

Abstract
Objective
Juvenile neuronal ceroid lipofuscinosis (JNCL), or juvenile Batten disease, is a pediatric lysosomal storage disease caused by autosomal recessive mutations in CLN3, typified by blindness, seizures, progressive cognitive and motor decline, and premature death. Currently, there is no treatment for JNCL that slows disease progression, which highlights the need to explore novel strategies to extend the survival and quality of life of afflicted children. Cyclic adenosine monophosphate (cAMP) is a second messenger with pleiotropic effects, including regulating neuroinflammation and neuronal survival. Here we investigated whether 3 phosphodiesterase‐4 (PDE4) inhibitors (rolipram, roflumilast, and PF‐06266047) could mitigate behavioral deficits and cell‐specific pathology in the Cln3Δex7/8 mouse model of JNCL.
Methods
In a randomized, blinded study, wild‐type (WT) and Cln3Δex7/8 mice received PDE4 inhibitors daily beginning at 1 or 3 months of age and continuing for 6 to 9 months, with motor deficits assessed by accelerating rotarod testing. The effect of PDE4 inhibitors on cAMP levels, astrocyte and microglial activation (glial fibrillary acidic protein and CD68, respectively), lysosomal pathology (lysosomal‐associated membrane protein 1), and astrocyte glutamate transporter expression (glutamate/aspartate transporter) were also examined in WT and Cln3Δex7/8 animals.
Results
cAMP levels were significantly reduced in the Cln3Δex7/8 brain, and were restored by PF‐06266047. PDE4 inhibitors significantly improved motor function in Cln3Δex7/8 mice, attenuated glial activation and lysosomal pathology, and restored glutamate transporter expression to levels observed in WT animals, with no evidence of toxicity as revealed by blood chemistry analysis.
Interpretation
These studies reveal neuroprotective effects for PDE4 inhibitors in Cln3Δex7/8 mice and support their therapeutic potential in JNCL patients. Ann Neurol 2016;80:909–923
Juvenile neuronal ceroid lipofuscinosis (JNCL; or CLN3 disease) is an autosomal recessive lysosomal storage disease resulting from CLN3 mutations, with an estimated incidence of 1 in 100,000 live births.1, 2 Children with JNCL appear healthy at birth, with progressive vision loss typically being the first sign of disease that presents around 4 to 7 years of age. Later, affected children develop motor dysfunction that manifests as Parkinson‐like symptoms, along with seizures and cognitive deterioration, with a life expectancy of late teens to 20s.3 Magnetic resonance imaging findings include cortical and cerebellar atrophy with abnormally high signal intensity in the white matter, reflecting moderate demyelination.4, 5 The protracted nature of the disease, coupled with the current lack of effective therapeutics, highlights the need to identify novel approaches to improve the longevity and quality of life for these patients.
In JNCL mouse models, reactive glia are apparent within 1 to 3 months of age and coincide with areas of eventual neuron loss, which is significantly delayed in comparison (ie, 12–18 months).6, 7 Current evidence supports non–cell‐autonomous influences of diseased glia in JNCL pathogenesis in addition to intrinsic neuronal deficits. For example, Cln3Δex7/8 microglia are primed to produce exaggerated levels of several inflammatory mediators in response to danger signals encountered in the JNCL brain.8 Likewise, molecules critical for astrocyte glutamate regulation are dramatically reduced in Cln3Δex7/8 mice, including glutamate/aspartate transporter (GLAST) and glutamine synthetase.6 These findings support the neurotransmitter imbalance described in CLN3 mouse models, namely excessive glutamate and reduced γ‐aminobutyric acid (GABA), which has been implicated in neurotoxicity.9, 10, 11
Cyclic adenosine monophosphate (cAMP) is a second messenger that can regulate a variety of signaling events involved in learning and memory and synaptic plasticity.12, 13 Reductions in cAMP can have broad effects, ranging from a generalized negative impact on neuronal homeostasis, to promotion of microglial proinflammatory activity, to reduced glutamate transporter expression in astrocytes.14, 15, 16 cAMP levels are regulated by a balance between synthesis and degradation, mediated by adenylyl cyclase and phosphodiesterases (PDEs), respectively. There are 11 PDE enzyme families, with PDE4 most prominently expressed in the central nervous system (CNS).17 PDE4 inhibitors have shown beneficial effects in a wide range of neurodegenerative disorders, by limiting neuronal apoptosis and neuroinflammation as well as augmenting glutamate transporter expression.14, 18, 19, 20 Based on our previous findings demonstrating reductions in GLAST expression in Cln3Δex7/8 astrocytes,6 exaggerated proinflammatory mediator production in Cln3Δex7/8 microglia,8 and intrinsic neuron pathology as shown by other groups,7, 21 we surmised that PDE4 inhibitors would represent an excellent means to target this triad of cellular dysfunction.
Here we show that cAMP levels are significantly reduced in several brain regions of Cln3Δex7/8 mice where neuropathology occurs. Treatment of Cln3Δex7/8 animals with 3 distinct PDE4 inhibitors (rolipram, roflumilast, or PF‐06266047) significantly improved motor function over a 6‐month period. PDE4 inhibitors also reduced glial activation and lysosomal pathology and restored impaired glutamate transporter expression in Cln3Δex7/8 mice to wild‐type (WT) levels. Importantly, the beneficial effects of PDE4 inhibitors were still observed when treatment was delayed until 3 months of age, suggesting that they are capable of improving more advanced disease. This is the first study demonstrating that PDE4 inhibitors are effective at reducing motor deficits and pathological attributes of JNCL, supporting their potential therapeutic utility.
Materials and Methods
Mice
This study was conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center (approval ID: 11‐074‐08‐EP). This study used male Cln3Δex7/8 knock‐in mice (C57BL/6 background), which harbor the same 1.02kb deletion in CLN3 that occurs in approximately 85% of mutated CLN3 alleles.22 Age‐matched male C57BL/6 mice were used as WT controls.
PDE4 Inhibitor Treatment
The PDE4 inhibitor rolipram (Sigma‐Aldrich, St Louis, MO) was reconstituted in dimethylsulfoxide and working stocks for subcutaneous injections were made weekly in sterile phosphate‐buffered saline (PBS). Based on studies of rolipram pharmacokinetics (PK),23 we selected doses of 0.5 and 5mg/kg/day for proof‐of‐principle studies. We calculated that a dose of 0.5mg/kg would yield a concentration of around 68nM in the brain 1 hour after administration, with a half‐life of 1 to 3 hours. By extension, 5mg/kg was expected to increase CNS doses 10‐fold (ie, ∼680mM). The US Food and Drug Administration (FDA) approved PDE4 inhibitor roflumilast (Daliresp) was provided by AstraZeneca (Wilmington, DE) and was tested at oral doses of 2.5, 5, and 10mg/kg/day. A roflumilast stock solution was prepared by dissolving in PEG400 (Sigma‐Aldrich), and working stocks for oral gavage were made weekly by diluting in Methocel E15 (Dow Chemical Company, Midland, MI). Based on the PK properties of roflumilast following oral delivery,24, 25 we calculated that the highest dose of 10mg/kg would lead to a concentration around 50nM in the brain 1 hour after treatment, with a half‐life of 1 to 4 hours, although its active metabolite, roflumilast‐N‐oxide, extends drug action to 8 hours. Of note, the half‐life of roflumilast is much longer in humans (18 hours), making it amenable to once daily dosing.24, 25 The PDE4 inhibitor PF‐06266047 (ABI‐4) was provided by Pfizer (Boston, MA) and evaluated at oral doses of 0.5 and 1mg/kg/day. A working solution of PF‐06266047 for oral gavage was prepared weekly by directly dissolving in Methocel A4M. Based on the PK properties of PF‐06266047, we calculated that the highest dose of 1mg/kg would lead to a concentration around 400nM (total) in the brain 1 hour after administration, with a half‐life of 7.5 hours.26 Importantly, because therapeutic levels of PF‐06266047 would persist in the brain for nearly 24 hours with 1mg/kg dosing, we also examined a lower dose of 0.5mg/kg, which equated to an approximate concentration of 200nM (total) in the brain 1 hour after treatment. In terms of selectivity, rolipram, roflumilast, and PF‐06266047 are specific inhibitors of all PDE4 isoforms (PDE4A‐D) in rodents and do not affect the function of any other PDE isoenzymes.24, 25 In addition, PF‐06266047 had no activity at 10 µM in > 100 off‐targets (which exceeds the highest dose examined in our study by ∼ 100‐fold; patent number: US 20140235612). Our strategy was to evaluate several concentrations of each PDE4 inhibitor to identify the minimal effective dose that could be considered for potential clinical use. Because roflumilast is currently available in oral tablet form and several preclinical studies with other PDE4 inhibitors have utilized oral delivery,24, 25 we administered roflumilast and PF‐06266047 via oral gavage to most closely model the mode of therapeutic delivery in JNCL patients.
Experiments were performed using the National Institute of Neurological Disorders and Stroke recommended guidelines to ensure the rigor of these preclinical studies.27 Briefly, age‐matched male Cln3Δex7/8 and C57BL/6 WT mice were randomized into treatment groups (WT vehicle, Cln3Δex7/8 vehicle, WT PDE4 inhibitor, and Cln3Δex7/8 PDE4 inhibitor; n = 8/group) and assigned numbered ear tags to ensure that all participating research personnel were blinded to mouse genotype and treatment group. The number of mice utilized for each experiment was determined in pilot studies by identifying a sufficient group size that demonstrated statistical significance for accelerating rotarod activity (0.85; alpha = 0.05). PDE4 inhibitor and vehicle stocks were prepared and assigned generic labels, so that treatment identity was masked to the individual administering the compounds. The genotype and treatment status of individual animals remained unknown until data analysis was completed, whereupon groups were deidentified. Daily PDE4 inhibitor treatment was initiated in either 1‐ or 3‐month‐old mice, with treatment continuing for 6 to 9 months. The rationale for treating 1‐month‐old mice is that this roughly approximates the age at which a genetic diagnosis of JNCL is usually made in children (ie, 5–10 years), recognizing the caveats associated with human–rodent age equivalent estimates.28 Delaying treatment until 3 months of age was performed to determine whether PDE4 inhibitors can impact advanced disease, because a positive diagnosis of JNCL in children is often delayed. Body weights were recorded on a weekly basis after drug treatment commenced until completion of the study, because PDE4 inhibitors have been associated with weight loss.25
Accelerating Rotarod
Motor activity of mice was monitored monthly with an AccuRotor 4‐Channel Rotarod (Omnitech Electronics, Columbus, OH). Mice were subjected to testing over 4 consecutive days with a training period in the morning and testing period in the afternoon that were separated by 2 hours. All trials were conducted during the same time of day for consistency. During the training period (1 trial in the morning), mice were placed on the rotarod, which was set to a constant speed of 4rpm for 5 minutes. For the testing period, the apparatus was set to accelerate from 0 to 40rpm over 5 minutes. The rotation speed of 40rpm was maintained after the 5‐minute acceleration phase, and the latency to fall was digitally recorded by the instrument. Each mouse was subjected to 3 afternoon trials daily with at least 20 minutes of rest between each run.
Blood Chemistry Analysis
To assess the safety profile of chronic PDE4 inhibitor dosing in juvenile mice, blood chemistry analysis was performed at 2‐ to 3‐month intervals throughout the study. Blood was collected from the facial vein into heparinized capillary tubes and transferred to Vetscan Comprehensive Diagnostic Panel rotors (Abaxis, Union City, CA), which measured alanine aminotransferase, albumin, alkaline phosphatase, amylase, total calcium (Ca2+), creatinine, globulin, glucose, phosphorus, potassium (K+), sodium (Na2+), total bilirubin, total protein, and urea nitrogen.
Histology and Quantitation of Ocular Inclusions
As another assessment of safety/toxicity of PDE4 inhibitors in juvenile mice, organs were collected following sacrifice for histopathological assessment by a board‐certified pathologist with expertise in mouse toxicology studies (Samuel M. Cohen). The following tissues were fixed in 10% formalin and sent to AML Laboratories (Baltimore, MD) for sectioning and hematoxylin and eosin staining: kidney, stomach, liver, lung, spleen, heart, cervical lymph nodes, testes, thymus, trachea, muscle, spinal cord, larynx, pancreas, adrenal gland, aorta, bladder, colon, esophagus, small intestine, and femur/bone marrow.
To evaluate ocular inclusions, eyes were dissected under a microscope to remove the cornea and lens. Dissected eye cups were washed in PBS and embedded in acrylamide, whereupon cryostat sections (8 µm) were prepared to include the optic nerve. The most central optic nerve section was chosen for each eye, where a series of sequentially overlapping ×10 images were taken covering the entire section using fluorescence microscopy (tetramethylrhodamine filter). Inclusions from each retinal layer were counted in a masked fashion using ImageJ software (NIH, Bethesda, MD).
cAMP Quantitation
Various brain regions that are known to be affected in JNCL mice (ie, somatosensory barrel field cortex [S1BF], visual cortex [VC], hippocampus [HPC], and thalamus [TH]) were dissected from vibratome sections of WT and Cln3Δex7/8 mice at 7 and 12 months of age (n = 5–10 per group), whereupon cAMP levels were quantified using a cAMP enzyme‐linked immunosorbent assay (ELISA) kit (Enzo, Farmingdale, NY). cAMP concentrations were normalized to the amount of total protein collected to account for differences in tissue size. In some experiments where only tissue sections were available, cAMP levels were assessed by immunofluorescence staining as described below.
Immunofluorescence and Confocal Microscopy
Coronal brain sections were prepared from vehicle or PDE4‐inhibitor–treated WT and Cln3Δex7/8 mice at the end of the 6‐month dosing period as previously described with minor modifications.6 Briefly, animals were deeply anesthetized using sodium pentobarbital and transcardially perfused with saline followed by 4% paraformaldehyde (PFA). The brain was removed, postfixed in 4% PFA overnight, and cryoprotected with 30% sucrose overnight prior to embedding in optimal cutting temperature medium. Free‐floating cryostat sections (30 µm) were incubated overnight at 4 °C with antibodies specific for cAMP (1:200; Santa Cruz Biotechnology, Dallas, TX), glial fibrillary acidic protein (GFAP; 1:500; Dako, Carpinteria, CA), GLAST (1:200), lysosomal‐associated membrane protein 1 (LAMP1; 1:250), or CD68 (1:200; all from Abcam, Cambridge, MA) followed by appropriate biotin‐streptavidin–conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) and detection with either streptavidin–Alexa Fluor 594 or streptavidin–Alexa Fluor 488 (Invitrogen, Carlsbad, CA). TrueBlack (Biotium, Hayward, CA) was used to quench autofluorescent inclusions in Cln3Δex7/8 tissues. Immunofluorescence staining was visualized using a Carl Zeiss (Oberkochen, Germany) LSM 710 META confocal microscope with a ×40 oil immersion objective in a 450 × 450 µm field of view (FOV; × 200 magnification) or 225 × 225 µm FOV (×400 magnification). Tile images were acquired to visualize the entire S1BF (810 × 1,080 μm) or TH (810 × 810 μm) of each section (2 tissue slices per brain region per mouse). Tile images for GLAST quantitation were acquired at 270 × 270 μm for greater resolution. For image quantification, 200 × 200 μm nonoverlapping regions of interest (ROIs) were positioned throughout the S1BF (total of 9 ROIs) and TH (total of 6 ROIs) using AxioVision software (Zeiss), whereupon total values from all 6 to 9 ROIs were averaged for each of the 2 sections per mouse. Individual slice values per mouse were analyzed using SPSS (IBM, Armonk, NY) to evaluate statistical significance using a mixed linear model approach. Results are reported as the mean intensity staining values (GFAP and GLAST) or area (CD68 and LAMP1) determined by a custom software program incorporating the Visual Basic function of AxioVision (developed by Dr Nikolay Karpuk, University of Nebraska Medical Center). To quantify cAMP expression levels in the brain, 6 nonoverlapping × 40 images in the TH, HPC, and VC from 5 mice per group were acquired. Images were processed as described above, and results are reported as the mean staining intensity values.
Statistics
All assays were blinded throughout the entirety of data collection and analysis. For body weight comparisons, weights were averaged for mice in each treatment group and were analyzed by repeated measures analysis of variance (ANOVA) followed by a post hoc Tukey test. Quantification of CD68, GFAP, LAMP1, and GLAST immunofluorescence staining was assessed using a mixed linear model, where the variables included group, animal, slice, and staining area or intensity. Statistical significance between groups for each brain region was analyzed in SPSS. For accelerating rotarod analysis, results from all trials were averaged for each mouse in each treatment group and were analyzed by a mixed linear model in SPSS. Quantification of cAMP was assessed by 1‐way ANOVA in Prism (GraphPad, La Jolla, CA).
Results
Brain cAMP Levels Are Significantly Reduced in Cln3Δex7/8 Mice
Reductions in cAMP‐mediated signaling have been shown to impact neurobehavioral and cognitive function in normal aging, as well as in neurodegenerative diseases.12, 13 In addition, decreased cAMP levels are associated with proinflammatory responses and reductions in glutamate transporter expression,14, 15 which agrees with our prior findings of increased inflammatory mediator production by Cln3Δex7/8 microglia and decreased GLAST expression in the Cln3Δex7/8 brain.6, 8 To determine whether these changes may be linked to alterations in cAMP, cAMP levels were quantitated by ELISA in brain regions where neuropathology occurs in Cln3Δex7/8 mice.7 cAMP was significantly reduced in the TH of Cln3Δex7/8 mice at 7 months, whereas the HPC and VC showed a similar trend but did not reach statistical significance (Fig 1, upper panel). At 12 months, significant reductions in cAMP were observed in the TH, HPC, and VC of Cln3Δex7/8 mice compared to WT animals (see Fig 1, lower panel). Immunofluorescence staining for cAMP confirmed the reductions in 7‐month‐old mice (data not shown). cAMP levels were similar in the S1BF of WT and Cln3Δex7/8 mice at both ages examined, emphasizing the region‐specific neuropathology that is observed in JNCL.
Figure 1.
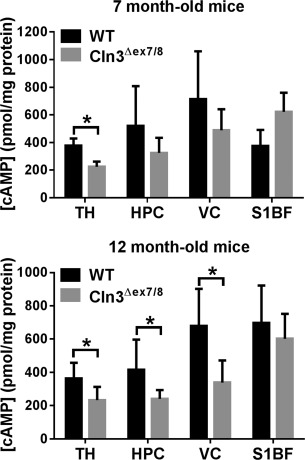
Cyclic adenosine monophosphate (cAMP) levels are decreased in vulnerable brain regions of Cln3Δex7/8 mice. Cln3Δex7/8 and wild‐type (WT) mice (n = 5–10/group) were sacrificed at 7 (top panel) or 12 months of age (bottom panel), whereupon cAMP levels were quantified in the thalamus (TH), hippocampus (HPC), visual cortex (VC), and somatosensory barrel field cortex (S1BF) by enzyme‐linked immunosorbent assay and normalized to total protein to account for differences in tissue size. Results are combined from 2 independent experiments, and significant differences between WT and Cln3Δex7/8 mice are denoted by asterisks (p < 0.05).
PDE4 Inhibitors Reverse Motor Deficits in Cln3Δex7/8 Mice
As previously mentioned, PDE4 inhibitors improve cognitive and motor function in various acute and chronic neurodegenerative diseases, including traumatic brain injury, spinal cord injury, Alzheimer disease, and Parkinson disease.18, 19, 20, 29, 30, 31 In our hands, Cln3Δex7/8 mice displayed significant impairments in motor function, as evident by reduced latency to fall on the accelerating rotarod (Fig 2),32 which has been attributed, in part, to excessive glutamatergic input.10, 33, 34, 35 We utilized this rotarod deficit to determine whether 3 distinct PDE4 inhibitors, including an early generation (rolipram),36 second generation (roflumilast),24 and third generation (PF‐06266047) inhibitor could improve motor function in Cln3Δex7/8 mice over a 6‐month treatment period. Administration of each PDE4 inhibitor was initiated in WT and Cln3Δex7/8 mice at 1 month of age, and motor activity was assessed by repeat testing at monthly intervals throughout the study.
Figure 2.
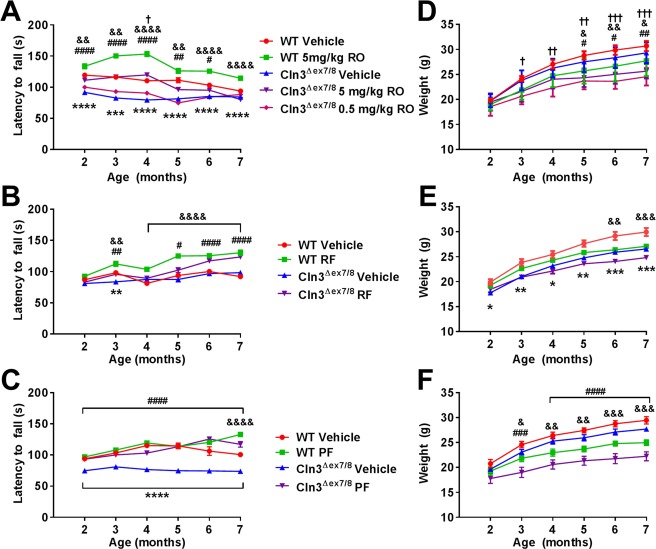
Phosphodiesterase 4 (PDE4) inhibitors improve motor function in Cln3Δex7/8 mice. Cln3Δex7/8 and wild‐type (WT) mice (n = 8/group) received vehicle or (A, D) rolipram (RO; 0.5 or 5mg/kg), (B, E) roflumilast (RF; 5mg/kg), or (C, F) PF‐06266047 (PF; 0.5mg/kg) beginning at 1 month of age, after which motor activity was assessed by accelerating rotarod testing at monthly intervals. Across all comparisons, significant differences between vehicle‐treated WT and Cln3Δex7/8 animals are denoted by asterisks. (A) Significant differences between Cln3Δex7/8 vehicle‐treated versus Cln3Δex7/8 5mg/kg/day rolipram are indicated by hash signs, Cln3Δex7/8 vehicle‐treated versus Cln3Δex7/8 0.5mg/kg/day rolipram are denoted by daggers, and WT vehicle‐treated versus WT 5mg/kg/day rolipram are indicated by ampersand signs. (B) Cln3Δex7/8 vehicle‐treated versus Cln3Δex7/8 roflumilast‐treated mice are indicated by hash signs, and WT vehicle‐treated versus WT roflumilast are indicated by ampersand signs. (C) Cln3Δex7/8 vehicle‐treated versus Cln3Δex7/8 PF‐06266047–treated mice are indicated by hash signs, and WT vehicle‐treated versus WT PF‐06266047–treated mice are indicated by ampersand signs. (D–F) Weights of Cln3Δex7/8 and WT mice during PDE4 inhibitor treatment. (D) Significant differences between Cln3Δex7/8 vehicle‐treated versus Cln3Δex7/8 5mg/kg/day rolipram are indicated by hash signs, Cln3Δex7/8 vehicle‐treated versus Cln3Δex7/8 0.5mg/kg/day rolipram are denoted by daggers, and WT vehicle‐treated versus WT 5mg/kg/day rolipram are indicated by ampersand signs. (E) Significant differences between WT vehicle‐treated and WT roflumilast are indicated by ampersand signs. (F) Significant differences between Cln3Δex7/8 vehicle‐treated versus Cln3Δex7/8 PF‐06266047–treated mice are indicated by hash signs, and WT vehicle‐treated versus WT PF‐06266047–treated mice are indicated by ampersand signs. For all comparisons, *, #, &, † reflect p < 0.05; **, ##, &&, †† denote p < 0.01; ***, ###, &&&, ††† reflect p < 0.001; and ****, ####, &&&& represent p < 0.0001.
We began our analysis with the first generation PDE4 inhibitor rolipram as proof of principle, because the compound has demonstrated beneficial effects in a wide range of neurological disorders but was halted from further clinical development because of its adverse side effects.18, 19, 20, 29, 30, 31 Rolipram exhibited a dose‐dependent effect on motor behavior in Cln3Δex7/8 mice, where the highest concentration examined (5mg/kg/day) significantly improved motor function within the first month of treatment, which extended out to 4 months (see Fig 2A). This benefit was negated with a lower drug concentration (0.5mg/kg/day). Interestingly, WT mice receiving 5mg/kg/day rolipram displayed increased rotarod performance as compared to vehicle‐treated WT animals at 2 to 6 months (see Fig 2A). A recent study demonstrated that long‐term rolipram treatment in WT mice enhanced mGluR‐mediated long‐term depression,37 which could account for our findings in WT animals, although this remains speculative. As mentioned above, rolipram was not used for subsequent mechanistic studies in the Cln3Δex7/8 model, because it is not a clinically viable agent due to its adverse side effects.
Roflumilast (Daliresp) is an FDA‐approved PDE4 inhibitor used to reduce the risk of exacerbations in patients with chronic obstructive pulmonary disease.25 Because roflumilast is FDA approved, this offered the possibility of repurposing the compound for JNCL, should efficacy be demonstrated. Cln3Δex7/8 mice were treated with 3 different concentrations of roflumilast (2.5, 5, and 10mg/kg) via daily oral gavage, with effects on motor function evaluated at monthly intervals. Only the highest doses of roflumilast tested (5 and 10mg/kg/day) led to significant improvements in rotarod activity, whereas the lower concentration (2.5mg/kg/day) was not effective (see Fig 2B and data not shown). The groups of WT and Cln3Δex7/8 vehicle‐treated mice used to assess 5mg/kg/day roflumilast did not differ from each other dramatically in accelerating rotarod performance, which was unusual (see Fig 2B). Nevertheless, treatment of Cln3Δex7/8 animals with 5mg/kg/day roflumilast did significantly improve motor performance compared to vehicle‐treated Cln3Δex7/8 mice. Compared to rolipram, the beneficial effects of roflumilast on motor activity were not observed until 3 months of treatment, which may be explained by the lower blood–brain barrier permeability of roflumilast.24, 25
The final compound examined was a third generation PDE4 inhibitor PF‐06266047 (patent number: US 20140235612). A phase 1 clinical trial has recently been completed to assess the safety and tolerability of PF‐06266047 in healthy human subjects (ClinicalTrials.gov identifier: NCT02539550). Unlike roflumilast, PF‐06266047 has superior blood–brain barrier permeability and better rodent PK, which translated into lower dosages for our in vivo studies. Cln3Δex7/8 and WT mice received 0.5 or 1mg/kg of PF‐06266047 via daily oral gavage beginning at 1 month of age, with monthly rotarod assessments performed. Cln3Δex7/8 mice treated with the lowest dose of PF‐06266047 (0.5mg/kg/day) displayed significant improvements in rotarod activity as early as 1 month post‐treatment, which was not observed with the higher dose (see Fig 2C and data not shown). One possible explanation for this dosage‐dependent effect is that the PK profile of PF‐06266047 indicated that therapeutic levels would be maintained for ∼24 hours at the 1mg/kg dose. Reducing the dose by a factor of 2 would inhibit enzyme activity for approximately 12 hours, which was considered more desirable to allow for some PDE4 activity that is likely needed to maintain homeostatic functions. Therefore, we interpreted the inability of the higher dose to provide benefit to be due to it completely preventing PDE4 action throughout a 24‐hour period. Based on this, PF‐06266047 was examined at 0.5mg/kg/day for all subsequent studies. We also demonstrated that this dose of PF‐06266047 restored cAMP in the TH, VC, and HPC of Cln3Δex7/8 mice to levels seen in WT animals (Fig 3).
Figure 3.
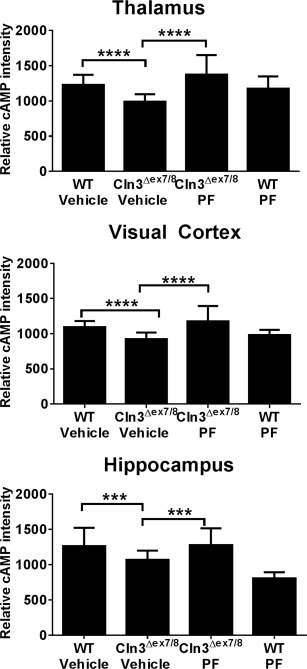
PF‐06266047 restores cyclic adenosine monophosphate (cAMP) levels in Cln3Δex7/8 mice. Cln3Δex7/8 and wild‐type (WT) mice received PF‐06266047 (PF; 0.5mg/kg) or vehicle via daily oral gavage beginning at 1 month of age, whereupon animals were sacrificed 6 months later for cAMP quantitation by immunofluorescence staining in the thalamus, visual cortex, and hippocampus (n = 5/group). Significant differences between groups are denoted by asterisks (***p < 0.001; ****p < 0.0001).
To the best of our knowledge, chronic PDE4 inhibitor administration has not been examined in any juvenile species. When considering the possible utility of PDE4 inhibitors in JNCL patients, safety/toxicity assessments are needed. To this end, weekly weight measurements and blood chemistry profiles (every 2–3 months) were conducted on WT and Cln3Δex7/8 mice receiving the various PDE4 inhibitors over the 6‐month treatment period, with histopathology on multiple organs at the time of sacrifice. Both WT and Cln3Δex7/8 mice that received PDE4 inhibitors weighed less than their vehicle‐treated counterparts; however, the rate of weight gain was similar between the groups (see Fig 2D–F). Blood chemistry analysis did not reveal any significant abnormalities in liver, kidney, or pancreatic function throughout the 6‐month treatment period (data not shown), indicating that the compounds do not induce overt toxicity when chronically administered to juvenile animals. In addition, histopathological assessments of numerous organs did not reveal any evidence of toxicity as assessed by a board‐certified pathologist (S. M. Cohen; data not shown).
PDE4 Inhibitors Attenuate Disease‐Associated Pathology in Cln3Δex7/8 Mice
Prior work from our laboratory and others has identified early microglial and astrocyte activation in Cln3Δex7/8 mice that precedes and predicts regions of eventual neuronal loss.6, 7 Additionally, Cln3Δex7/8 astrocytes show reduced expression of the glutamate transporter GLAST and glutamine synthetase, indicating altered glutamate recycling capacity, which supports the neurotransmitter imbalance associated with JNCL.6, 9, 10, 11 We have also reported that Cln3Δex7/8 microglia are primed toward a proinflammatory phenotype when exposed to danger signals present in the Cln3Δex7/8 brain.8 Given these findings and prior reports documenting the utility of PDE4 inhibitors to dampen microglial proinflammatory activity and enhance glutamate transporter expression in astrocytes,15, 38 we examined whether PDE4 inhibitors affected glial activation and glutamate transporter expression in Cln3Δex7/8 mice. We limited our analysis to roflumilast and PF‐06266047 for these studies, because both agents demonstrated beneficial effects on motor function in Cln3Δex7/8 animals and are viable therapeutics to consider for use in JNCL patients (see Fig 2B, C).
Cln3Δex7/8 and WT mice were sacrificed after the 6‐month PDE4 inhibitor treatment period, whereupon effects on microglial and astrocyte activation were examined by immunofluorescence staining for CD68 and GFAP, respectively. CD68 reactivity was significantly elevated in the S1BF and TH of Cln3Δex7/8 animals, which was attenuated following roflumilast treatment (Fig 4A, B). PF‐06266047 also led to significant reductions in microglial activation (see Fig 4C, D). GFAP immunoreactivity was significantly enhanced in the S1BF and TH of Cln3Δex7/8 mice as compared to WT controls, in agreement with previous observations from us and others.6, 7 Roflumilast significantly reduced GFAP immunoreactivity in both brain regions of Cln3Δex7/8 mice (Fig 5A, B). Similar findings were obtained with PF‐06266047, which significantly attenuated GFAP levels in the S1BF and TH of Cln3Δex7/8 animals (see Fig 5C, D). Importantly, neither roflumilast nor PF‐06266047 altered CD68 or GFAP expression in WT mice, supporting specific disease‐modifying effects.
Figure 4.
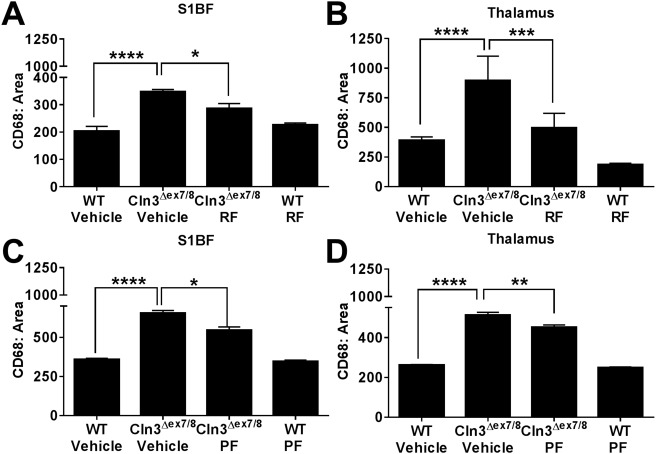
Phosphodiesterase 4 inhibitors attenuate microglial activation in Cln3Δex7/8 mice. Cln3Δex7/8 and wild‐type (WT) mice (n = 4–8/group) received vehicle, (A, B) 5mg/kg roflumilast (RF), or (C, D) 0.5mg/kg PF‐06266047 (PF) via daily oral gavage beginning at 1 month of age, whereupon animals were sacrificed 6 months later for quantitative assessments of microglial activation in the somatosensory barrel field cortex (S1BF) and thalamus by immunofluorescence staining for CD68. Significant differences between groups are denoted by asterisks (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Figure 5.
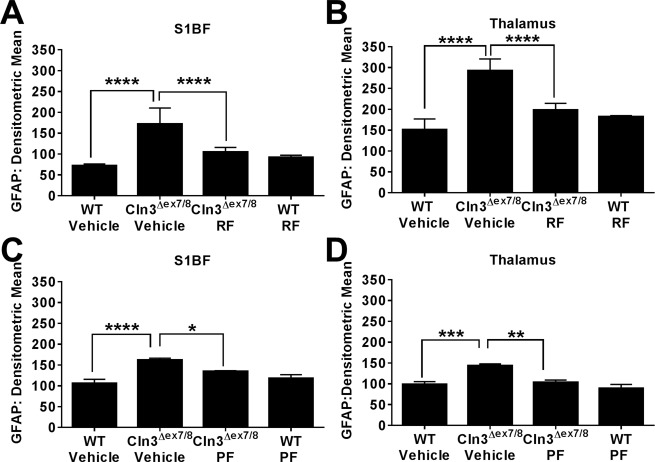
Phosphodiesterase 4 inhibitors reduce astrocyte activation in Cln3Δex7/8 mice. Cln3Δex7/8 and wild‐type (WT) mice (n = 4–8/group) received vehicle, (A, B) 5mg/kg roflumilast (RF), or (C, D) 0.5mg/kg PF‐06266047 (PF) via daily oral gavage beginning at 1 month of age, whereupon animals were sacrificed 6 months later for quantitative assessments of astrocyte activation in the somatosensory barrel field cortex (S1BF) and thalamus by immunofluorescence staining for glial fibrillary acidic protein (GFAP). Significant differences between groups are denoted by asterisks (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
JNCL is characterized by progressive lysosomal dysfunction, which is most pronounced in neurons.39, 40 LAMP1 is widely used as a readout of lysosomal pathology and is often considered a more sensitive indicator compared to inclusion burdens.41, 42 Therefore, we evaluated the effects of PDE4 inhibitors on lysosomal pathology by LAMP1 immunostaining. As has been shown for other lysosomal storage disorders,41, 43, 44 LAMP1 expression was increased in vehicle‐treated CLN3Δex7/8 mice compared to WT animals in the S1BF and TH (Fig 6). Similar to what was observed for glial activation, both roflumilast and PF‐06266047 significantly reduced LAMP1 levels (see Fig 6A, B and C, D, respectively), reflecting reduced lysosomal pathology. Interestingly, neither roflumilast nor PF‐06266047 had any effect on autofluorescent inclusions in either the brain or the retina (data not shown). This could be explained by the drug treatment not ensuing until significant inclusion burdens had already developed at 1 month of age, and although the observed reductions in LAMP1 suggest improvements in lysosomal biology, this was not sufficient to clear established inclusion material. However, the contribution of inclusions to neuronal death remains uncertain, because many inclusion‐positive neurons are not lost in the disease.1, 45, 46
Figure 6.
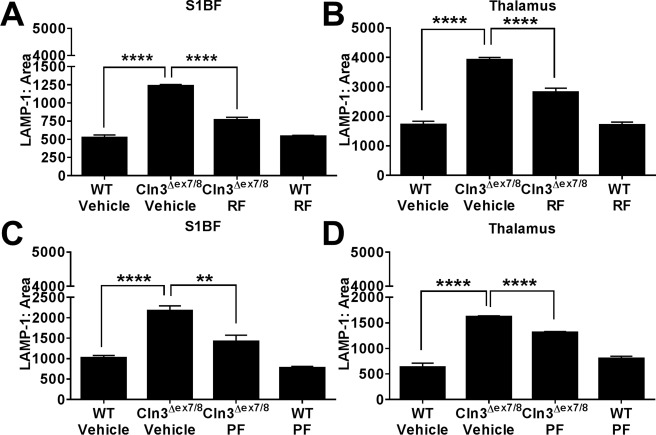
Phosphodiesterase 4 inhibitors attenuate lysosomal pathology in the Cln3Δex7/8 brain. Cln3Δex7/8 and wild‐type (WT) mice (n = 4–8/group) received vehicle, (A, B) 5mg/kg roflumilast (RF), or (C, D) 0.5mg/kg PF‐06266047 (PF) via daily oral gavage beginning at 1 month of age, whereupon animals were sacrificed 6 months later for quantitative assessments of lysosomal‐associated membrane protein 1 (LAMP‐1) expression in the somatosensory barrel field cortex (S1BF) and thalamus. Significant differences between groups are denoted by asterisks (**p < 0.01; ****p < 0.0001).
As previously mentioned, we recently reported significant reductions in the astrocytic glutamate transporter GLAST in 3‐month‐old Cln3Δex7/8 mice,6 which could represent one mechanism accounting for excessive glutamate levels in JNCL that have been implicated in neuron excitotoxicity.10, 33, 34, 35 This finding was extended in the current study, where significant reductions in GLAST expression were observed in the S1BF and TH of Cln3Δex7/8 mice at 7 months of age (Fig 7). Importantly, treatment of Cln3Δex7/8 animals with either roflumilast or PF‐06266047 beginning at 1 month of age restored GLAST expression in both brain regions to levels similar to those seen in WT mice (see Fig 7A, B and C, D, respectively). Recent studies attributed elevated glutamate levels to rotarod deficits in Cln3Δex7/8 mice11, 34 and our results support a potential link between increasing the expression of molecules critical for glutamate homeostasis and improvements in motor activity in Cln3Δex7/8 animals. Importantly, neither roflumilast nor PF‐06266047 altered LAMP1 or GLAST expression in WT mice, supporting specific disease‐modifying effects. Collectively, these results demonstrate the ability of two distinct PDE4 inhibitors to reverse some pathological features of Cln3Δex7/8 mice, which when coupled with improvements in motor behavior suggests that these compounds may offer therapeutic benefit in JNCL patients.
Figure 7.
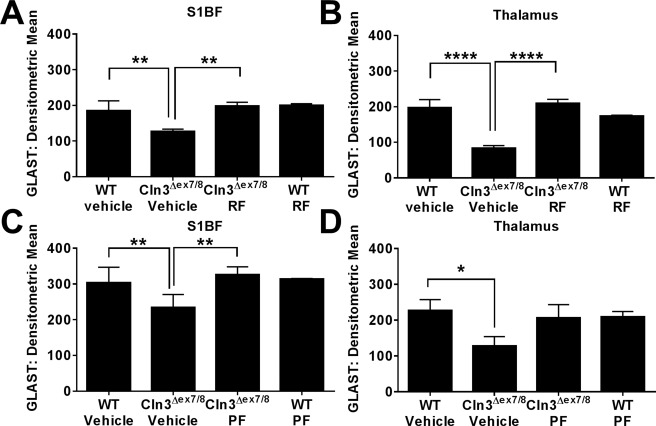
Phosphodiesterase 4 inhibitors restore the loss of astrocytic glutamate/aspartate transporter (GLAST) expression in Cln3Δex7/8 mice. Cln3Δex7/8 and wild‐type (WT) mice (n = 4–8/group) received vehicle, (A, B) 5mg/kg roflumilast (RF), or (C, D) 0.5mg/kg PF‐06266047 (PF) via daily oral gavage beginning at 1 month of age, whereupon animals were sacrificed 6 months later for quantitative assessments of GLAST expression in the somatosensory barrel field cortex (S1BF) and thalamus. Significant differences between groups are denoted by asterisks (*p < 0.05; **p < 0.01; ****p < 0.0001).
PDE4 Inhibitors Are Capable of Improving Motor Activity in Cln3Δex7/8 Mice with Advanced Disease
Although roflumilast and PF‐06266047 were capable of improving motor activity and disease‐associated features in Cln3Δex7/8 mice when treatment was initiated at 1 month of age, we next examined whether delayed administration would be efficacious. This is an important issue in terms of translational potential, because a JNCL diagnosis can often be delayed and patients would have already experienced increasing disease burden. To this end, we began PDE4 inhibitor treatment in 3‐month‐old animals, which display more advanced disease in terms of increased lysosomal pathology and motor deficits. Delayed treatment with both roflumilast (5mg/kg/day) and PF‐06266047 (0.5mg/kg/day) significantly improved rotarod performance in Cln3Δex7/8 mice, with activity restored to near levels observed in WT animals (Fig 8A, B, respectively). Both WT and Cln3Δex7/8 mice receiving delayed PDE4 inhibitor treatment weighed less than their vehicle counterparts; however, weight gain occurred in all groups (see Fig 8C, D). One interesting difference was observed with early versus delayed roflumilast treatment of Cln3Δex7/8 mice, with 3 months of treatment needed to demonstrate an effect when drug administration began at 1 month of age (see Fig 3), whereas in animals with more advanced disease (ie, 3 months), roflumilast improved motor function in Cln3Δex7/8 animals after only 1 month of treatment (see Fig 8). The reason for this difference is not known; however, it could result from altered blood–brain barrier permeability with increasing disease burden at the time of treatment or PK differences in juvenile versus adult animals. Collectively, these findings demonstrate the utility of PDE4 inhibitors to treat advanced disease, suggesting that they may offer therapeutic benefit to JNCL patients at later stages of the disease process.
Figure 8.
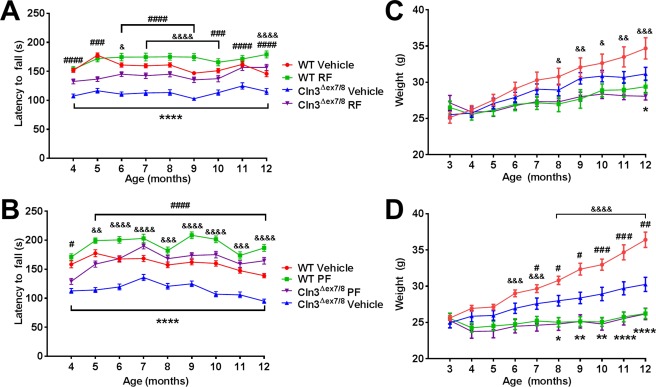
Delayed treatment with phosphodiesterase 4 (PDE4) inhibitors improves motor function in Cln3Δex7/8 mice. Cln3Δex7/8 and wild‐type (WT) mice (n = 8/group) received vehicle, (A, C) 5mg/kg roflumilast (RF), or (B, D) 0.5mg/kg PF‐06266047 (PF) via daily oral gavage beginning at 3 months of age, after which motor activity was assessed by accelerating rotarod testing at monthly intervals. Across all comparisons, significant differences between vehicle‐treated WT and Cln3Δex7/8 animals are denoted by asterisks, Cln3Δex7/8 vehicle‐treated versus Cln3Δex7/8 PDE4 inhibitor‐treated mice are indicated by hash signs, and WT vehicle‐treated versus WT PDE4 inhibitor‐treated mice are indicated by ampersand signs. (C, D) Weights of Cln3Δex7/8 and WT mice receiving delayed PDE4 inhibitor treatment. Significant differences between Cln3Δex7/8 vehicle‐treated versus Cln3Δex7/8 PDE4 inhibitor‐treated mice are indicated by hash signs, and WT vehicle‐treated versus WT PDE4 inhibitor‐treated mice are indicated by ampersand signs. For all comparisons, *, #, & reflect p < 0.05; **, ##, && denote p < 0.01; ###, &&& reflect p < 0.001; and ****, ####, &&&& represent p < 0.0001.
Discussion
Motor defects are a hallmark of JNCL,47 some of which are recapitulated in the Cln3Δex7/8 mouse model,32 as demonstrated by the rotarod deficits described in the current study. Prior reports using CLN3 knockout mice revealed heightened glutamate levels and reduced GABA in the cortex and cerebellum, and N‐methyl‐D‐aspartate or a‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor blockade could improve motor function in these animals, lending support to the excitotoxic theory of neuronal loss in JNCL.10, 11, 33 Our recent work also supports this possibility, in that the expression of key molecules involved in glutamate clearance/detoxification from the synaptic cleft (GLAST and glutamine synthetase, respectively) were significantly reduced in the Cln3Δex7/8 brain.6 In addition, we recently demonstrated transient hemichannel opening in Cln3Δex7/8 mice, which could exacerbate extracellular glutamate accumulation and promote excitotoxic neuronal death.6 A second potential insult that may contribute to neuronal loss in JNCL is aberrant inflammatory activity that our laboratory has described in Cln3Δex7/8 microglia.8 These potential non–cell‐autonomous effects of dysfunctional astrocytes and microglia in JNCL are expected to synergize with intrinsic neuronal defects to culminate in cell loss.48 These changes led to our interest in investigating the utility of PDE4 inhibitors, because prior studies have shown their efficacy in attenuating microglial proinflammatory activity, augmenting astrocyte glutamate transporter expression, and delivering prosurvival signals to neurons.15, 16, 38 Our findings support a key role for cAMP in dictating motor dysfunction, glial activation, and lysosomal pathology in the Cln3Δex7/8 brain, and support the feasibility of PDE4 inhibitors as potential therapeutics for the treatment of JNCL patients.
To our knowledge, this is the first report demonstrating significant cAMP reductions in the Cln3Δex7/8 brain in vivo. cAMP is a critical second messenger involved in regulating synaptic activity, glutamate homeostasis, and bioenergetic functions.13, 16 cAMP influences transcriptional events by binding to cAMP‐responsive element binding protein, a ubiquitous transcription factor that controls the expression of a variety of genes, including synaptic proteins and glutamate receptors that are involved in glutamate release.16, 49 Therefore, it is possible that generalized reductions in cAMP could affect downstream events responsible for optimal synaptic activity, behavior, and neuronal survival in JNCL. This was supported by our findings, where 3 distinct PDE4 inhibitors (rolipram, roflumilast, and PF‐06266047) were capable of restoring motor activity of Cln3Δex7/8 mice to performance levels near or equal to WT animals. Because prior work has implicated excessive glutamate stimulation as a major factor responsible for rotarod deficits in CLN3 knockout mice,11, 33 this suggests that PDE4 inhibitors may be modulating glutamatergic pathways. This was supported by our findings where chronic treatment of Cln3Δex7/8 animals with PDE4 inhibitors returned GLAST expression to levels observed in WT mice, which would be expected to help clear extracellular glutamate from the synapse and improve motor coordination. Importantly, both roflumilast and PF‐06266047 were capable of improving motor activity in Cln3Δex7/8 mice when treatment was delayed until 3 months of age, revealing the ability of both compounds to benefit more advanced disease. With regard to roflumilast dosing, the group of WT and Cln3Δex7/8 vehicle‐treated mice used to assess 5mg/kg/day roflumilast (see Fig 2B) did not differ from each other dramatically in accelerating rotarod performance, which was unusual. Nevertheless, treatment of Cln3Δex7/8 animals with 5mg/kg/day roflumilast did significantly improve motor performance compared to vehicle‐treated Cln3Δex7/8 mice, and when considered in conjunction with its ability to attenuate microglial and astrocyte activation and lysosomal pathology, this indicates that roflumilast exerts beneficial effects on CNS pathology at this dose. Increasing roflumilast to 10mg/kg/day improved accelerating rotarod performance even further in Cln3Δex7/8 mice, which still reduced glial activation (data not shown); however, 10mg/kg in mice is nearing the maximal tolerated dose in humans of 500 µg/day24, 25 and resulted in more weight loss in both WT and Cln3Δex7/8 animals compared to 5mg/kg/day (data not shown). This suggests that the dose required to improve motor activity may fall slightly above that needed to attenuate glial activation and lysosomal pathology and would require regular clinical assessments of JNCL patients to identify the optimal dose to improve motor coordination while minimizing known side effects of the compound (ie, nausea and weight loss). To date, roflumilast is only FDA approved for adult use; however, one report examined the PK properties of a single roflumilast dose in children and adolescents with stable mild to moderate asthma and found that the drug was well tolerated with PK parameters similar to those of adults.50 Chronic roflumilast dosing in children has not yet been performed, but the authors speculated that this would be well‐tolerated, which is supported by our findings, where chronic dosing of juvenile animals was not associated with severe adverse events.
One interesting observation was that both roflumilast and PF‐06266047 also increased motor performance in WT animals. The reason for this is not clear, but may be explained by compensation in cAMP signaling. For example, the neuronal network maintains an optimal range for cAMP signaling in the brain.51 Therefore, chronic cAMP elevations in WT mice, which would occur with continued PDE4 inhibitor treatment, likely increased cAMP‐mediated signaling above the optimal range. This could result in an altered set point and manifest as improved motor performance, which was observed in the current study. Although WT mice demonstrated improved accelerating rotarod activity with chronic PDE4 inhibitor administration, this did not translate to changes in glial activation, glutamate transporter expression, or lysosome biology, which were identical between WT animals treated with vehicle or PDE4 inhibitors. In contrast, all 3 PDE4 inhibitors reduced disease‐associated pathology in Cln3Δex7/8 mice across all of the neurological readouts examined in this study, which suggests that they are disease‐modifying rather than symptomatic treatment. From a therapeutic perspective, PDE4 inhibitors would not be used in healthy individuals, but the inclusion of WT animals in the current study design was for completeness and to evaluate the potential toxicity of PDE4 inhibitors in juvenile animals, which has not yet been examined over a prolonged (6‐month) interval. This was an important issue to address, because treatment of JNCL patients would require chronic dosing to impact disease progression. To this end, we did not observe any significant differences in blood chemistry values or tissue histopathology after a 6‐month dosing period, although animals receiving PDE4 inhibitors weighed less than vehicle controls, but still showed weight gain throughout the study.
Another feature of JNCL is neuronal loss, which is more limited in Cln3 mouse models compared to human disease.7, 52 A prior publication reported thalamic neuron loss in Cln3Δex7/8 mice on a mixed 129Sv/Ev/CD1 background at 12 months7; however, this reduction was relatively modest and we have not observed any differences in neuron counts of Nissl‐stained sections in a 1 in 6 series using unbiased stereology between vehicle‐treated WT and Cln3Δex7/8 animals on a C57BL/6 background, which is known to harbor milder disease phenotypes. That we could not demonstrate evidence of neuronal loss in Cln3Δex7/8 mice at this time point precluded our ability to assess the effects of PDE4 inhibitors on promoting neuronal survival. Therefore, our analysis has instead focused on other quantifiable pathological outcomes, including glial activation and lysosomal pathology. Nevertheless, at the conclusion of each dosing trial, brains were weighed and normalized to total body weight to account for weight loss resulting from chronic PDE4 inhibitor treatment. There were no significant changes in brain:body weight ratios either between vehicle‐treated WT and Cln3Δex7/8 mice or with any of the 3 PDE4 inhibitors tested, demonstrating that chronic PDE4 inhibitor dosing did not adversely affect neuron survival as measured by total brain weight.
In addition to augmenting glutamate transporter expression, it is also possible that the beneficial effects of PDE4 inhibitors in Cln3Δex7/8 mice may be mediated, in part, by dampening neuroinflammatory responses that are suggested to occur in JNCL,6, 7, 8, 53 because PDE4 inhibitors have been shown to reduce inflammation in other neurological conditions.17, 18, 19, 24, 25, 26 The ability of PDE4 inhibitors to significantly reduce astrocyte and microglial activation in Cln3Δex7/8 mice supports this tenet. Neuroinflammation could be a risk factor in JNCL, because Cln3Δex7/8 microglia were shown to produce numerous proinflammatory cytokines (including tumor necrosis factor‐α [TNF‐α] and interleukin‐1β [IL‐1β]) when exposed to danger‐associated molecular patterns (DAMPs) encountered during the disease process.8 Another possibility is that Cln3Δex7/8 microglia could release DAMPs that induce reactive astrocytosis and impair glutamate homeostasis and other astrocyte functions.48 Glial crosstalk could also occur via TNF‐α and IL‐1β production by Cln3Δex7/8 microglia that are known to induce astrocyte hemichannel opening,54 which we have also shown occurs in Cln3Δex7/8 mice and can lead to extracellular glutamate release.6, 55
Although lysosomal inclusions form throughout the body in JNCL due to the ubiquitous expression of CLN3, the primary site of disease manifestation is within the CNS. However, cardiac dysfunction is also observed in children as the disease progresses,56 and it remains unknown what systemic effects PDE4 inhibitors may exert in this context. Of note, besides the CNS, PDE4 isoforms are expressed in the cardiovascular system, the reproductive organs, the gastrointestinal tract, and the immune system, where PDE4 inhibition has been associated with anti‐inflammatory effects.57 Because inflammation has been linked to pathology in JNCL, it is likely that PDE4 inhibitors may dampen inflammation, which would be desirable in the CNS to limit deleterious inflammation in response to dying neurons and autoantibody formation that is characteristic of the disease.58, 59 We did not examine potential systemic effects of PDE4 inhibitors in the current report, because there are no peripheral biomarkers that have been identified in JNCL and the few systemic changes reported to be altered in Cln3Δex7/8 mice are modest in nature.60 However, we did not observe any adverse effects on blood chemistry values throughout the 6‐month duration of PDE4 inhibitor treatment in either Cln3Δex7/8 or WT animals. Importantly, clinical studies have not revealed any severe adverse effects of roflumilast on the cardiac, reproductive, or gastrointestinal systems.24, 25
In summary, this study is the first to demonstrate the utility of PDE4 inhibitors to limit disease‐associated attributes of JNCL. In particular, PDE4 inhibitors were found to improve motor function as well as mitigate astrocyte and microglial activation, reduce lysosomal pathology, and restore glutamate transporter expression in Cln3Δex7/8 mice. Although the precise mechanisms whereby PDE4 inhibitors exert these beneficial effects are not known, targeting PDE4 represents a novel therapeutic strategy for JNCL patients.
Author Contributions
T.K. was responsible for study conception and design and wrote the manuscript. A.A., M.E.B., M.B., K.V.R.R., R.F., J.O., J.D.C., and A.V.D. were responsible for acquisition and analysis of data. A.A. and M.E.B. contributed equally to this work.
Potential Conflicts of Interest
T.K. is the principle investigator for a clinical study with Pfizer that is unrelated to the present work. Pfizer provided a drug that was tested in this study. T.K. holds a patent for using PDE4 inhibitors in juvenile Batten disease (serial No.: 14/066,506).
Acknowledgment
This work was supported by the NIH National Institute of Neurological Disorders and Stroke (1R21NS084392‐01A1, T.K.), the Batten Disease Support and Research Administration (T.K.), and a University of Nebraska Medical Center Graduate Fellowship (M.E.B.).
We thank Dr S. M. Cohen for histopathological analysis, Dr C. Heim and K. Schuberth for unmasking the treatment groups, Dr N. Karpuk for designing the custom software used to quantitate immunofluorescence staining from confocal images, and Dr T. Chappie for critical review of the revised manuscript.
References
- 1. International Batten Disease Consortium . Isolation of a novel gene underlying Batten disease, CLN3. Cell 1995;82:949–957. [DOI] [PubMed] [Google Scholar]
- 2. Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology 2012;79:183–191. [DOI] [PubMed] [Google Scholar]
- 3. Schultz ML, Tecedor L, Chang M, Davidson BL. Clarifying lysosomal storage diseases. Trends Neurosci 2011;34:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Autti T, Hamalainen J, Aberg L, et al. Thalami and corona radiata in juvenile NCL (CLN3): a voxel‐based morphometric study. Eur J Neurol 2007;14:447–450. [DOI] [PubMed] [Google Scholar]
- 5. Autti TH, Hamalainen J, Mannerkoski M, et al. JNCL patients show marked brain volume alterations on longitudinal MRI in adolescence. J Neurol 2008;255:1226–1230. [DOI] [PubMed] [Google Scholar]
- 6. Burkovetskaya M, Karpuk N, Xiong J, et al. Evidence for aberrant astrocyte hemichannel activity in juvenile neuronal ceroid lipofuscinosis (JNCL). PLoS One 2014;9:e95023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pontikis CC, Cotman SL, MacDonald ME, Cooper JD. Thalamocortical neuron loss and localized astrocytosis in the Cln3Deltaex7/8 knock‐in mouse model of Batten disease. Neurobiol Dis 2005;20:823–836. [DOI] [PubMed] [Google Scholar]
- 8. Xiong J, Kielian T. Microglia in juvenile neuronal ceroid lipofuscinosis are primed toward a pro‐inflammatory phenotype. J Neurochem 2013;127:245–258. [DOI] [PubMed] [Google Scholar]
- 9. Brockmann K, Pouwels PJ, Christen HJ, et al. Localized proton magnetic resonance spectroscopy of cerebral metabolic disturbances in children with neuronal ceroid lipofuscinosis. Neuropediatrics 1996;27:242–248. [DOI] [PubMed] [Google Scholar]
- 10. Pears MR, Cooper JD, Mitchison HM, et al. High resolution 1H NMR‐based metabolomics indicates a neurotransmitter cycling deficit in cerebral tissue from a mouse model of Batten disease. J Biol Chem 2005;280:42508–42514. [DOI] [PubMed] [Google Scholar]
- 11. Kovacs AD, Pearce DA. Attenuation of AMPA receptor activity improves motor skills in a mouse model of juvenile Batten disease. Exp Neurol 2008;209:288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davis RL, Cherry J, Dauwalder B, et al. The cyclic AMP system and Drosophila learning. Mol Cell Biochem 1995;149–150:271–278. [DOI] [PubMed] [Google Scholar]
- 13. Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB‐1, CREB‐2, and CPEB. Mol Brain 2012;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tawfik VL, Lacroix‐Fralish ML, Bercury KK, et al. Induction of astrocyte differentiation by propentofylline increases glutamate transporter expression in vitro: heterogeneity of the quiescent phenotype. Glia 2006;54:193–203. [DOI] [PubMed] [Google Scholar]
- 15. Mizuno T, Kurotani T, Komatsu Y, et al. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology 2004;46:404–411. [DOI] [PubMed] [Google Scholar]
- 16. Nicol X, Gaspar P. Routes to cAMP: shaping neuronal connectivity with distinct adenylate cyclases. Eur J Neurosci 2014;39:1742–1751. [DOI] [PubMed] [Google Scholar]
- 17. Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev 2011;91:651–690. [DOI] [PubMed] [Google Scholar]
- 18. Atkins CM, Oliva AA Jr, Alonso OF, et al. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp Neurol 2007;208:145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nikulina E, Tidwell JL, Dai HN, et al. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U S A 2004;101:8786–8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sommer N, Martin R, McFarland HF, et al. Therapeutic potential of phosphodiesterase type 4 inhibition in chronic autoimmune demyelinating disease. J Neuroimmunol 1997;79:54–61. [DOI] [PubMed] [Google Scholar]
- 21. Fossale E, Wolf P, Espinola JA, et al. Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BMC Neurosci 2004;5:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cotman SL, Vrbanac V, Lebel LA, et al. Cln3(Deltaex7/8) knock‐in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum Mol Genet 2002;11:2709–2721. [DOI] [PubMed] [Google Scholar]
- 23. Krause W, Kuhne G. Pharmacokinetics of rolipram in the rhesus and cynomolgus monkeys, the rat and the rabbit. Studies on species differences. Xenobiotica 1988;18:561–571. [DOI] [PubMed] [Google Scholar]
- 24. Hatzelmann A, Morcillo EJ, Lungarella G, et al. The preclinical pharmacology of roflumilast—a selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm Pharmacol Ther 2010;23:235–256. [DOI] [PubMed] [Google Scholar]
- 25. Rabe KF. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br J Pharmacol 2011;163:53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hedde J, Semproni A, Graf R, et al. The phosphodiesterase‐4 inhibitor, ABI‐4, attenuates the increases in brain cytokines and translocator protein binding caused by lipopolysaccharide. Schizophr Bull 2015;41(suppl 1):S1–S341. 26305006 [Google Scholar]
- 27. Landis SC, Amara SG, Asadullah K, et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature 2012;490:187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geifman N, Rubin E. The mouse age phenome knowledgebase and disease‐specific inter‐species age mapping. PLoS One 2013;8:e81114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pearse DD, Pereira FC, Marcillo AE, et al. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med 2004;10:610–616. [DOI] [PubMed] [Google Scholar]
- 30. Gong B, Vitolo OV, Trinchese F, et al. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest 2004;114:1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Myeku N, Clelland CL, Emrani S, et al. Tau‐driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP‐PKA signaling. Nat Med 2016;22:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bosch ME, Aldrich A, Fallet R, et al. Self‐complementary AAV9 gene delivery partially corrects pathology associated with juvenile neuronal ceroid lipofuscinosis (CLN3). J Neurosci 2016;36:9669–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kovacs AD, Saje A, Wong A, et al. Age‐dependent therapeutic effect of memantine in a mouse model of juvenile Batten disease. Neuropharmacology 2012;63:769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kovacs AD, Saje A, Wong A, et al. Temporary inhibition of AMPA receptors induces a prolonged improvement of motor performance in a mouse model of juvenile Batten disease. Neuropharmacology 2011;60:405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salek RM, Pears MR, Cooper JD, et al. A metabolomic comparison of mouse models of the neuronal ceroid lipofuscinoses. J Biomol NMR 2011;49:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Griswold DE, Webb EF, Breton J, et al. Effect of selective phosphodiesterase type IV inhibitor, rolipram, on fluid and cellular phases of inflammatory response. Inflammation 1993;17:333–344. [DOI] [PubMed] [Google Scholar]
- 37. Choi CH, Schoenfeld BP, Bell AJ, et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res 2011;1380:106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tawfik VL, Regan MR, Haenggeli C, et al. Propentofylline‐induced astrocyte modulation leads to alterations in glial glutamate promoter activation following spinal nerve transection. Neuroscience 2008;152:1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haltia M. The neuronal ceroid‐lipofuscinoses. J Neuropathol Exp Neurol 2003;62:1–13. [DOI] [PubMed] [Google Scholar]
- 40. Williams RE, Aberg L, Autti T, et al. Diagnosis of the neuronal ceroid lipofuscinoses: an update. Biochim Biophys Acta 2006;1762:865–872. [DOI] [PubMed] [Google Scholar]
- 41. Meikle PJ, Brooks DA, Ravenscroft EM, et al. Diagnosis of lysosomal storage disorders: evaluation of lysosome‐associated membrane protein LAMP‐1 as a diagnostic marker. Clin Chem 1997;43(8 pt 1):1325–1335. [PubMed] [Google Scholar]
- 42. Usenovic M, Tresse E, Mazzulli JR, et al. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha‐synuclein accumulation, and neurotoxicity. J Neurosci 2012;32:4240–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ryazantsev S, Yu WH, Zhao HZ, et al. Lysosomal accumulation of SCMAS (subunit c of mitochondrial ATP synthase) in neurons of the mouse model of mucopolysaccharidosis III B. Mol Genet Metab 2007;90:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saftig P, Hartmann D, Lullmann‐Rauch R, et al. Mice deficient in lysosomal acid phosphatase develop lysosomal storage in the kidney and central nervous system. J Biol Chem 1997;272:18628–18635. [DOI] [PubMed] [Google Scholar]
- 45. Bronson RT, Lake BD, Cook S, et al. Motor neuron degeneration of mice is a model of neuronal ceroid lipofuscinosis (Batten's disease). Ann Neurol 1993;33:381–385. [DOI] [PubMed] [Google Scholar]
- 46. Finn R, Kovacs AD, Pearce DA. Altered sensitivity of cerebellar granule cells to glutamate receptor overactivation in the Cln3(Deltaex7/8)‐knock‐in mouse model of juvenile neuronal ceroid lipofuscinosis. Neurochem Int 2011;58:648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aberg L, Liewendahl K, Nikkinen P, et al. Decreased striatal dopamine transporter density in JNCL patients with parkinsonian symptoms. Neurology 2000;54:1069–1074. [DOI] [PubMed] [Google Scholar]
- 48. Rama Rao KV, Kielian T. Neuron‐astrocyte interactions in neurodegenerative diseases: role of neuroinflammation. Clin Exp Neuroimmunol 2015;6:245–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discov 2006;5:660–670. [DOI] [PubMed] [Google Scholar]
- 50. Neville KA, Szefler SJ, Abdel‐Rahman SM, et al. Single‐dose pharmacokinetics of roflumilast in children and adolescents. J Clin Pharmacol 2008;48:978–985. [DOI] [PubMed] [Google Scholar]
- 51. Sato T, Tanaka K, Ohnishi Y, et al. Inhibitory effects of group II mGluR‐related drugs on memory performance in mice. Physiol Behav 2004;80:747–758. [DOI] [PubMed] [Google Scholar]
- 52. Pontikis CC, Cella CV, Parihar N, et al. Late onset neurodegeneration in the Cln3‐/‐ mouse model of juvenile neuronal ceroid lipofuscinosis is preceded by low level glial activation. Brain Res 2004;1023:231–242. [DOI] [PubMed] [Google Scholar]
- 53. Bosch ME, Kielian T. Neuroinflammatory paradigms in lysosomal storage diseases. Front Neurosci 2015;9:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Retamal MA, Froger N, Palacios‐Prado N, et al. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci 2007;27:13781–13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bosch M, Kielian T. Hemichannels in neurodegenerative diseases: is there a link to pathology? Front Cell Neurosci 2014;8:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ostergaard JR, Rasmussen TB, Molgaard H. Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology 2011;76:1245–1251. [DOI] [PubMed] [Google Scholar]
- 57. Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res 2007;100:309–327. [DOI] [PubMed] [Google Scholar]
- 58. Castaneda JA, Pearce DA. Identification of alpha‐fetoprotein as an autoantigen in juvenile Batten disease. Neurobiol Dis 2008;29:92–102. [DOI] [PubMed] [Google Scholar]
- 59. Chattopadhyay S, Ito M, Cooper JD, et al. An autoantibody inhibitory to glutamic acid decarboxylase in the neurodegenerative disorder Batten disease. Hum Mol Genet 2002;11:1421–1431. [DOI] [PubMed] [Google Scholar]
- 60. Staropoli JF, Haliw L, Biswas S, et al. Large‐scale phenotyping of an accurate genetic mouse model of JNCL identifies novel early pathology outside the central nervous system. PLoS One 2012;7:e38310. [DOI] [PMC free article] [PubMed] [Google Scholar]