing tumor cells while producing minimal systemic toxicity. The first example of an approved ADC, gemtuzumab ozogamicin (Mylotarg®), was launched by Wyeth (subsequently Pfizer) in 2000 for the treatment of acute myeloid leukaemia,94 although it has since been withdrawn.95 Currently two ADCs are approved for cancer therapy, with over 55 others presently in early and late stage clinical development.91 Brentuximab vedotin (Adcetris®), developed by Seattle Genetics Inc in collaboration with Takeda Oncology, targets the CD30 antigen, carries an auristatin payload and is used in patients with anaplastic large cell lymphoma (ALCL) and Hodgkin's lymphoma. In the latter disease it provides an overall response rate of 75 %, with approximately 35 % and 40 % complete and partial responses, respectively. Similar results are obtained in ALCL, with an overall 86 % response rate, and 53 % and 33 % complete and partial responses, respectively.96 The second ADC currently approved is ado‐trastuzumab emtansine (Kadcyla®), developed by Genentech Inc, a subsidiary of Roche. This ADC, which targets the HER2 receptor and carries a maytansinoid payload, is used to treat metastatic breast cancer.97 In the clinic it has provided an overall survival (OS) time of 30.9 months compared to 25.1 months for treatment with a combination of lapatinib and capecitabine.
Figure 14.

Schematic diagram of an ADC showing the general structure of a typical antibody used for ADC construction. The antibody is comprised of heavy (blue) and light (green) chains, and contains antigen‐binding sites at the N‐terminus engineered to recognize antigens associated with a tumor cell. The cytotoxic payloads are connected via linkers to the antibody, with the example shown having two payloads attached via cysteine residues in the hinge region. When the ADC arrives at the tumor cell, the whole construct is typically internalized and the linkers cleaved, usually by proteases, to release the payload molecules which subsequently kill the cell.
Since the approval of gemtuzumab ozogamicin (Mylotarg®) in 2000, there has been a constant search for new highly‐cytotoxic molecules suitable for use as chemical payloads for ADCs. The quest for novel payload molecules is driven by a number of factors including the need to enhance cytotoxic potency, avoid the development of resistance and improve physicochemical properties, the availability and ease of manufacture, and the novelty of the molecule and thus the ability to protect by patenting. For example, the enediyne‐based payload of Mylotarg® is particularly complex and expensive to manufacture. Therefore, research in the 1990s and 2000s led to payloads based on the auristatins and maytansines which led to the commercially successful ADCs brentuximab vedotin and ado‐trastuzumab emantasine, respectively, although the IP space around these two payloads is now very restricted.
The PBD dimers were first studied as potential payloads for ADCs in a research collaboration between Spirogen Ltd and Seattle Genetics Inc in the mid to late 2000s. This led to two PBD‐based ADCs, SGN‐CD33A98 and SGN‐CD70A99 (Figure 15), which are currently in Phase III and Phase I clinical trials, respectively. This work also led to Seattle Genetics sub‐licencing its PBD dimer ADC technology to Abbvie Inc. Both Seattle Genetics ADCs utilize a simple valine–alanine (Val–Ala) dipeptide specifier unit between the PBD dimer (SGD‐1882) and the antibody, attached through thiol residues (i.e., Cys239 groups) in the hinge region of the antibody via a maleimidocaproyl (mc) linker. In the case of SGN‐CD33A, its antibody component is targeted to CD33 receptors on the surface of AML cells, and SGN‐CD70A is targeted to the CD70 antigen which is expressed on both renal cell carcinoma and non‐Hodgkin lymphoma cells but with limited expression on normal cells. In both ADCs, the PBD payload (SGD‐1882) is an unsymmetrical C2/C2′‐bis‐aryl C2‐C3/C2′‐C3′‐endo‐unsaturated C8/C8′‐linked PBD dimer, containing a C2′‐para‐methoxyphenyl substituent on the unconnected end, and a para‐aminophenyl C2‐substituent on the other end through which the Val–Ala specifier is attached.100 For SGN‐CD33A, conjugation is achieved through a serine‐to‐cysteine mutation at position 239 of the heavy chain of the antibody (a solvent accessible location), resulting in selective payload attachment with an average drug–antibody ratio (DAR) of 1.9.
Figure 15.

Structures of SGN‐CD33A (Vadastuximab talirine; one of the first PBD‐based ADCs to be developed), SGN‐CD70A and SGN‐CD123A, all constructed from the same Val–Ala dipeptide specifier unit and PBD dimer payload (SGD‐1882) linked to CD‐33‐, CD‐70‐ and CD123‐targeting antibodies, respectively, via a maleimidocaproyl (mc) moiety. SGN‐CD33A was developed through a research collaboration between Seattle Genetics Inc and Spirogen Ltd, and is presently in Phase III clinical trials.
In pre‐clinical studies SGN‐CD33A demonstrated significant in vitro and in vivo activity against a broad panel of AML cell lines and in pre‐clinical AML mouse models,19 respectively. For example, it had a mean IC50 of 22 ng mL−1 across 12 different AML cell lines, and was active in 15 out of 18 patient‐derived primary AML samples with a mean IC50 of 8 ng mL−1. Crucially, it had negligible activity in cell lines and patient‐derived samples having no or negligible CD33 expression, thus confirming the selectivity for this antigen. Other experiments demonstrated CD33‐dependent and dose‐dependent effects on the formation of CFU‐GMs (colony forming unit granulocytes/macrophages) in an ex vivo bone marrow colony formation assay. Furthermore, in experiments designed to confirm that the PBD dimer was released in target cells with subsequent DNA interstrand cross‐link formation, AML cells were treated for up to 48 hours with SGN‐CD33A followed by evaluation with the γ‐H2AX foci assay. A dose‐dependent increase in γ‐H2AX foci formation was observed from 16 to 24 hours after ADC exposure, and in related Western blot experiments SGN‐CD33A was shown to generate a dose‐dependent down‐regulation of p53, Chk1, Chk2 and caspase‐3 in HEL 92.1.7 AML cells. Finally, cell cycle analysis in AML cell lines using flow‐cytometry revealed that G2/M arrest occurred after treatment with SGN‐CD33A, providing further evidence for successful release of the PBD dimer from the ADC. SGN‐CD33A was also evaluated in several subcutaneous AML mouse xenograft models based on HL‐60, TF‐1α and HEL 92.1.7 tumors in which complete and persistent anti‐leukaemic responses were observed in all animals treated with a single dose of 100 μg kg−1 or 1000 μg kg−1,98 although reduced cell proliferation was also observed at the lower dose of 30 μg kg−1 compared to the non‐treated control group. Furthermore, in tumor models resistant to the CD33‐targeting gemtuzumab ozogamicin (e.g., HEL 92.1.7), SGN‐CD33A had similar potent anti‐leukaemic activity with complete responses at a single dose of 1000 μg kg−1, and with 3 out of 7 mice still tumor‐free at the end of the study.
These encouraging pre‐clinical results led to the initiation of Phase I clinical trials in 2013 to evaluate the safety, tolerability, PK and clinical activity of SGN‐CD33A in acute myelogenous, myeloid and promyelocytic leukaemia patients.101 The first trial involved 40 patients (48 % female) of average age 75 years, evaluating dose levels of 5 μg kg−1 (n=3), 10 μg kg−1 (n=3), 20 μg kg−1 (n=13), 40 μg kg−1 (n=18) and 60 μg kg−1 (n=3). A rapid and marked decrease of bone marrow blasts was observed at the dose levels of 40 and 60 μg kg−1 in 19 out of 21 patients, and complete disease remission occurred at the 5 (one patient), 10 (one patient) and 20 (two patients) μg kg−1 dose levels. Preliminary PK data showed an elevation in plasma concentration of the agent with increasing dose levels followed by rapid clearance, and a MTD of 40 μg kg−1 was established.101 Two DLTs of Grade 3 pulmonary embolism (at 20 μg kg−1) and Grade 4 hypocellular marrow (at 40 μg kg−1) were observed after 28 days. Other less serious side effects were encountered including Grade 3 febrile neutropenia, fatigue and malaise, GI effects (e.g., diarrhoea, constipation), lung toxicities (e.g., pleural effusion, cough and dyspnoea), epistaxis, peripheral oedema and hypokalaemia. Overall, all ADRs were manageable, and no treatment‐related deaths occurred. A Phase III combination trial was initiated by Seattle Genetics in May 2016 to evaluate SGN‐CD33A in combination with a hypomethylating agent (i.e., either azacitidine or decitabine) in older patients with newly diagnosed AML.
A second PBD‐based ADC developed by Seattle Genetics, SGN‐CD70A, uses an identical payload–specifier–linker assembly to SGN‐CD33A but is conjugated to the anti‐CD70 antibody h1F6 (Figure 15).102 As with SGN‐CD33A, conjugation was achieved through the Cys‐239 positions of the heavy chains of the antibody to produce an average DAR of 1.9. In vitro evaluation of both the ADC and PBD dimer payload alone gave nearly equivalent cytotoxicity in an ACHN cell line, and in vivo studies in BALB/c mice demonstrated that SGN‐CD70A was well tolerated at 2.5 mg kg−1 (single dose) with no weight loss. Human tumor xenograft mouse models demonstrated pronounced antitumor activity at doses as low as 0.1 mg kg−1. For example, in the ACHN model, a cure rate of 100 % was observed based on two weekly doses of 0.1 mg kg−1. Furthermore, a non‐Hodgkin's lymphoma model in SCID mice was very sensitive to this ADC, with 100 % cures after two weekly 0.1 mg kg−1 doses. Other experiments were carried out to confirm that the biological activity of SGN‐CD70A was consistent with the known mechanism of action of PBD dimers,103 such as activation of DNA damage pathways and promotion of cell death through G2 cell cycle arrest.99 For example, an immunofluorescence assay in which antibodies specific to double‐strand break repair proteins (i.e., Rad50 and Mre11) were used to monitor DNA damage foci demonstrated that foci were present after exposure to either SGN‐CD70A or the PBD dimer payload alone. Evidence for DNA strand breakage was obtained through observation of increased levels of phosphorylated Chk1 and Chk2 proteins (which both co‐ordinate DNA damage response) within 4 hours of treatment, along with increased levels of pBRCA1.103 The relevance of the DNA damage pathway to mechanism of action was also confirmed through experiments with small‐molecule inhibitors of DNA damage‐sensing kinases such as ATM, ATR and members of the Chk family, which were all synergistic with SGN‐CD70A. Similarly, a Mps1 inhibitor capable of shortening mitosis and abrogating the mitotic checkpoint was also synergistic.99 Based on these promising pre‐clinical results, a multi‐center, dose‐escalation Phase I clinical trial was initiated in 2014 to establish the MTD of SGN‐CD70A, and to evaluate its safety, PK and efficacy in patients with renal cell carcinoma, Mantle‐cell lymphoma and diffuse large B‐cell lymphoma.
Seattle Genetics has recently disclosed a third PBD‐based ADC, SGN‐CD123A, consisting of a humanized anti‐CD123 antibody targeted to CD123 (the alpha subunit of the IL‐3 receptor expressed by the tumor cells of the majority of AML patients) conjugated to the same dipeptide specifier and PBD dimer payload (i.e., SGD‐1882) through cysteines on each heavy chain of the antibody (Figure 15) to provide a DAR of approximately 2.104 It was initially assessed in 12 AML cell lines, four of which were MDR‐positive, and was highly active in 11 of these with a mean IC50 of 6 ng mL−1. It was also assessed in 23 patient‐derived primary AML samples with varying cytogenic profiles, and was shown to be active in 20 of these with a mean IC50 of 0.8 ng mL−1. Next, in vivo efficacy was investigated in AML xenograft models based on both MDR‐negative and MDR‐positive CD123‐positive tumor cell lines, and a single dose resulted in significant antitumor activity in all models. For example, in the xenograft based on the CD123‐positive/MDR‐negative cell line Molm‐13, a single dose of 10 μg kg−1 resulted in complete cures and significant survival compared to untreated or control ADC groups.104
A fourth Seattle Genetics ADC (SGN‐CD352A) targeting multiple myeloma has recently been disclosed.105 CD352A (SLAMF6) was found to be expressed in 87 % of multiple myeloma patient samples evaluated, thereby representing a viable ADC target. Studies have shown that conjugation of an anti‐CD352A antibody to an identical payload complex to that used in SGN‐CD33A (see Figure 15), results in an ADC with potent in vitro cytotoxicity (i.e., EC50 of 2–7 pm) in both multiple myeloma (e.g., MM.1R) and non‐Hodgkin's lymphoma (e.g., Ramos) cell‐lines. Furthermore, apoptotic cell death is activated in 48 hours, but the ADC does not affect the viability of normal resting human lymphocytes. SGN‐CD352A has also been shown to have potent in vivo activity in both multiple myeloma and non‐Hodgkin's lymphoma xenograft models, where durable responses were evident at single dose levels of between 30–100 μg kg−1.
Seattle Genetics has also reported studies on the bystander effect associated with PBD‐containing ADCs,106 demonstrating that an ADC comprised of a CD30+‐targeted antibody conjugated to a MC‐Val‐Ala‐PBD dimer construct (identical to that used in SGN‐CD33A, SGN‐CD70A and SGN‐CD123A) can produce substantial bystander killing effects in neighbouring antigen CD30− cells in comparison to a less membrane permeable CD30+‐targeted ADC carrying a MMAF payload. In these experiments, an admixed in vivo tumor model based on both Karpas‐35R (CD30−, and CD30‐VC‐MMAE resistant) and Karpas‐299 (CD30+) cells was developed. It was shown that when the level of Karpas‐299 CD30+ cells was reduced to 34 % in the admixed tumor model, treatment with the CD30+ PBD ADC caused tumor regression while the CD30‐vcMMAE ADC gave only growth delay. These results suggest that PBD‐based ADCs appear to require the presence of fewer antigen‐positive tumor cells to mediate bystander killing in vivo compared to an MMAE‐based ADC. This may be due to the superior potency of PBD dimers, and/or their ability to diffuse across cell membranes at a greater rate than MMAE payloads. In more recent work,132 Ma and co‐workers at Genentech have investigated the degree of DNA–PBD dimer adduct formation in tumour cells in mouse xenograft models. This involved collection of tumor and organ tissues from mice treated with the ADC, followed by DNA isolation and payload recovery. This was achieved through the use of Nuclease P1 to cleave the DNA to the level of individual adducts which could then be quantitated using a LC‐MS technique. The results suggested that adduct formation in tumour cells was highly efficient, with 80 % of the ADC entering tumors leading to DNA‐drug adducts after 96 hours. Furthermore, the number of adducts in tumor tissues was found to be 70‐fold greater than in healthy tissues (i.e., lung and liver) at a similar time point. The results also allowed the researchers to calculate that there were 41 PBD dimer adducts per million base pairs in tumor cells, compared to only 1 PBD dimer per million base pairs in lung and liver cells (and less in other organs such as the kidney). Importantly, these findings mirror clinical data for PBD‐containing ADCs, and highlight the advantages of PBD payloads including clean release, direct DNA targeting and DNA adduct stability.132
Along with SGN‐CD33A developed by Seattle Genetics, the DLL3‐targeted ADC SC16LD6.5 (also known as rovalpituzumab tesirine, or Rova‐T) developed through a collaboration between Stemcentrx Inc (recently acquired by Abbvie) and Spirogen Ltd was one of the first PBD dimer‐based ADCs to enter the clinic. DLL3 is an antigen found in the Notch signalling pathway, and is prevalent in neuroendocrine tumors. Rova‐T consists of a PBD dimer conjugated to a SC16 antibody through a cathepsin‐B cleavable Val–Ala specifier coupled to a PEG8 linker (Figure 16). Attached to the hinge‐region thiols of the antibody through maleimide coupling, the conjugate has a DAR of 2. The C8/C8′‐coupled PBD dimer payload used in this ADC contains a central pentamethylene linker and is C2‐C3/C2′‐C3′‐endo‐unsaturated with C2/C2′‐bis‐methyl substitution. These modifications to the PBD dimer were introduced in an effort to enhance isohelicity with the minor groove, and to reduce hydrophobicity.107 The payload is joined to the specifier via a para‐aminobenzyl alcohol (PAB) self‐immolative spacer unit attached to one of the N10‐atoms of the PBD through a carbamate linkage, a strategy previously disclosed in the patent literature by Genentech Inc and Spirogen Ltd.108
Figure 16.
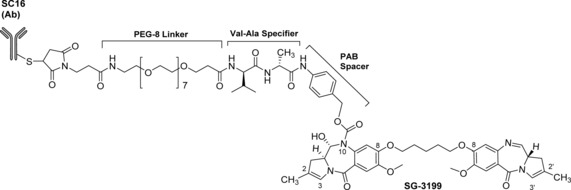
Structure of SC16LD6.5 (also known as rovalpituzumab tesirine, or Rova‐T), a DLL3‐targeted PBD‐containing ADC developed by Stemcentrx Inc through a collaboration with Spirogen Ltd. The PBD dimer payload is connected to the antibody through one of its N10‐positions via a self‐immolative para‐aminobenzyl alcohol (PAB) spacer unit, a cathepsin‐B‐cleavable Val–Ala dipeptide specifier and a PEG8 linker. Conventional maleimide conjugation was used to provide a DAR of 2. Rova‐T has recently entered Phase III clinical trials for patients with small‐cell lung cancer (SCLC).109
In pre‐clinical studies110 the naked antibody itself, SC16, was not cytotoxic toward either DLL3‐null HEK‐293T or DLL3‐expressing HEK‐293T.hDLL3 cell lines, whereas the PBD dimer alone and the full ADC (Rova‐T) both had significant cytotoxicity toward HEK‐293T.hDLL3 cells, with EC50 values of 46.9 pm and 7.8 pm, respectively. In addition, Rova‐T was shown to be more effective than standard agents in these cell lines109 but had no cytotoxicity in the stat‐null HEK‐293 cell line. Similarly, in the small‐cell lung cancer cell line LU64, Rova‐T had an EC50 of 8.3 pm, whereas the PBD dimer alone only achieved cell kill at concentrations of 500 pm or above, demonstrating a significant enhancement in cytotoxic activity through antibody conjugation. Successful knockdown of DLL3 expression by this ADC was also demonstrated in flow cytometry experiments based on patient‐derived LU37 large cell neuroendocrine carcinoma (LCNEC) cells transduced with a lentivirus expressing a DLL3‐targeted short hairpin RNA. Follow‐on in vivo studies110 involved mouse models implanted with patient‐derived small cell lung carcinoma (SCLC) or LCNEC cells with tumors grown to 140–200 mm before treatment. After three cycles of 1 mg kg−1 of Rova‐T administered intraperitoneally every four days (i.e., q4D×3), complete responses were obtained with no recurring tumors up to 144 days post‐dosing. To demonstrate payload‐dependent activity, similar xenografts treated with doses of up to 30 mg kg−1 of non‐conjugated antibody, or free PBD dimer corresponding to the concentration of payload on the ADC, provided no significant inhibition of tumor growth. Finally, in LU64 tumor‐bearing mice, treatment with Rova‐T provided a complete response after the tumors had rebounded from initial treatment with a cisplatin/etoposide combination. Based on these encouraging pre‐clinical data, a Phase I/II trial was initiated to establish its safety and tolerability at different dose levels in SCLC patients.109 Preliminary results, presented at an American Society of Clinical Oncology (ASCO) Annual Meeting in 2016, showed that 11 of 60 (18 %) evaluable patients dosed with Rova‐T experienced tumor shrinkage, and 41 (68 %) received associated clinical benefit. Interestingly, DLL3‐specific activity was observed in patients experiencing the best responses (i.e., 39 %; 10 of 26 patients) in the DLL3‐positive sub‐set. In these patients, median overall survival rate was 5.8 months, with 32 % of patients alive after 12 months. On the basis of these encouraging Phase I/II results, Rova‐T recently entered Phase III clinical trials in SCLC patients.
The same PBD dimer payload (i.e., a symmetrical C8/C8′‐coupled C2‐C3/C2‐C3′‐unsaturated C2/C2′‐dimethyl PBD dimer) has been used by Allozyne Inc. to generate a Her2/neu‐targeted ADC with a DAR of 1.9 (4D5‐PBD, Figure 17). 4D5‐PBD is unique in that it was constructed using “click” chemistry to enable site‐specific conjugation of the PBD dimer to the antibody through one of its N10‐positions via a Val–Ala specifier and self‐immolative PAB spacer. This was achieved through introduction of a non‐natural amino acid containing an azide moiety at defined places within the antibody which could react under very mild conditions with an alkyne group at the terminus of the linker‐payload assembly to form a stable heterocyclic triazole linkage between the payload and antibody. The click reaction involved a copper‐catalyzed cycloaddition process to attach the payload to both H274 positions of the antibody. During the design phase of this ADC, four potential conjugation sites were chosen based on analysis of the IgG1 crystal structure, and were selected for favorable characteristics such as a position distal to the antigen binding site and hinge regions, a location in a solvent‐exposed region and outwardly orientated to optimize ADC synthesis. This click chemistry approach was also used by the same researchers to produce an ADC based on the monomethyl auristatin F payload.
Figure 17.

Structure of 4D5‐PBD, a PBD‐containing HER2‐neu‐targeted ADC developed by Allozyne Inc. This ADC is unique in the use of click chemistry to form a stable heterocyclic triazole linkage between azide functionalities engineered into the antibody and an alkyne group at the terminus of a linker–specifier–PAB–PBD Dimer assembly.
4D5‐PBD was shown to have sub‐nanomolar potency across a number of relevant cell‐lines (e.g., EC50 of 0.061 nm and 0.504 nm in SKBR3 and SKOV3, respectively), but was marginally less potent in the same cells than a directly equivalent MMAF‐containing ADC (4D5‐AF). Other studies showed that 4D5‐PBD was less potent in some cell lines than trastuzumab‐PBD, however this could be partly explained by a DAR of 3 for the trastuzumab‐PBD conjugate, whereas 4D5‐PBD has a lower DAR of 1.8. Follow‐on in vivo studies showed little difference in activity between 4D5‐PBD and trastuzumab‐PBD, with both being similarly effective in inducing sustained tumor regression in nude mice implanted with BT474 tumors. Interestingly, the MMAF‐containing ADC 4D5‐AF did not provide curative effects at the same dose level in the same model.
Developed by ImmunoGen Inc., IMGN779 (Figure 18) is another PBD‐related ADC that has reached the clinic.111 This ADC, like SGN‐CD33A, targets CD33 receptors on the surface of AML cells, but contains an indolinobenzodiazepine moiety (referred to as an “IGN” by the company) as the cytotoxic payload. There are four unique features of this cytotoxic payload that distinguish it from the PBD dimers. First, the IGN structure DGN462 is based on two coupled indolinobenzodiazepine structures comprising two PBD skeletons with each containing an additional D‐ring attached to the C‐ring. The D‐ring is thought to act as an improved bioisosteric equivalent to C2/C3‐endo or C2‐exo‐unsaturation within the PBD skeleton (which is known to improve DNA‐binding affinity and cytotoxicity). For example, a difference in cytotoxicity was observed in comparisons of the cytotoxicity of SJG‐136 (a C2‐exo‐unsaturated PBD dimer) with a bis‐alkylating IGN dimer, where the presence of the D‐ring of the IGN (i.e., replacement of the pyrrolo C‐ring of the PBD with an indolino moiety) conferred an approximately 10‐fold higher potency in vitro compared to SJG‐136.111 It has also been suggested that introduction of the D‐ring may improve the electrophilicity and reactivity of the N10–C11 imine functionality through an electronic effect, although this has not been proven.112
Figure 18.

Structure of IMGN779, an indolinobenzodiazepine‐based ADC developed by ImmunoGen Inc. It is distinguished by four unique structural features: 1) the two additional D‐rings attached to the C‐rings of the PBD skeletons (i.e., to create indolobenzodiazepines or “IGNs”), 2) the conversion of one N10–C11 imine to a non‐electrophilic secondary amine to create a payload that mono‐alkylates rather than cross‐links DNA, 3) the central aniline ring within the C8/C8′‐linker to which the antibody is attached, a concept originally developed by Sanofi, and 4) joining of the DGN462 payload to the antibody via a cleavable disulfide linker.
A second unique feature of IMGN779 is that, although its payload is similar in overall format to a standard C8/C8′‐coupled PBD dimer structure, only one of the indolinobenzodiazepine moieties contains an electrophilic N10–C11 imine functionality capable of alkylating a guanine base in the DNA minor groove. The second imine functionality is reduced to a N10–C11 secondary amine which is non‐electrophilic, and so this indolinobenzodiazepine dimer arrangement is only capable of mono‐alkylating DNA and not cross‐linking. Although IMGN779 is the most clinically advanced IGN‐containing ADC to date (presently in Phase I clinical trials), it was preceded by studies of an ADC based on a bis‐alkylating payload (i.e., the bis‐imine IGN1 dimer, Figure 19) which was capable of cross‐linking DNA.113
Figure 19.

Structures of the bis‐imine indolobenzodiazepine (“IGN”) payload IGN1, and the isolated DNA‐binding component of DGN462. The bis‐imine IGN1 dimer is a symmetrical IGN dimer containing two electrophilic imines capable of producing DNA cross‐links in a similar manner to standard C8/C8′‐linked PBD dimers, whereas the DGN462 payload contains only one imine moiety, with the second reduced to a non‐electrophilic secondary amine. This payload is only capable of mono‐alkylating rather than cross‐linking DNA, and so ADCs containing it may produce less off‐target toxicity than IGN1 or other related cross‐linking PBD dimers.115
During the development of IMGN779, three IGN‐based bis‐alkylating ADCs were generated (i.e., an anti‐EPCAM‐IGN ADC, an anti‐CD‐33‐IGN ADC and an anti‐FRα‐IGN ADC) linked through amide bonds to lysine residues within the antibodies.109 All three of these ADCs had a DAR of 3 and were attached to the antibody via a non‐cleavable linker requiring lysosomal degradation of the antibody to release the lysine‐linked IGN. These constructs were found to be highly potent with an antigen‐dependent cell‐killing effect and the hindered disulfide linker providing good bystander killing of proximal cancer cells. For example, the anti‐CD‐33‐IGN ADC killed cells of the low CD‐33 antigen‐expressing leukaemia cell‐line NB4 with an IC50 value of 3 pm, whereas the antigen‐negative cell‐line Namalwa was >100‐fold less sensitive to the same ADC. Although these in vitro data were promising, a troubling pattern started to emerge during human tumor xenograft studies. Although displaying potent dose‐dependent antitumor activity in a human tumor xenograft of the epidermoid carcinoma KB starting with a single i.v. dose (activity was observed at concentrations as low as 0.5 mg kg−1), the anti‐FRα ADC caused prolonged loss in body weight, and in some cases delayed lethality in non‐tumor bearing CD‐1 mice at a dose of 3.75 mg kg−1 (qw×3, i.e., once per week for three weeks). In order to explore the SAR of the series, the IGN bis‐imine dimer was partially reduced to form a mono‐alkylating molecule (i.e., DGN462, Figure 19). Biophysical evaluation (e.g., Comet assay and thermal denaturation studies) suggested that the mono‐imine and bis‐imine IGNs have different mechanisms of action (i.e., mono‐alkylation and cross‐linking), and that the bis‐imine molecule forms a more stable DNA adduct. Remarkably, when the mono‐imine IGN was conjugated to an identical set of antibodies to the bis‐imine IGN to form three new ADCs (i.e., anti‐EPCAM, anti‐CD‐33 and anti‐FRα‐ADC), potent cytotoxicity was observed (i.e., IC50 values of 2–60 pm), and these ADCs were only 1.3‐ to 6‐fold less potent than the equivalent bis‐imine cross‐linking IGN ADCs. Furthermore, in vivo studies of the anti‐FRα mono‐imine IGN ADC produced dose‐dependent antitumor activity similar to the bis‐imine IGN ADC in the KB model, but with ∼2‐fold higher dosing (i.e., 7.5 mg kg−1, qw×3). Non‐tumor‐bearing CD‐1 mice treated on an identical schedule (i.e., 7.5 mg kg−1, qw×3) with the anti‐FRα mono‐imine IGN ADC showed no evidence of prolonged or delayed toxicity. The MTD of the the anti‐FRα mono‐imine IGN ADC in non‐tumor bearing mice was estimated to be 10 mg kg−1. As the minimum effective dose (MED) was estimated to be 1 mg kg−1, this provided an in vivo therapeutic index of approximately 10.
The design concept of mono‐alkylation (i.e., IMGN779) may have the advantage of producing lower toxicity compared to indolinobenzodiazepines or PBD dimers (e.g., bis‐imine IGN dimers or SG3199) that contain two imine moieties, as it is now apparent that ADCs containing traditional PBD dimer payloads that cross‐link DNA can lead to a number of off‐target delayed toxicities including bone marrow suppression.101
A third innovation relates to the concept of joining the indolinobenzodiazepine dimer to the antibody through a central aromatic ring within the C8/C8′‐linker. In this arrangement, three carbons of the central aromatic ring act as bioisosteres for the central three methylenes of the C8/C8′‐dioxypentanyl linker found in many traditional PBD dimers. Finally, the entire ADC construct (IMGN779), which has a DAR of approximately three, has the indolinobenzodiazepine mono‐alkyating dimer joined to the antibody via a cleavable disulfide linker which is thought to help avoid some of the toxicities observed with non‐cleavable linker formats.114 After obtaining positive pre‐clinical data, ImmunoGen has now started a Phase I open‐label study of IMGN779 in patients with relapsed/refractory CD33‐positive AML.
Genentech Inc is also developing PBD dimer based ADCs, and their method of attaching the PBD payload to the antibody is similar to that used by Allozyne Inc and Stemcentrx Inc, involving a self‐immolative PAB unit joined through one of the N10‐positions of the PBD dimer (Figure 20).116 One example provided in a Genentech patent consists of a C2/C2′‐exo‐methylene‐substituted PBD dimer with a C8/C8′‐dioxypentamethylene linker (i.e., SG2057) joined to a CD79b‐targeting antibody (e.g., huMA79bv28 or huMA79bv32) to treat lymphomas through the CD79b antigen found on B‐cells.117 The huMA79bv28 AI HC antibody, used for proof of concept studies, possesses an AI‐18C mutation in the heavy chain that includes a conjugatable thiol group for attachment to the linker‐payload assembly. The payload and antibody were joined through either Phe–HomoLys (Figure 20 A) or Val–Cit (Figure 20 B) specifiers with both connecting to one of the N10‐positions of the PBD dimer via a self‐immolative PAB spacer. The Phe‐HomoLys‐containing construct (Figure 20 A) also included a polyglycol (i.e., PEG) linker, whereas the Val‐Cit‐containing ADC (Figure 20 B) had a simple methylene linker.
Figure 20.

Schematic diagram of the PBD‐containing CD79b ADC conjugates developed by Genentech Inc. In these conjugates, the PBD dimer (SG2057) is connected to the antibody–linker–specifier–PAB assembly through one of its N10‐positions in a similar manner to the ADCs developed by Allozyne Inc and Stemcentrx Inc. The payload SG2057 is similar to SJG‐136 in having C2/C2′‐exo‐methylene substituents, but differs in that the two PBD units are connected via a C8/C8′‐dioxypentamethylene linker. The two ADC constructs shown above are similar except that one has a Phe–HomoLys specifier and a PEG‐based linker (A), whereas the other utilizes a Val–Cit specifier and a simple methylene linker (B).
In vivo studies involving the CD79b ADC shown in Figure 20 B, and the free PBD dimer SG2057 (at an equivalent dose to the ADC itself) were undertaken in which groups of mice transplanted with CD79b‐expressing WSU‐DLCL2 cells were treated with a single intravenous dose of 0.5 or 2.0 mg ADC/kg or 12.9 μg kg−1 of free PBD dimer. A control ADC consisting of an antibody non‐specific for the antigens expressed on WSU‐DLCL2 cells was included in the study. In a 35‐day time course study, compared to controls, the CD79b ADC produced inhibition of tumor growth at the 2.0 mg kg−1 dose level with complete responses. Importantly, growth inhibition was greater than for an ADC based on the same huMA79bv28 antibody but conjugated to the auristatin payload MMAE. Interestingly, the PBD dimer (SG2057) alone did not provide any inhibition of tumor growth under the same conditions.
Similar constructs to those in Figure 20 but based on anti‐CD33 antibodies have also been patented by Genentech Inc.116a Their efficacy was investigated using mouse xenograft models based on cell lines of the AML subtypes M2 (HL‐60) and M4a (EOL‐1). The animals were first dosed intraperitoneally with an excess of an anti‐dF control antibody to ensure non‐specific antibody binding sites were blocked. After this, substantial tumor growth inhibition was observed in both models after administration of the anti‐CD33 ADCs at both the 1.0 mg kg−1 and 20.0 μg m−2 dose levels.
The unsymmetrical PBD dimer approach, initially established by ImmunoGen Inc with their mono‐alkylating indolinobenzodiazepine‐containing IMGN779 ADC, has been adopted by others in the field. For example, Spirogen Ltd has filed a patent on PBD dimers containing unsymmetrical C2/C2′‐substitution patterns.118 A number of other PBD‐based ADCs are either in late‐stage pre‐clinical development or in early clinical trials, although information is not always available in the public domain. For example, in 2015 and 2016, MedImmune/Spirogen announced that it had sub‐licensed its PBD payload technology to Tanabe Research Labs and Regeneron Inc, respectively, although no details of planned ADCs were disclosed by either licensee, and other companies such as Kolltan and Pierre Fabre are also investigating PBD dimers as ADC payloads. Later stage PBD‐containing ADCs include ADCT‐301, developed by ADC Therapeutics Ltd, which comprises a CD25‐targeting antibody (i.e., HuMax‐TAC from Genmab Inc) and a traditional PBD dimer payload. This conjugate has shown potent dose‐dependent cytotoxicity in CD25‐positive cell lines at low doses, with significant activity against some drug‐resistant tumor types. It entered Phase I clinical trials in early 2015 for the treatment of lymphoma. ADC Therapeutics is also developing ADCT‐401 which contains a traditional PBD dimer attached to an antibody (i.e., J591) targeted to the prostate‐specific membrane antigen (PSMA). Although in vitro and in vivo data have yet to be disclosed, the conjugate is expected to enter clinical trials in the near future with a focus on hormone‐refractory prostate cancer. Another analogue, ADCT‐402, has also been disclosed by ADC Therapeutics, and has received regulatory approval to enter clinical trials. This ADC contains a PBD‐dimer coupled to a CD19 antibody through a maleimide–Val–Ala specifier assembly, and in vivo studies have demonstrated dose‐dependent antitumor activity in a human Burkitt's lymphoma‐derived Ramos xenograft model. For example, a single dose of 0.33 mg kg−1 resulted in a significant delay of tumor growth compared to vehicle treatment alone, with 4/10 and 10/10 tumor‐free survivors at doses of 0.66 mg kg−1 and 1 mg kg−1, respectively.119 Furthermore, doses of up to 2 mg kg−1 were well tolerated with no adverse side‐effects.119 Recently, two other ADCs utilizing tesirine have also been disclosed by ADC Therapeutics. Both ADCT‐502 and ADCT‐602 are currently at the pre‐clinical stage. ADCT‐502 has been developed to target HER2‐expressing tumors whereas ADCT‐602 is a CD‐22 targeting agent. Similarly, efforts are continuing to explore alternative linking positions to the C2‐ and N10‐positions for PBD dimers, and MedImmune/Spirogen have recently disclosed the use of the C7‐position for this purpose.
Feng and co‐workers at the NCI have recently reported studies involving a PBD‐based CD56‐targeted ADC.120 CD56 is over‐expressed in a number of different cancer types including multiple myeloma, acute myeloid leukaemia and pancreatic cancer. Using a phage display technique, two high‐affinity anti‐CD56 monoclonal antibodies were initially identified, M900 and M906, that bind to membrane proximal fibronectin Type III‐like domains and the N‐terminal IgG‐like domain, respectively. A C2/C2′‐bis‐aryl‐substituted PBD dimer connected to a MC–Val–Ala linker construct through the C2‐arylamino position was conjugated to both antibodies with a DAR of 4. The PBD dimer payload was specifically chosen over other payload families, as many childhood tumors (especially in children having received prior treatment) are resistant to ADCs containing MMAE and MMAF payloads.121 Furthermore, it has been shown that in some tumors with high multi‐drug resistance (e.g., acute myeloid leukaemia models),19 PBD dimers can maintain potency. In the experiments reported to date, the M906‐based ADC produced more potent in vitro activity (i.e., IC50 of 0.05–1.7 pm) compared to the M900 version (i.e., IC50∼100 pm), and in vivo studies are presently underway.
The first generation of PBD‐based ADCs have utilized G–G cross‐linking PBD dimers. However, a recent conference publication has disclosed that Genentech is actively investigating PBD‐based G–A cross‐linkers as potential ADC payloads.123 Three types of these molecules have been reported in the literature to date,124, 125, 126 each comprising a G‐alkylating PBD connected to either a CBI or CI A‐alkylating unit. Denny and co‐workers reported125 an extensive study of PBD–CBI hybrids which contained simple methylene linkers between the CBI and PBD units (Figure 21), and these molecules are now being developed as ADC payloads by Genentech. For example, 27eS, was found to have a CL50 value of approximately 0.01 μm toward pcDNA3 plasmid DNA, and an IC50 as low as 0.0078 nm in some tumor cell lines (i.e., EMT6). The molecule was also found to have antitumor activity in human tumor xenograft mouse models. When conjugated to the humanized anti‐HER2 antibody 7C2 (hu7C2) via a disulfide linkage, the resulting ADC had potent activity in vitro and in vivo, and the payload alone was found to be a poor substrate for Pgp and MDR1.123 An MTD of 15 mg kg−1 (single dose) was observed in rats for the PBD/CBI‐containing ADC, and this compared favorably to values of <5 mg kg−1 and 10 mg kg−1 observed for the stand‐alone CBI dimer (A–A cross‐linker) and PBD dimer payloads, respectively. Both the PBD dimer and PBD–CBI dimer ADCs contained an identical linker system (i.e., disulfide), whereas the CBI dimer was based on a non‐cleavable linker format.
Figure 21.

Parent structure of the PBD–CBI hybrid dimer series produced by Denny and co‐workers.122 27eS (containing a pentamethylene linker) was found to be most potent, and has been licensed for use as an ADC payload by Genentech Inc.
Finally, other companies working on PBD‐based ADCs have focussed on modifications to the central C8/C8′‐linker. For example, Hangzhou DAC Biotech Ltd has patented a PBD dimer with C2/C2′‐exo‐methylene substituents and a thio‐oxophosphane moiety at the center of the C8/C8′‐linker (Figure 22 A).127 The thio‐ethane substituent on the central oxophosphane acts as a novel linking point for antibody attachment. However, another important feature is that both the N10–C11 and N10′–C11′ imine moieties have been converted to their sodium bisulfite addition products. Although this type of modification can produce a prodrug form of a PBD dimer with significantly enhanced water solubility, it makes purification more challenging. This approach was used by Spirogen Ltd to synthesize the PBD dimer prodrug SG2285 (Figure 22 B).128
Figure 22.
Structure of the novel PBD dimer payload patented by Hangzhou DAC Biotech Ltd127 (A), and the standalone PBD dimer SG2285 synthesized by Spirogen Ltd (B). The dimer produced by Hangzhou contains a central oxophosphane moiety as part of its C8/C8′‐linker, with an appended thio‐ethane substituent through which an antibody can be attached. The molecule has the same C2/C2′‐exo‐methylene substitution as SJG‐136 but is unique for a PBD ADC payload in having the N10–C11/N10–C11′ imine functionalities converted to their sodium bisulfite addition products, thus producing a highly water soluble prodrug form. This prodrug approach was also used by Spirogen Ltd to produce the highly water soluble PBD dimer SG2285 shown in B.123
6. Conclusion
It has taken 50 years from the discovery of the first pyrrolobenzodiazepine (PBD) molecule, anthramycin, by William Leimgruber in the mid‐1960s2 for the first PBD‐based cancer chemotherapeutic agents to make an impact in the clinic. In the interim, a vast amount of data on both the SAR and DNA sequence selectivity of PBDs has been accumulated, contributing to a broad understanding of their mechanism of action. Although the PBD dimer SJG‐136 reached the clinic as a single agent and is still being evaluated, the first PBD dimer‐based agents to make a clinical impact have been ADCs containing PBD dimers as chemical payloads (e.g., Seattle Genetics’ SGN‐CD33A,101 and Stemcentrx's SC16LD6.5,110 both of which are now in Phase III clinical trials). Although anthramycin, sibiromycin and neothramycin were all investigated in previous decades,5 examples of PBD monomers have not reached the clinic in recent years. However, ImmunoGen Inc. has spearheaded a resurgence of interest in molecules analogous to PBD monomers by developing a PBD dimer‐type ADC payload (DGN462) with one active N10–C11 imine functionality disabled (i.e., by conversion to a non‐electrophilic secondary amine) resulting in an ADC (i.e., IMGN779) which has just entered the clinic,115 and this promising technology was sub‐licenced by Takeda in late 2015. Other groups are now developing PBD monomers as stand‐alone anticancer agents (particularly as transcription factor inhibitors7, 129), and as antibacterial agents.130 A further interesting development, pioneered by Sanofi S.S. and ImmunoGen Inc, is attachment of the antibody to a PBD dimer‐type payload through a substituted aromatic ring in the center of the C8/C8′‐linker that joins the two DNA‐interactive units together. This has the advantage that the PBD and PBD‐analog units themselves can have maximum diversity in their substitution patterns as they do not have to contain an antibody‐linking site.
In conclusion, of the 63 ADCs currently commercialized or in development, 7 (11 %) contain a PBD‐based payload, with auristatin (17, 27 %) and maytansine (13, 21 %) payloads used in the majority. However, given the performance of the PBD dimers as ADC payloads in the clinic to date, and their structural simplicity and ease of synthesis compared to many other payload types, it is likely that the percentage of PBD‐containing ADCs will increase significantly in the next decade, although their wider availability may be restricted by IP considerations. The concept of potentially reducing systemic toxicity by using mono‐alkylating PBD‐type molecules (i.e., IGNs), if proven in the clinic, may enhance the attractiveness of these molecules as payloads still further.
Overall, the success in the clinic and the current level of interest in the ADC area shown by pharmaceutical companies suggests that the ADC field will be a rapid growth area in the next decade. There are still a number of challenges to overcome such as widening the therapeutic window131 and the optimization of ADCs that work in solid tumor types as well as haematological cancers, but the success of ado‐trastuzumab emtansine (Kadcyla©), and the number of ADCs targeted to solid tumors currently in development, suggest that this will be a significant growth area in the future. Also, in the next decade, the development of novel and more‐sophisticated conjugation strategies and linker systems to join payloads to antibodies may lead to major improvements in terms of well‐defined DAR values for consistence of manufacture, improved stability of the final ADC products and more efficient release of payloads at the tumor site. Emerging linker technologies also raise the exciting possibility of attaching different payloads to the same antibody to improve efficacy. For example, in the case of PBD‐based payloads, combinations with DNA repair or P‐gp inhibitors, or payloads with different mechanisms of action could lead to improvements in efficacy and reduction in the development of resistance.
Biographical Information
Julia Mantaj graduated as a pharmacist from the Technical University of Braunschweig (Germany), and then completed her Ph.D. at King's College London with studies on the interaction of DNA minor‐groove binding agents with DNA and their effects on gene expression. She is currently employed as a biologist by the ADC payload company Femtogenix Ltd.

Biographical Information
Paul J. M. Jackson holds a degree in pharmaceutical sciences (Liverpool John Moores University), an MSc in Biosystems and Informatics (University of Liverpool) and a Ph.D. in Computational Drug Discovery (King's College London). Both his Ph.D. and post‐doctoral research has involved the development of computational chemistry methods to study the SAR of DNA‐interactive agents, and the computational design of DNA sequence‐selective molecules. He is a co‐founder of Transcriptogen Ltd and Femtogenix Ltd, for which he works as Project Manager.

Biographical Information
Khondaker Miraz Rahman is a Senior Lecturer in Medicinal Chemistry at the Institute of Pharmaceutical Science at King's College London. His research activities are focused on the application of synthetic medicinal chemistry and chemical biology techniques to the design, synthesis and evaluation of drug‐like chemical scaffolds as anticancer and anti‐infective agents. He completed his Ph.D. studies at the UCL School of Pharmacy (UK) followed by three years as a Cancer Research UK research fellow at the same institute. Prior to his Ph.D., he worked for three years as a R&D pharmacist and also held a number of academic positions. He is a co‐founder of Transcriptogen Ltd and Femtogenix Ltd.

Biographical Information
David E. Thurston is Professor of Drug Discovery at King's College London. He has a first degree in pharmacy and a Ph.D. in synthetic medicinal chemistry, and has previously worked in Schools of Pharmacy at the University of Kentucky and University of Texas at Austin, and the Universities of Portsmouth, Nottingham and UCL. He is one of the scientific co‐founders of Spirogen Ltd, and acted as CSO for the company from its formation in 2000 until 2011. He is also a co‐founder of Transcriptogen Ltd and Femtogenix Ltd, and Editor‐in‐Chief of the “Drug Discovery” book series of the Royal Society of Chemistry.

Acknowledgements
This review is dedicated to Professor Laurence H. Hurley (College of Pharmacy, University of Arizona, USA) whose early work on, and enthusiasm for, both naturally occurring and synthetic pyrrolobenzodiazepines inspired many generations of students working in his laboratory, ultimately leading to the PBD‐containing ADCs presently in the clinic.
J. Mantaj, P. J. M. Jackson, K. M. Rahman, D. E. Thurston, Angew. Chem. Int. Ed. 2017, 56, 462.
References
- 1.
- 1a. Thurston D. E., Bose D. S., Chem. Rev. 1994, 94, 433–465; [Google Scholar]
- 1b. Thurston D. E. in Molecular Aspects of Anticancer Drug-DNA Interactions, Vol. 1 (Eds.: S. Neidle, M. J. Waring), Macmillan, London, 1993, pp. 54–88; [Google Scholar]
- 1c. Khan G. S., Shah A., Rehman Z., Barker D., J. Photochem. Photobiol. B 2012, 115, 105–118; [DOI] [PubMed] [Google Scholar]
- 1d. Gerratana B., Med. Res. Rev. 2012, 32, 254–293; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Cipolla L., Araujo A. C., Airoldi C., Bini D., Anti-Cancer Agents Med. Chem. 2009, 9, 1–31; [DOI] [PubMed] [Google Scholar]
- 1f. Antonow D., Thurston D. E., Chem. Rev. 2011, 111, 2815–2864; [DOI] [PubMed] [Google Scholar]
- 1g. Dervan P. B., Bioorg. Med. Chem. 2001, 9, 2215–2235; [DOI] [PubMed] [Google Scholar]
- 1h. Neidle S., Nat. Prod. Rep. 2001, 18, 291–309; [DOI] [PubMed] [Google Scholar]
- 1i. Smellie M., Bose D. S., Thompson A. S., Jenkins T. C., Hartley J. A., Thurston D. E., Biochemistry 2003, 42, 8232–8239. [DOI] [PubMed] [Google Scholar]
- 2. Leimgruber W., Stefanovic V., Schenker F., Karr A., Berger J., J. Am. Chem. Soc. 1965, 87, 5791–5793. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Wells G., Martin C. R. H., Howard P. W., Sands Z. A., Laughton C. A., Tiberghien A., Woo C. K., Masterson L. A., Stephenson M. J., Hartley J. A., Jenkins T. C., Shnyder S. D., Loadman P. M., Waring M. J., Thurston D. E., J. Med. Chem. 2006, 49, 5442–5461; [DOI] [PubMed] [Google Scholar]
- 3b. Thurston D. E., Vassoler H., Jackson P. J., James C. H., Rahman K. M., Org. Biomol. Chem. 2015, 13, 4031–4040; [DOI] [PubMed] [Google Scholar]
- 3c. Thurston D. E., Chemistry and Pharmacology of Anticancer Drugs, CRC, Taylor & Francis, Boca Raton, 2006. [Google Scholar]
- 4. Rahman K. M., Vassoler H., James C. H., Thurston D. E., ACS Med. Chem. Lett. 2010, 1, 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Korman S., Tendler M. D., J. New Drugs 1965, 5, 275–284. [DOI] [PubMed] [Google Scholar]
- 6. Varvounis G., Molecules 2016, 21, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rahman K. M., Jackson P. J., James C. H., Basu B. P., Hartley J. A., de la Fuente M., Schatzlein A., Robson M., Pedley R. B., Pepper C., Fox K. R., Howard P. W., Thurston D. E., J. Med. Chem. 2013, 56, 2911–2935. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Farmer J. D., Rudnicki S. M., Suggs J. W., Tetrahedron Lett. 1988, 29, 5105–5108; [Google Scholar]
- 8b. J. D. Farmer, Jr. , Gustafson G. R., Conti A., Zimmt M. B., Suggs J. W., Nucleic Acids Res. 1991, 19, 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bose D. S., Thompson A. S., Ching J. S., Hartley J. A., Berardini M. D., Jenkins T. C., Neidle S., Hurley L. H., Thurston D. E., J. Am. Chem. Soc. 1992, 114, 4939–4941. [Google Scholar]
- 10. Gregson S. J., Howard P. W., Hartley J. A., Brooks N. A., Adams L. J., Jenkins T. C., Kelland L. R., Thurston D. E., J. Med. Chem. 2001, 44, 737–748. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Gregson S. J., Howard P. W., Thurston D. E., Bioorg. Med. Chem. Lett. 2003, 13, 2277–2280; [DOI] [PubMed] [Google Scholar]
- 11b. Reddy B. S., Damayanthi Y., Lown J. W., Anti-Cancer Drug Des. 2000, 15, 225–238. [PubMed] [Google Scholar]
- 12. Kamal A., Prabhakar S., Shankaraiah N., Reddy C. R., Reddy P. V., Tetrahedron Lett. 2008, 49, 3620–3624. [Google Scholar]
- 13. Kopka M. L., Goodsell D. S., Baikalov I., Grzeskowiak K., Cascio D., Dickerson R. E., Biochemistry 1994, 33, 13593–13610. [DOI] [PubMed] [Google Scholar]
- 14. Rahman K. M., Thompson A. S., James C. H., Narayanaswamy M., Thurston D. E., J. Am. Chem. Soc. 2009, 131, 13756–13766. [DOI] [PubMed] [Google Scholar]
- 15. Gregson S. J., Howard P. W., Gullick D. R., Hamaguchi A., Corcoran K. E., Brooks N. A., Hartley J. A., Jenkins T. C., Patel S., Guille M. J., Thurston D. E., J. Med. Chem. 2004, 47, 1161–1174. [DOI] [PubMed] [Google Scholar]
- 16. Hartley J. A., Spanswick V. J., Brooks N., Clingen P. H., McHugh P. J., Hochhauser D., Pedley R. B., Kelland L. R., Alley M. C., Schultz R., Hollingshead M. G., Schweikart K. M., Tomaszewski J. E., Sausville E. A., Gregson S. J., Howard P. W., Thurston D. E., Cancer Res. 2004, 64, 6693–6699. [DOI] [PubMed] [Google Scholar]
- 17. Rahman K. M., Rosado H., Moreira J. B., Feuerbaum E.-A., Fox K. R., Stecher E., Howard P. W., Gregson S. J., James C. H., de la Fuente M., Waldron D. E., Thurston D. E., Taylor P. W., J. Antimicrob. Chemother. 2012, 67, 1683–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Janjigian Y. Y., Lee W., Kris M. G., Miller V. A., Krug L. M., Azzoli C. G., Senturk E., Calcutt M. W., Rizvi N. A., Cancer Chemother. Pharmacol. 2010, 65, 833–838; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Hochhauser D., Meyer T., Spanswick V. J., Wu J., Clingen P. H., Loadman P., Cobb M., Gumbrell L., Begent R. H., Hartley J. A., Jodrell D., Clin Cancer Res. 2009, 15, 2140–2147. [DOI] [PubMed] [Google Scholar]
- 19. Sutherland M. S. K., Walter R. B., Jeffrey S. C., Burke P. J., Yu C., Kostner H., Stone I., Ryan M. C., Sussman D., Lyon R. P., Zeng W., Harrington K. H., Klussman K., Westendorf L., Meyer D., Bernstein I. D., Senter P. D., Benjamin D. R., Drachman J. G., McEarchern J. A., Blood 2013, 122, 1455–1463. [DOI] [PubMed] [Google Scholar]
- 20. Reid J. M., Buhrow S. A., Kuffel M. J., Jia L., Spanswick V. J., Hartley J. A., Thurston D. E., Tomaszewski J. E., Ames M. M., Cancer Chemother. Pharmacol. 2011, 68, 777–786. [DOI] [PubMed] [Google Scholar]
- 21. Puvvada M. S., Hartley J. A., Jenkins T. C., Thurston D. E., Nucleic Acids Res. 1993, 21, 3671–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Puvvada M. S., Forrow S. A., Hartley J. A., Stephenson P., Gibson I., Jenkins T. C., Thurston D. E., Biochemistry 1997, 36, 2478–2484. [DOI] [PubMed] [Google Scholar]
- 23. Shameem M., Kumar R., Krishna S., Kumar C., Siddiqi M. I., Kundu B., Banerjee D., Chem.-Biol. Interact. 2015, 237, 115–124. [DOI] [PubMed] [Google Scholar]
- 24. Baraldi P. G., Cacciari B., Guiotto A., Romagnoli R., Spalluto G., Leoni A., Bianchi N., Feriotto G., Rutigliano C., Mischiati C., Gambari R., Nucleosides Nucleotides Nucleic Acids 2000, 19, 1219–1229. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Kotecha M., Kluza J., Wells G., O'Hare C. C., Forni C., Mantovani R., Howard P. W., Morris P., Thurston D. E., Hartley J. A., Hochhauser D., Mol. Cancer Ther. 2008, 7, 1319–1328; [DOI] [PubMed] [Google Scholar]
- 25b. Brucoli F., Hawkins R. M., James C. H., Wells G., Jenkins T. C., Ellis T., Hartley J. A., Howard P. W., Thurston D. E., Bioorg. Med. Chem. Lett. 2011, 21, 3780–3783. [DOI] [PubMed] [Google Scholar]
- 26. Rahman K. M., Jackson P. J., James C. H., Basu B. P., Hartley J. A., de la Fuente M., Schatzlein A., Robson M., Pedley R. B., Pepper C., J. Med. Chem. 2013, 56, 2911–2935. [DOI] [PubMed] [Google Scholar]
- 27. Chou Y.-W., Senadi G. C., Chen C.-Y., Kuo K.-K., Lin Y.-T., Wang J.-J., Lee J.-H., Wang Y.-C., Hu W.-P., Eur. J. Med. Chem. 2016, 109, 59–74. [DOI] [PubMed] [Google Scholar]
- 28. Hsieh M.-C., Hu W.-P., Yu H.-S., Wu W.-C., Chang L.-S., Kao Y.-H., Wang J.-J., Toxicol. Appl. Pharmacol. 2011, 255, 150–159. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Kohn K. W., Spears C. L., J. Mol. Biol. 1970, 51, 551–572; [DOI] [PubMed] [Google Scholar]
- 29b. Kohn K. W., Glaubiger D., Spears C. L., Biochim. Biophys. Acta 1974, 361, 288–302. [DOI] [PubMed] [Google Scholar]
- 30. Hurley L. H., Gairola C., Zmijewski M., Biochim. Biophys. Acta 1977, 475, 521–535. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Antonow D., Jenkins T. C., Howard P. W., Thurston D. E., Bioorg. Med. Chem. 2007, 15, 3041–3053; [DOI] [PubMed] [Google Scholar]
- 31b. Kamal A., Ramesh G., Laxman N., Ramulu P., Srinivas O., Neelima K., Kondapi A. K., Sreenu V. B., Nagarajaram H. A., J. Med. Chem. 2002, 45, 4679–4688. [DOI] [PubMed] [Google Scholar]
- 32. Malhotra R. K., Ostrander J. M., Hurley L. H., McInnes A. G., Smith D. G., Walter J. A., Wright J. L., J. Nat. Prod. 1981, 44, 38–44. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Reddy B. S., Sharma S. K., Lown J. W., Curr. Med. Chem. 2001, 8, 475–508; [DOI] [PubMed] [Google Scholar]
- 33b. Reddy B. S., Sharma S. K., Lown J. W., Curr. Med. Chem. 2001, 8, 475–508. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Graves D. E., Stone M. P., Krugh T. R., Biochemistry 1985, 24, 7573–7581; [DOI] [PubMed] [Google Scholar]
- 34b. Krugh T. R., Graves D. E., Stone M. P., Biochemistry 1989, 28, 9988–9994. [DOI] [PubMed] [Google Scholar]
- 35. Boyd F. L., Stewart D., Remers W. A., Barkley M. D., Hurley L. H., Biochemistry 1990, 29, 2387–2403. [DOI] [PubMed] [Google Scholar]
- 36. Hurley L. H., Reck T., Thurston D. E., Langley D. R., Holden K. G., Hertzberg R. P., Hoover J. R., G. Gallagher, Jr. , Faucette L. F., Mong S. M. et al., Chem. Res. Toxicol. 1988, 1, 258–268. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Jenkins T. C., Hurley L. H., Neidle S., Thurston D. E., J. Med. Chem. 1994, 37, 4529–4537; [DOI] [PubMed] [Google Scholar]
- 37b. Hopton S. R., Thompson A. S., Biochemistry 2011, 50, 4720–4732. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Adams L. J., Jenkins T. C., Banting L., Thurston D. E., Pharm. Pharmacol. Commun. 1999, 5, 555–560; [Google Scholar]
- 38b. Remers W. A., Mabilia M., Hopfinger A. J., J. Med. Chem. 1986, 29, 2492–2503; [DOI] [PubMed] [Google Scholar]
- 38c. Jackson P. J. M., James C. H., Jenkins T. C., Rahman K. M., Thurston D. E., ACS Chem. Biol. 2014, 9, 2432–2440. [DOI] [PubMed] [Google Scholar]
- 39. Barkley M. D., Cheatham S., Thurston D. E., Hurley L. H., Biochemistry 1986, 25, 3021–3031. [DOI] [PubMed] [Google Scholar]
- 40. Thurston D. E., Bose D. S., Howard P. W., Jenkins T. C., Leoni A., Baraldi P. G., Guiotto A., Cacciari B., Kelland L. R., Foloppe M. P., Rault S., J. Med. Chem. 1999, 42, 1951–1964. [DOI] [PubMed] [Google Scholar]
- 41.
- 41a. Brucoli F., Hawkins R. M., James C. H., Jackson P. J. M., Wells G., Jenkins T. C., Ellis T., Kotecha M., Hochhauser D., Hartley J. A., Howard P. W., Thurston D. E., J. Med. Chem. 2013, 56, 6339–6351; [DOI] [PubMed] [Google Scholar]
- 41b. Chou Y.-W., Senadi G. C., Chen C.-Y., Kuo K.-K., Lin Y.-T., Wang J.-J., Lee J.-H., Wang Y.-C., Hu W.-P., Eur. J. Med. Chem. 2016, 109, 59–74. [DOI] [PubMed] [Google Scholar]
- 42. Antonow D., Jenkins T. C., Howard P. W., Thurston D. E., Bioorg. Med. Chem. 2007, 15, 3041–3053. [DOI] [PubMed] [Google Scholar]
- 43.
- 43a. Wu J., Clingen P. H., Spanswick V. J., Mellinas-Gomez M., Meyer T., Puzanov I., Jodrell D., Hochhauser D., Hartley J. A., Clin. Cancer Res. 2013, 19, 721–730; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43b. Gregson S. J., Howard P. W., Hartley J. A., Brooks N. A., Adams L. J., Jenkins T. C., Kelland L. R., Thurston D. E., J. Med. Chem. 2001, 44, 737–748. [DOI] [PubMed] [Google Scholar]
- 44. Borkovec A. B., Chang S. C., Horwitz S. B., J. Econ. Entomol. 1971, 64, 983–984. [DOI] [PubMed] [Google Scholar]
- 45. Jackson P. J. M., Rahman K. M., Thurston D. E., Cancer Res. 2014, 74, 5329. [Google Scholar]
- 46. Arima K., Kosaka M., Tamura G., Imanaka H., Sakai H., J. Antibiot. 1972, 25, 437–444. [DOI] [PubMed] [Google Scholar]
- 47. Brazhnikova M. G., Konstantinova N. V., Mesentsev A. S., J. Antibiot. 1972, 25, 668–673. [DOI] [PubMed] [Google Scholar]
- 48. Hisamatsu T., Uchida S., Takeuchi T., Ishizuka M., Umezawa H., Gann 1980, 71, 308–312. [PubMed] [Google Scholar]
- 49.
- 49a. Kaneko T., Wong H., Doyle T. W., J. Antibiot. 1984, 37, 300–302; [DOI] [PubMed] [Google Scholar]
- 49b. Konishi M., Hatori M., Tomita K., Sugawara M., Ikeda C., Nishiyama Y., Imanishi H., Miyaki T., Kawaguchi H., J. Antibiot. 1984, 37, 191–199. [DOI] [PubMed] [Google Scholar]
- 50. Walton M. I., Goddard P., Kelland L. R., Thurston D. E., Harrap K. R., Cancer Chemother. Pharmacol. 1996, 38, 431–438. [DOI] [PubMed] [Google Scholar]
- 51. Osada H., Ishinabe K., Yano T., Kajikawa K., Isono K., Agric. Biol. Chem. 1990, 54, 2875–2881. [PubMed] [Google Scholar]
- 52. Horwitz S. B., Chang S. C., Grollman A. P., Borkovec A. B., Science 1971, 174, 159–161. [DOI] [PubMed] [Google Scholar]
- 53.
- 53a. Thurston D. E., Jones G. B., Davis M. E., J. Chem. Soc. Chem. Commun. 1990, 874–876; [Google Scholar]
- 53b. Jackson P. J. M., Procopiou G., Veillard N., Mantaj J., Rahman K. M., Thurston D. E., Cancer Research 2016, 76, 4779. [Google Scholar]
- 54. Thurston D. E., Rahman K. M., Jackson P. J. M., WO 2015166289, 2015.
- 55. Bose D. S., Thompson A. S., Smellie M., Berardini M. D., Hartley J. A., Jenkins T. C., Neidle S., Thurston D. E., J. Chem. Soc. Chem. Commun. 1992, 1518–1520. [Google Scholar]
- 56. Morris S. J., Thurston D. E., Nevell T. G., J. Antibiot. 1990, 43, 1286–1292. [DOI] [PubMed] [Google Scholar]
- 57. Alley M. C., Hollingshead M. G., Pacula-Cox C. M., Waud W. R., Hartley J. A., Howard P. W., Gregson S. J., Thurston D. E., Sausville E. A., Cancer Res. 2004, 64, 6700–6706. [DOI] [PubMed] [Google Scholar]
- 58.
- 58a. Rahman K. M., James C. H., Thurston D. E., Nucleic Acids Res. 2011, 39, 5800–5812; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58b. Rahman K. M., James C. H., Bui T. T., Drake A. F., Thurston D. E., J. Am. Chem. Soc. 2011, 133, 19376–19385. [DOI] [PubMed] [Google Scholar]
- 59. Rahman K. M., James C. H., Thurston D. E., Nucleic Acids Res. 2011, 39, 5800–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hartley J. A., Hochhauser D., Curr. Opin. Pharmacol. 2012, 12, 398–402. [DOI] [PubMed] [Google Scholar]
- 61. Kamal A., Ramulu P., Srinivas O., Ramesh G., Bioorg. Med. Chem. Lett. 2003, 13, 3955–3958. [DOI] [PubMed] [Google Scholar]
- 62. Jackson P. J. M., Doctoral Thesis, King's College London, London, 2014. [Google Scholar]
- 63. Hartley J. A., Berardini M. D., Souhami R. L., Anal. Biochem. 1991, 193, 131–134. [DOI] [PubMed] [Google Scholar]
- 64. Lenglet G., David-Cordonnier M.-H., J. Nucleic Acids 2010, 290935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.
- 65a. Kelland L. R., Barnard C. F., Mellish K. J., Jones M., Goddard P. M., Valenti M., Bryant A., Murrer B. A., Harrap K. R., Cancer Res. 1994, 54, 5618–5622; [PubMed] [Google Scholar]
- 65b. Alley M. C., Scudiero D. A., Monks A., Hursey M. L., Czerwinski M. J., Fine D. L., Abbott B. J., Mayo J. G., Shoemaker R. H., Boyd M. R., Cancer Res. 1988, 48, 589–601. [PubMed] [Google Scholar]
- 66. Martin C., Ellis T., McGurk C. J., Jenkins T. C., Hartley J. A., Waring M. J., Thurston D. E., Biochemistry 2005, 44, 4135–4147. [DOI] [PubMed] [Google Scholar]
- 67. Hartley J. A., Hamaguchi A., Suggitt M., Gregson S. J., Thurston D. E., Howard P. W., Invest. New Drugs 2012, 30, 950–958. [DOI] [PubMed] [Google Scholar]
- 68. Ledermann J. A., Gabra H., Jayson G. C., Spanswick V. J., Rustin G. J., Jitlal M., James L. E., Hartley J. A., Clin. Cancer Res. 2010, 16, 4899–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Spanswick V. J., Hartley J. M., Ward T. H., Hartley J. A., Methods Mol. Med. 1999, 28, 143–154. [DOI] [PubMed] [Google Scholar]
- 70. Furuta T., Takemura H., Liao Z. Y., Aune G. J., Redon C., Sedelnikova O. A., Pilch D. R., Rogakou E. P., Celeste A., Chen H. T., Nussenzweig A., Aladjem M. I., Bonner W. M., Pommier Y., J. Biol. Chem. 2003, 278, 20303–20312. [DOI] [PubMed] [Google Scholar]
- 71. Wu J., Clingen P. H., Spanswick V. J., Mellinas-Gomez M., Meyer T., Puzanov I., Jodrell D., Hochhauser D., Hartley J. A., Clin. Cancer Res. 2013, 19, 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Arnould S., Spanswick V. J., Macpherson J. S., Hartley J. A., Thurston D. E., Jodrell D. I., Guichard S. M., Mol. Cancer Ther. 2006, 5, 1602–1609. [DOI] [PubMed] [Google Scholar]
- 73. Guichard S. M., Macpherson J. S., Thurston D. E., Jodrell D. I., Eur. J. Cancer 2005, 41, 1811–1818. [DOI] [PubMed] [Google Scholar]
- 74. Fojo A. T., Ueda K., Slamon D. J., Poplack D. G., Gottesman M. M., Pastan I., Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Narayanaswamy M., Griffiths W. J., Howard P. W., Thurston D. E., Anal. Biochem. 2008, 374, 173–181. [DOI] [PubMed] [Google Scholar]
- 76. Hartley J. A., Spanswick V. J., Brooks N., Clingen P. H., McHugh P. J., Hochhauser D., Pedley R. B., Kelland L. R., Alley M. C., Schultz R., Hollingshead M. G., Schweikart K. M., Tomaszewski J. E., Sausville E. A., Gregson S. J., Howard P. W., Thurston D. E., Cancer Res. 2004, 64, 6693–6699. [DOI] [PubMed] [Google Scholar]
- 77. Mantaj J., Jackson P. J. M., Karu K., Rahman K. M., Thurston D. E., PLoS One 2016, 11, e0152303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rahman K. M., James C. H., Thurston D. E., Org. Biomol. Chem. 2011, 9, 1632–1641. [DOI] [PubMed] [Google Scholar]
- 79. Rahman K. M., Mussa V., Narayanaswamy M., James C. H., Howard P. W., Thurston D. E., Chem. Commun. 2009, 227–229. [DOI] [PubMed] [Google Scholar]
- 80.See Ref. [77].
- 81. Monks A., Scudiero D. A., Johnson G. S., Paull K. D., Sausville E. A., Anti-Cancer Drug Des. 1997, 12, 533–541. [PubMed] [Google Scholar]
- 82. Paull K. D., Shoemaker R. H., Hodes L., Monks A., Scudiero D. A., Rubinstein L., Plowman J., Boyd M. R., J. Natl. Cancer Inst. 1989, 81, 1088–1092. [DOI] [PubMed] [Google Scholar]
- 83. Hollingshead M. G., Alley M. C., Camalier R. F., Abbott B. J., Mayo J. G., Malspeis L., Grever M. R., Life Sci. 1995, 57, 131–141. [DOI] [PubMed] [Google Scholar]
- 84. Wilkinson G. P., Taylor J. P., Shnyder S., Cooper P., Howard P. W., Thurston D. E., Jenkins T. C., Loadman P. M., Invest. New Drugs 2004, 22, 231–240. [DOI] [PubMed] [Google Scholar]
- 85. Cheung A., Struble E., He J., Yang C., Wang E., Thurston D. E., Liu P., J. Chromatogr. B 2005, 822, 10–20. [DOI] [PubMed] [Google Scholar]
- 86. Webley S. D., Francis R. J., Pedley R. B., Sharma S. K., Begent R. H., Hartley J. A., Hochhauser D., Br. J. Cancer 2001, 84, 1671–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Parchment R. E., Volpe D. A., LoRusso P. M., Erickson-Miller C. L., M. J. Murphy, Jr. , Grieshaber C. K., J. Natl. Cancer Inst. 1994, 86, 273–280. [DOI] [PubMed] [Google Scholar]
- 88. Buhrow S. A., Reid J. M., Jia L., McGovern R. M., Covey J. M., Kobs D. J., Grossi I. M., Ames M. M., J. Chromatogr. B 2006, 840, 56–62. [DOI] [PubMed] [Google Scholar]
- 89. Calcutt M. W., Lee W., Puzanov I., Rothenberg M. L., Hachey D. L., J. Mass Spectrometry 2008, 43, 42–52. [DOI] [PubMed] [Google Scholar]
- 90. Puzanov I., Lee W., Chen A. P., Calcutt M. W., Hachey D. L., Vermeulen W. L., Spanswick V. J., Liao C. Y., Hartley J. A., Berlin J. D., Rothenberg M. L., Clin Cancer Res. 2011, 17, 3794–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chari R. V., Miller M. L., Widdison W. C., Angew. Chem. Int. Ed. 2014, 53, 3796–3827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3872–3904. [Google Scholar]
- 92. Weiner L. M., Surana R., Wang S., Nat. Rev. Immunol. 2010, 10, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ducry L., Stump B., Bioconjugate Chem. 2010, 21, 5–13. [DOI] [PubMed] [Google Scholar]
- 94. Sievers E. L., Blood Cells Mol. Dis. 2003, 31, 7–10. [DOI] [PubMed] [Google Scholar]
- 95. Peters C., Brown S., Biosci. Rep. 2015, 35, e00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.
- 96a. Katz J., Janik J. E., Younes A., Clin. Cancer Res. 2011, 17, 6428–6436; [DOI] [PubMed] [Google Scholar]
- 96b. Younes A., Bartlett N. L., Leonard J. P., Kennedy D. A., Lynch C. M., Sievers E. L., Forero-Torres A., N. Engl. J. Med. 2010, 363, 1812–1821. [DOI] [PubMed] [Google Scholar]
- 97. Verma S., Miles D., Gianni L., Krop I. E., Welslau M., Baselga J., Pegram M., Oh D.-Y., Diéras V., Guardino E., N. Engl. J. Med. 2012, 367, 1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sutherland M. S. K., Walter R. B., Jeffrey S. C., Burke P. J., Yu C., Kostner H., Stone I., Ryan M. C., Sussman D., Lyon R. P., Blood 2013, 122, 1455–1463. [DOI] [PubMed] [Google Scholar]
- 99. Sandall S. L., McCormick R., Miyamoto J., Biechele T., Law C.-L., Lewis T. S., Cancer Res. 2015, 75, 946. [Google Scholar]
- 100. Kolakowski R. V., Young T. D., Howard P. W., Jeffrey S. C., Senter P. D., Tetrahedron Lett. 2015, 56, 4512–4515. [Google Scholar]
- 101.E. M. Stein, A. Stein, R. B. Walter, A. T. Fathi, J. E. Lancet, T. J. Kovacsovics, A. S. Advani, D. J. DeAngelo, M. M. O'Meara, B. Zhao, D. A. Kennedy, H. P. Erba, in 56th ASH Annual Meeting and Exposition, Moscone Centre, San Francisco, 2014.
- 102. Jeffrey S. C., Burke P. J., Lyon R. P., Meyer D. W., Sussman D., Anderson M., Hunter J. H., Leiske C. I., Miyamoto J. B., Nicholas N. D., Okeley N. M., Sanderson R. J., Stone I. J., Zeng W., Gregson S. J., Masterson L., Tiberghien A. C., Howard P. W., Thurston D. E., Law C.-L., Senter P. D., Bioconjugate Chem. 2013, 24, 1256–1263. [DOI] [PubMed] [Google Scholar]
- 103. Sandall S., Anderson M., Jonas M., Nesterova A., Miyamoto J., Stone I. J., Zeng W., Law C.-L., Lewis T. S., Cancer Res. 2014, 74, 2647. [Google Scholar]
- 104. Sutherland M. S. K., Yu C., Walter R. B., Westendorf L., Valliere-Douglass J., Pan L., Sussman D., Anderson M., Zeng W., Stone I., Klussman K., Ulrich M., Jonas M., Senter P., Drachman J. G., Benjamin D., Blood 2015, 126, 330. [Google Scholar]
- 105. Lewis T., Olson D. J., Gordon K. A., Sandall S. L., Miyamoto J., Westendorf L., Linares G., Leiske C., Kostner H., Stone I., Anderson M., Nesterova A., Jonas M., Law C.-L., Cancer Research 2016, 76, 1195–1195. [Google Scholar]
- 106. Li F., Emmerton K. K., Jonas M., Zhang X., Miyamoto J. B., Setter J. R., Nicholas N. D., Okeley N. M., Lyon R. P., Benjamin D. R., Law C.-L., Cancer Res. 2016, 76, 2710–2719. [DOI] [PubMed] [Google Scholar]
- 107.A. C. Tiberghien, J.-N. Levy, L. A. Masterson, N. V. Patel, L. R. Adams, S. Corbett, D. G. Williams, J. A. Hartley, P. W. Howard, ACS Med. Chem. Lett 2016, ASAP. [DOI] [PMC free article] [PubMed]
- 108. Howard P. W., Masterson L., Tiberghien A., Flygare J. A., Gunzner J. L., Polakis P., Polson A., Raab H. E., Spencer S. D., Vol. WO2011130598 A1, 2011.
- 109.SC16LD6.5 in Recurrent Small Cell Lung Cancer In: ClinicalTrials.gov [Internet]. 2013- [cited 2016 Mar 01]. Available from: http://clinicaltrials.gov/show/NCT01901653 NLM Identifier: NCT01901653, 2013.
- 110. Saunders L. R., Bankovich A. J., Anderson W. C., Aujay M. A., Bheddah S., Black K., Desai R., Escarpe P. A., Hampl J., Laysang A., Liu D., Lopez-Molina J., Milton M., Park A., Pysz M. A., Shao H., Slingerland B., Torgov M., Williams S. A., Foord O., Howard P., Jassem J., Badzio A., Czapiewski P., Harpole D. H., Dowlati A., Massion P. P., Travis W. D., Pietanza M. C., Poirier J. T., Rudin C. M., Stull R. A., Dylla S. J., Sci. Transl. Med. 2015, 7, 302ra136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Miller M. L., Fishkin N. E., Li W., Whiteman K. R., Kovtun Y., Reid E. E., Archer K. E., Maloney E. K., Audette C. A., Mayo M. F., Wilhelm A., Modafferi H. A., Singh R., Pinkas J., Goldmacher V., Lambert J. M., Chari R. V., Mol. Cancer. Ther. 2016, 15, 1870–1878 [DOI] [PubMed] [Google Scholar]
- 112.W. Li, N. E. Fishkin, M. Miller, B. Leece, E. Reid, K. Archer, E. Maloney, G. Jones, Y. Kovtun, R. Singh, R. V. J. Chari, in 239th ACS National Meeting, San Francisco, California, 2013.
- 113. Miller M., Fishkin N., Li W., Leece B., Mayo M., Jones G., Reid E., Archer K., Maloney E., Kovtun Y., Pinkas J., Singh R., Chari R., Mol. Cancer Ther. 2009, 8, B126. [DOI] [PubMed] [Google Scholar]
- 114. Polson A. G., Calemine-Fenaux J., Chan P., Chang W., Christensen E., Clark S., de Sauvage F. J., Eaton D., Elkins K., Elliott J. M., Frantz G., Fuji R. N., Gray A., Harden K., Ingle G. S., Kljavin N. M., Koeppen H., Nelson C., Prabhu S., Raab H., Ross S., Stephan J.-P., Scales S. J., Spencer S. D., Vandlen R., Wranik B., Yu S.-F., Zheng B., Ebens A., Cancer Res. 2009, 69, 2358–2364. [DOI] [PubMed] [Google Scholar]
- 115. Whiteman K., Audette C., Dandeneau A., Ellis M., Fishkin N., Harvey L., Johnson H., Kovtun Y., Maloney E., Miller M., Wilhelm A., Chari R., Cancer Res. 2014, 74, 2644. [Google Scholar]
- 116.
- 116a. Yu S. F., Liang W. C., Wu Y., Leong S., Polson A., WO 2015089344 A1, 2015;
- 116b. Asundi J., Clark S., Polakis P., WO 2014022679 A2, 2014;
- 116c. Leong S. R., Polson A., Polakis P., Wu Y., Liang W. C., Firestein R., WO 2014159835 A1, 2014.
- 117. Polakis P., Polson A., Spencer S. D., Yu S. F., WO 2014011519 A1, 2014.
- 118. Howard P. W., Tiberghien A., Jeffrey S., Burke P., WO 2013053873 A1, 2013.
- 119. Zammarchi F., Williams D. G., Adams L., Havenith K., Chivers S., D′Hooge F., Howard P. W., Hartley J. A., van Berkel P. H., Blood 2015, 126, 1564. [Google Scholar]
- 120. Feng Y., Wang Y., Zhu Z., Sussman R. T., Randall M., Bosse K. R., Maris J. M., Dimitrov D. S., Li W., mAbs 2016, 8, 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Loganzo F., Tan X., Sung M., Jin G., Myers J. S., Melamud E., Wang F., Diesl V., Follettie M. T., Musto S., Lam M. H., Hu W., Charati M. B., Khandke K., Kim K. S., Cinque M., Lucas J., Graziani E., Maderna A., O'Donnell C. J., Arndt K. T., Gerber H. P., Mol. Cancer Ther. 2015, 14, 952–963. [DOI] [PubMed] [Google Scholar]
- 122. Tercel M., Stribbling S. M., Sheppard H., Siim B. G., Wu K., Pullen S. M., Botting K. J., Wilson W. R., Denny W. A., J. Med. Chem. 2003, 46, 2132–2151. [DOI] [PubMed] [Google Scholar]
- 123. Lewis Phillips G. D., Li G., Guo J., Lau J., Yu S.-F., Pillow T., Lee B.-C., Sadowsky J., Schutten M., Fields C., Sliwkowski M. X., Cancer Res. 2016, 76, 1207. [Google Scholar]
- 124. Zhou Q., Duan W. H., Simmons D., Shayo Y., Raymond M. A., Dorr R. T., Hurley L. H., J. Am. Chem. Soc. 2001, 123, 4865–4866. [DOI] [PubMed] [Google Scholar]
- 125. Tercel M., Stribbling S. M., Sheppard H., Siim B. G., Wu K., Pullen S. M., Botting K. J., Wilson W. R., Denny W. A., J. Med. Chem. 2003, 46, 2132–2151. [DOI] [PubMed] [Google Scholar]
- 126. Purnell B., Sato A., O′Kelley A., Price C., Summerville K., Hudson S., O′Hare C., Kiakos K., Asao T., Lee M., Hartley J. A., Bioorg. Med. Chem. Lett. 2006, 16, 5677–5681. [DOI] [PubMed] [Google Scholar]
- 127. Zhao R. Y., WO 2015028850 A1, 2015.
- 128. Hartley J. A., Hamaguchi A., Coffils M., Martin C. R. H., Suggitt M., Chen Z. Z., Gregson S. J., Masterson L. A., Tiberghien A. C., Hartley J. M., Pepper C., Lin T. T., Fegan C., Thurston D. E., Howard P. W., Cancer Res. 2010, 70, 6849–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.
- 129a. Rahman K. M., Jackson P. J. M., James C. H., Basu B. P., Hartley J. A., de la Fuente M., Schatzlein A., Robson M., Pedley B., Pepper C., Fox K. R., Howard P. W., Thurston D. E., Cancer Res. 2013, 73, 1129; [Google Scholar]
- 129b. Jackson P. J., Rahman K. M., Thurston D. E., Cancer Res. 2014, 74, 5370. [Google Scholar]
- 130. Rahman K. M., Rosado H., Moreira J. B., Feuerbaum E. A., Fox K. R., Stecher E., Howard P. W., Gregson S. J., James C. H., de la Fuente M., Waldron D. E., Thurston D. E., Taylor P. W., J. Antimicrob. Chemother. 2012, 67, 1683–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. de Goeij B. E. C. G., Lambert J. M., Curr. Opin. Immunol. 2016, 40, 14–23. [DOI] [PubMed] [Google Scholar]
- 132.Y. Ma, S. C. Khojasteh, C. E. Hop, H. K. Erickson, A. Polson, T. H. Pillow, S. F. Yu, H. Wang, P. S. Dragovich, D. Zhang, Drug. Metabol. Dispos 2016, [epub ahead of print]. [DOI] [PubMed]