

Abstract
In the last decade, indoleamine 2,3‐dioxygenase 1 (IDO1) has attracted a great deal of attention being recognized as key regulator of immunosuppressive pathways in the tumor immuno‐editing process. Several classes of inhibitors have been developed as potential anticancer agents, but only few of them have advanced in clinical trials. Hence, the quest of novel potent and selective inhibitors of the enzyme is still active and mostly pursued by structure‐based drug design strategies based on early and more recent crystal structures of IDO1. Combining docking studies and molecular dynamic simulations, in this work we have comparatively investigated the structural features of each crystal structure of IDO1. The results pinpoint different features in specific crystal structures of the enzyme that may benefit the medicinal chemistry arena aiding the design of novel potent and selective inhibitors of IDO1.
Keywords: Tryptophan, IDO, Inhibitor, Antiviral, Antifungal, Antibacterial, Cancer, Drug Design
1. Introduction
Indoleamine 2,3‐dioxygenase (IDO; EC: 1.13.11.52) is a heme containing enzyme that catalyzes the oxidative cleavage of L‐Tryptophan (L‐Trp) in the first and rate‐limiting step of the kynurenine pathway to produce N‐formyl‐kynurenine.1 Two isoforms of this enzyme have been characterized, namely IDO1 and IDO2, sharing ∼43 % sequence identity and being endowed with distinct biochemical features.2 Although another structurally unrelated enzyme, tryptophan 2,3‐dioxygenase (TDO), has also been reported to be able to catalyze the same reaction, it is IDO1 that has attracted a great deal of attention since its discovery as key regulator of immunosuppressive pathways in maternal tolerance toward the allogenic fetus and in tumor immuno‐editing process.3 This interest has fostered the development of several classes of IDO1 inhibitors as anticancer agents with the aim of enhancing the efficacy of current chemotherapeutic drugs in combination therapies.4
Of these, few IDO1 inhibitors have nowadays reached the clinical stage of development including NLG919 (1, Figure 1), INCB024360 (2) and D‐1‐Methyl‐Trp (3)5 albeit this latter is no more considered a bona fide IDO1 inhibitor.6
Figure 1.
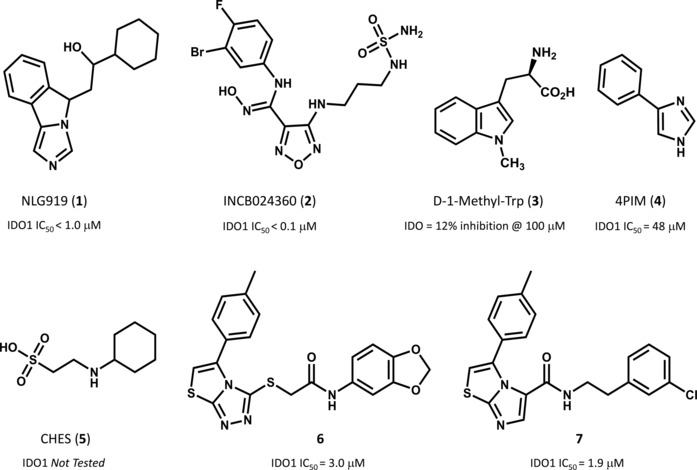
Chemical structures of selected IDO1 inhibitors and relative inhibition potencies as reported in references [1] and [4].
In 2006, the results of crystallographic studies on human IDO1 disclosed for the first time the structural features of the enzyme and provided clues on the binding mode of inhibitors (pdb code: 2D0T).7 Specifically, IDO1 folds into two distinct domains: the large domain and the small domain. The former is composed of fifteen α‐helices and contains the catalytic site, wherein the heme group is bound to residue His346 by the fifth coordination site of the iron atom. A long flexible loop (360–380, not solved in the crystal structures) is located at the entrance of the catalytic site defining a channel that runs parallel to the heme plane. Coarse graining simulation studies suggested the involvement of this loop in controlling the shuttling of substrate and products into the catalytic pocket of IDO1.8 The small domain of the enzyme is composed of nine α‐helices and two β‐sheets, and contains two immunoreceptor tyrosine‐based inhibitory motifs (ITIMs) located at residues 103–120 and 241–258, respectively. ITIMs are substrates of kinase‐mediated phosphorylation, promoting the interaction with the suppressor of cytokine signaling‐3 (SOCS3) and tagging IDO1 to the ubiquitin‐proteasome mediated degradation.9 The small domain is connected to the large domain by a flexible loop (residues 260–265) that also contributes to define the shape of the catalytic site.
Of note, the crystal structure of IDO1 in complex with 4‐phenylimidazole (4PIM, 4) showed the inhibitor binding to the sixth coordination site of the iron‐heme, as well as the presence of two additional molecules from the crystallization buffer, namely 2‐(N‐cyclohexylamino)ethane sulfonic acid (CHES, 5), binding to the enzyme (Figure 2A). The presence of these molecules was suggested to define an accessory site wherein substrate, enhancer or uncompetitive inhibitors could bind to the protein.8
Figure 2.
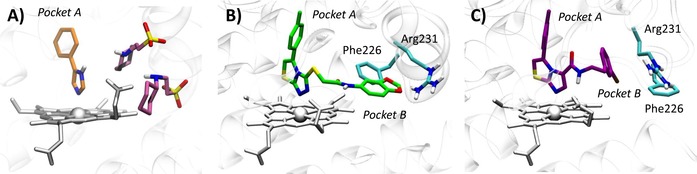
Features of the catalytic cleft of IDO1 in 2D0T (A), 4PK5 (B) and 4PK6 (C).
The above structure of IDO1 was instrumental to develop several structure‐based drug design (SBDD) approaches as well as to guide hit to lead (H2L) optimization efforts of inhibitors.10,11 Recently, additional crystallographic studies have been carried out on IDO1 bound to larger inhibitors than 4PIM (pdb codes: 4PK5, 4PK6).12 The results have shown additional features of the enzyme, including the existence of two additional pockets into the catalytic cleft that were in part anticipated as result of previous structure‐activity relationship studies of IDO1 inhibitors.11 Specifically, the two accessory binding pockets result from inhibitor‐induced conformational rearrangements of the enzyme which further expand the volume of the catalytic site (Figure 2B–C). The first pocket (pocket A) is localized above the sixth coordination site of the iron‐heme protruding into the small domain, whereas the second pocket (pocket B) is located at the entrance of the channel to the catalytic site including residues that were recognized fundamental for IDO1 activity by mutagenesis experiments such as Arg231, Phe226 and Phe227.7 Although this observation suggests that multiple conformations of the enzyme may affect to different extent the molecular recognition of inhibitors by shaping the volume of the catalytic site of IDO1, the conformational properties of IDO1 have only in part been investigated with coarse graining simulations or molecular dynamic (MD) simulations mostly used to study the catalytic mechanism of the enzyme.
Since the structure of IDO1 bound to 4PIM (4) was almost exclusively used for SBDD and H2L approaches, the aim of this work was to assess the extent of value that recent crystal structures of IDO1 may bring in the medicinal chemistry arena for designing novel potent inhibitors of the enzyme. Specifically, in the first part of the work, we used self‐ and cross‐docking studies to comparatively investigate the ability to predict the correct binding modes of co‐crystallized IDO1 inhibitors. In the second part, we used molecular dynamic (MD) simulations to investigate the stability of the binding poses resulting from docking studies in the three crystal structures, and then evaluate whether the structural features of the catalytic cleft observed in the more recent 4PK5 and 4PK6 entries could be somehow anticipated using the early structure of the enzyme in complex with 4PIM (4).
2. Materials and Methods
2.1. Docking Studies
Three IDO1 crystal structures (PDB codes: 2D0T, resolution=2.30 Å; 4PK5, resolution=2.79 Å; 4PK6, resolution=3.45 Å)7,12 were downloaded from the Protein Data Bank,13 and their chain A was prepared (addition of hydrogens, ionization states at pH 7.0, optimization and minimization of the structure) with the Protein Preparation Wizard, tool of Maestro 10.1 (Schrödinger Inc.). The unsolved loop 361–379 was reconstructed with Prime 3.9.14 The iron was set as FeIII because of experimental suggestions indicating that imidazole‐based molecules have greater affinity for the oxidized form of the enzyme.15 Ligands were prepared with LigPrep 3.3 generating all ionization states at pH 7±2.17 Docking studies and induced fit docking (IFD) studies were carried out using Glide 6.6 and standard precision (SP) mode, storing the best ten scored binding poses for each molecule as output. In both docking and IFD studies, grids were defined in the same way, generating one grid for each crystal with the centre located on the centre of mass of the co‐crystallized ligand. The inner grid box was sized 12×12×12 Å. Only for 2D0T, a second different grid was generated retaining the two crystallized molecules of CHES (5). All rotatable groups of residues inside the outer box were taken into account.16 In the IFD procedure, the side chains of binding site residues within 5 Å of the co‐crystallized ligand were selected for conformational searches. All other docking and IFD parameters were set on the relative default values. All stored binding poses from docking and IFD studies were inspected in order to find the most reliable ones, evaluating the root mean square deviation (RMSD) of heavy atoms from the relative experimental ligand binding pose and distances of interaction to the iron. Solvent accessible surface areas (SASA) of binding sites in 2D0T, 4PK5 and 4PK6 were calculated rolling a sphere of radius 1.4 Å (Connolly surface) around binding site residues defined as those residues falling within 5 Å of co‐crystallized ligand. Figure 3 was generated using VMD surf drawing method on binding site residues of 2D0T, 4PK5 and 4PK6.
Figure 3.
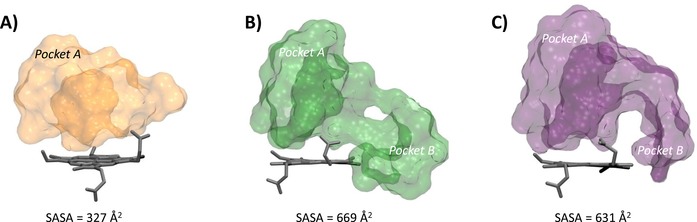
Molecular shapes and solvent accessible surfaces (SASA) of the catalytic cleft of IDO1 in 2D0T (A), 4PK5 (B) and 4PK6 (C).
2.2. Molecular Dynamic Simulations
MD simulations were run using ACEMD (Accelera Ltd)18 and CHARMM36 force field. The most reliable binding poses from docking studies were selected as starting structure. Atomic charges of the heme group were calculated using quantum mechanics with Jaguar 8.7, DFT theory and 6‐31G* basis set. During these calculations, FeIII‐heme bound to the basic nitrogen of methyl‐imidazole was used to mimic the heme‐His346 interaction.19 Ligands were parameterized with ParamChemTool.20 The protein‐ligand complex was solvated in a cubic box using TIP3P water molecules, extended 12 Å away from any protein atom. The system was neutralized adding a concentration of 0.15 M of chlorine and sodium ions and periodic boundary conditions were used. Cut‐off of non‐boded interactions was set to the value of 9 Å. Energy minimization of 1000 iterations was conducted before 1 ns positional constrained equilibration. Then, 1.5 ns equilibration without constraints was performed using slow increasing of temperature (10 degrees every 50 ps, 0 to 300 °K) to avoid jumps of the “hinge” loop (260–265). Finally, a 20 ns simulation was run, in NPT condition, sampling every 500 ps (200 total frames). The results were visualized with VMD software and analysed with tcl scripting.21 In particular, hydrophobic contacts were considered for hydrophobic side chain falling within 4 Å of aromatic and/or aliphatic carbons of the ligand, while hydrogen bonds were calculated using the H‐bonds plugin of VMD defining the following distance and angle cut‐off criteria: cut‐off distance value of 3 Å, cut‐off angle value of 30 degree.
3. Results and Discussion
3.1. Docking Studies
Self‐ and cross‐docking studies of 4PIM (4), thiazolotriazole derivative (6) and imidazothiazole derivative (7) with and without the induced fit docking procedure (IFD) were carried out into the crystal structures of IDO1 (pdb codes: 2D0T, 4PK5, 4PK6) using Glide as reported in the method section. Table 1 reports the best results in terms of scoring function (G‐score), root mean square deviation (RMSD‐xray) from the experimental binding pose, and distance of the coordinating nitrogen atom to the iron‐heme (dN‐Fe) among the top ten ranked solutions (see supplementary materials, Table S1–S10). As a general consideration, the inspection of the table reveals that self‐ and cross‐IFD studies do not provide any better solution than self‐ and cross‐docking in terms of closeness to the experimental binding pose.
Table 1.
Best docked poses in terms of G‐score, RMSD‐xray, and dN‐Fe along with averages and standard deviations of RMSD and dN‐Fe from MD simulations.
| Complex | Self/Cross‐Docking | G‐score (kcal/mol) | RMSD‐xray | dN‐Fe | Rank | RMSD | dN‐Fe |
|---|---|---|---|---|---|---|---|
| (Å) | (Å) | (Å) | (Å) | ||||
| (4)/2D0T*xray | x‐ray | – | 0.00 | 2.13 | – | 0.25±0.07 | 1.83±0.14 |
| (4)/2D0Tself | Self | −5.06 | 0.70 | 2.53 | 6th | 5.97±1.40 | 10.89±1.49 |
| (4)/2D0T*self | Self | −4.14 | 0.34 | 2.29 | 7th | 0.19±0.06 | 2.31±0.19 |
| (6)/2D0Tcross | Cross | −4.31 | 5.09 | 6.49 | 2nd | 1.48±0.40 | 7.18±0.42 |
| (7)/2D0Tcross‐IFD | Cross‐IFD | −8.72 | 5.73 | 4.08 | 1st | 2.27±0.67 | 11.81±1.43 |
| (6)/4PK5xray | x‐ray | – | 0.00 | 2.11 | – | 1.97±0.27 | 2.93±0.60 |
| (4)/4PK5cross | Cross | −5.22 | 1.13 | 2.80 | 6th | 6.97±2.14 | 9.33±1.90 |
| (6)/4PK5self | Self | −7.22 | 0.31 | 2.41 | 2nd | 1.09±0.55 | 3.58±1.08 |
| (7)/4PK5cross | Cross | −7.99 | 1.36 | 2.54 | 1st | 1.43±0.98 | 2.66±0.29 |
| (7)/4PK6xray | x‐ray | – | 0.00 | 2.21 | – | 0.57±0.17 | 2.46±0.41 |
| (4)/4PK6cross | Cross | −5.25 | 1.27 | 2.77 | 8th | 4.32±1.08 | 7.36±1.22 |
| (6)/4PK6cross | Cross | −8.86 | 2.51 | 2.82 | 1st | 2.13±0.27 | 3.81±0.47 |
| (7)/4PK6self | Self | −9.37 | 0.32 | 2.46 | 1st | 0.94±0.42 | 2.68±0.24 |
*2D0T including CHES (5) molecules.
Docking studies into 2D0T provide a reliable binding pose only for 4PIM (self‐docking approach; 4 RMSD‐xray=0.70 Å, dN‐Fe=2.53 Å), while a remote binding pose is obtained for thiazolotriazole derivative (cross‐docking approach; 6 RMSD‐xray=5.09 Å, dN‐Fe=6.49 Å) and solutions are found for the imidazothiazole derivative (7) only when using the IFD procedure (cross‐IFD approach), albeit with a poor RMSD‐xray value (Figure 4). This observation is very likely ascribed to the smaller size of the catalytic pocket observed in 2D0T with respect to 4PK5 and 4PK6 (Figure 3), which hampers the fit of the large molecular shape of the thiazolotriazole derivative (6, MW=425) and the imidazothiazole derivative (7, MW=396) by generating steric clashes with the pocket.
Figure 4.
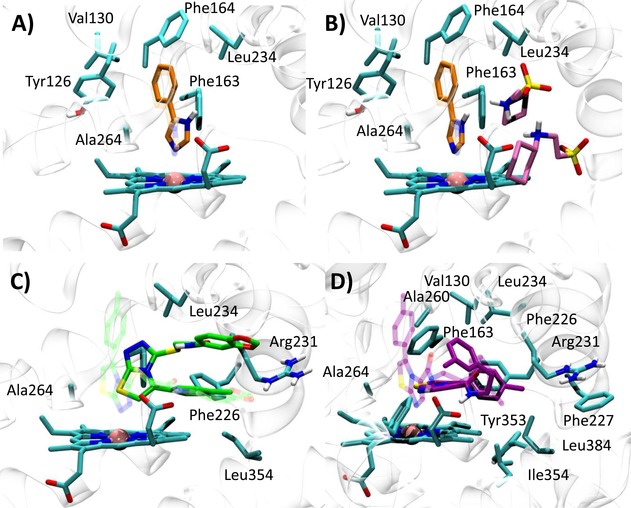
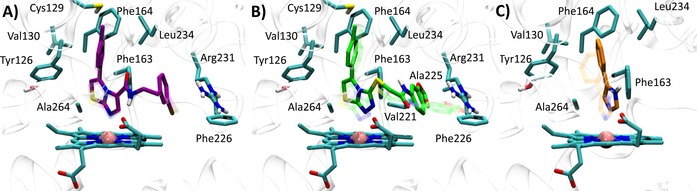
Best docked poses of 4PIM (4) into 2D0T (A, self‐docking), 2D0T including CHES (B, self‐docking), and best docked pose of the thiazolotriazole derivative (6) into 2D0T (C, cross‐docking) and imidazothiazole derivative (7) into 2D0T (D, cross‐IFD). Experimental binding poses are shown in transparency.
Next, we investigated whether the presence of the two CHES molecules observed in 2D0T could affect the binding pose of 4PIM (4) in docking studies. As a result, it is found that the docking of compound 4 into the catalytic cleft of IDO1 containing CHES molecules provides a more reliable binding pose of the inhibitor, with lower dN‐Fe (2.29 Å) and improved RMSD value (RMSD‐xray=0.34 Å). In agreement with the interactions observed in 2D0T, the distal nitrogen of the imidazole ring of 4PIM is here placed above the sixth coordination site of the iron‐heme, with the phenyl group making hydrophobic contacts with Val130, Leu234 and Ala264, and π‐π interactions with Tyr126, Phe163 and Phe164 (Figure 4). Interestingly, CHES molecules (5) seem to favour the prediction of the correct binding pose of 4PIM (4) to the enzyme, with one of them (NHE502) packing the small inhibitor (4, MW=144) into the catalytic cleft in hydrophobic contacts.
Docking of compounds 4, 6 and 7 into 4PK5 and 4PK6 yield solutions for all of the inhibitors, in line with the larger size of the catalytic pocket (Figure 3). In the case of 4PK5, results of compound 6 show a good reproduction of the experimental binding pose (self‐docking approach; 6 RMSD‐xray=0.31 Å, dN‐Fe=2.41 Å). In particular, the distal nitrogen atom of the thiazolotriazole moiety is placed above the sixth coordination site of the iron‐heme, with the p‐toluene group protruding into a hydrophobic cage of pocket A which is formed by the side chains of residues Tyr126, Cys129, Val130, Phe163, Phe164, Leu234 and Ala264. Moreover, an extended conformation of the benzodioxane side chain engages Phe226, Phe227, Ile354 and Leu384 with hydrophobic contacts and Arg231 with a hydrogen bond in pocket B (Figure 5A). Docking studies of 4PIM (4) and 7 yield binding poses that are slightly different from the experimental ones (cross‐docking approach; 4 RMSD‐xray=1.13 Å, dN‐Fe=2.80 Å; 7 RMSD‐xray=1.36 Å, dN‐Fe=2.54 Å). Specifically, 4PIM is still placed with its distal nitrogen of the imidazole ring above the sixth coordination site of the iron‐heme, while the phenyl moiety mostly interacts with Val130, Tyr126, Phe163, Phe164, Leu234 and Ala264 (Figure 5B). Compound 7 engages the sixth coordination site of the iron‐heme with its basic nitrogen of the imidazothiazole moiety and occupies the hydrophobic pocket A with the p‐toluene group. According to this docked pose, an extended conformation of the m‐chlorine benzyl side chain projects towards pocket B, making a π‐π interaction with Phe226 and hydrophobic contacts with Leu384 (Figure 5C).
Figure 5.
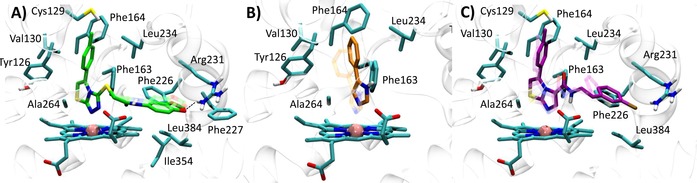
Best docked pose of the thiazolotriazole derivative (6) into 4PK5 (A, self‐docking), 4PIM (4) into 4PK5 (B, cross‐docking), and imidazothiazole derivative (7) into 4PK5 (C, cross‐docking). Experimental binding poses are shown in transparency.
Using 4PK6, compound 7 shows the binding pose closest to the experimental binding mode (self‐docking approach; 7 RMSD‐xray=0.32 Å, dN‐Fe=2.46 Å), whereas compounds 4 again displays a binding pose that is slightly different from the experimental one (cross‐docking approach; 4 RMSD‐xray value=1.27 Å, dN‐Fe=2.77 Å). Of note, the docked pose of compound 6 is markedly different from the binding mode observed in the relative crystal structure (cross‐docking approach; 6 RMSD‐xray=2.51 Å, dN‐Fe=2.82 Å). It is noteworthy that specific conformational rearrangements of binding site residues Phe226 and Arg231 in 4PK6 may force compound 6 to adopt a diverse binding pose in docking studies, thereby accounting for the higher RMSD‐xray value.
Hence, in agreement with crystallographic data, the distal nitrogen atom of the imidazothiazole moiety of 7 is correctly placed above the sixth coordination site of the iron‐heme, whereas the p‐toluene group is inserted into pocket A interacting with Tyr126, Cys129, Val130, Phe163, Phe164, Leu234 and Ala264. The m‐chlorine benzyl side chain is harboured into pocket B through a π‐cation interaction with Arg231 (Figure 6A). Establishing a π‐cation interaction, the specific conformation of Arg231 in 4PK6 constrains the benzodioxane side chain of 6 into a folded conformation that is not in agreement with the experimental data of 4PK5 (Figure 6B). 4PIM (4) is again placed with its imidazole ring on the sixth coordination site of the iron‐heme, albeit the phenyl moiety adopts a slightly different orientation into pocket A with respect to the one observed in 2D0T (Figure 6C).
Figure 6.
Best docked pose of the imidazothiazole derivative (7) into 4PK6 (A, self‐docking), thiazolotriazole derivative (6) into 4PK6 (B, cross‐docking), and 4PIM (4) into 4PK6 (C, cross‐docking). Experimental binding poses are shown in transparency.
3.2. Molecular Dynamics
MD simulations were carried out to assess the stability of selected binding poses as obtained from self‐ and cross‐docking studies. Results were also compared to the stability of the experimental binding poses of compounds 4, 6 and 7 as observed in 2D0T, 4PK5 and 4PK6, respectively.
Accordingly, a total of 13 MD trajectories were generated and analyzed calculating the RMSD of heavy atoms of the ligands from the relative starting pose and the distance of the coordinating nitrogen atom to the iron‐heme (dN‐Fe) over the 20 ns MD simulations (Table 1). Stable binding modes were deemed poses showing average RMSD<2.0 Å and dN‐Fe<4.0 Å along the 20 ns trajectories. Interaction patterns resulting from stable calculated and experimental binding modes of ligands 4, 6 and 7 are shown in Table 2.
Table 2.
Occupancies of key interactions in the catalytic cleft of IDO1 from MD simulations.
| Complex | Pocket A | Pocket B | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Y126 | V130 | F163 | F164 | L234 | A264 | R231 | F226 | I354 | L384 | |
| (4)/2D0T*xray | 81 % | 86 % | 98 % | 95 % | 62 % | 85 % | – | – | – | – |
| (4)/2D0T*self | 68 % | 95 % | 98 % | 86 % | 78 % | 97 % | – | – | – | – |
| (6)/4PK5xray | 91 % | – | 83 % | 31 % | 71 % | ‐ | 22 % [b] | – | – | 78 % |
| (6)/4PK5self | 59 % | 63 % | 89 % | 63 % | 48 % | 66 % | 29 % [a] | 56 % | 75 % | 41 % |
| (7)/4PK6xray | 99 % | 52 % | 97 % | 38 % | 88 % | 95 % | 62 % [b] | 74 % | 32 % | – |
| (7)/4PK5cross | 98 % | 26 % | 94 % | 40 % | 88 % | 83 % | 57 % [b] | 70 % | 55 % | – |
| (7)/4PK6self | 99 % | 43 % | 98 % | 40 % | 82 % | 94 % | 22 % [b] | 52 % | 94 % | – |
*2D0T including CHES (5) molecules; [a] Hydrogen bond interaction; [b] π‐cation interaction.
As a general consideration, MD simulations of crystal structures reveal strong stability of the experimental binding modes for compounds 4, 6 and 7, suggesting that the adopted force field (CHARMM36), atomic charge parameterization and length of simulation (20ns) are adequate to study ligands interacting with the heme‐containing binding site of IDO1.
More in detail, results of 2D0T simulations show that thiazolotriazole (6) and imidazothiazole (7) derivatives are not able to get closer to the experimental binding pose, with average distances between the coordinating nitrogen and the iron‐heme maintaining high values along the entire trajectories (Table 1, 6 dN‐Fe=7.18±0.42 Å; 7 dN‐Fe=11.81±1.43 Å). The binding pose of 4PIM (4) obtained using docking studies into 2D0T is fully unstable, with the ligand that loses its key interaction with the iron‐heme at the beginning of the simulation (Table 1, dN‐Fe=10.89±1.49 Å, RMSD=5.97±1.40 Å). Conversely, it is more stable the experimental binding pose of 4 in presence of CHES molecules (Table 1, dN‐Fe=1.83±0.14 Å, RMSD=0.25±0.07 Å), as well as the binding pose obtained when 4PIM (4) was docked into the binding cleft of 2D0T containing CHES molecules (Table 1, dN‐Fe=2.31±0.19 Å, RMSD=0.19±0.06 Å). This observation supports the packing effect of CHES (NHE502) on this small inhibitor. Specifically, 4PIM engages Tyr126, Phe163 and Phe164 with hydrophobic and π‐stacking interactions (Table 2, occupancies 81 %–68 %, 98 % and 95 %–86 %), and Val130, Leu234 and Ala264 with favorable van der Waals contacts along the 20 ns MD simulations (Table 2, occupancies 86 %–95 %, 62 %–78 %, and 85 %–97 %).
Results of 4PK5 simulations pinpoint again the lack of stability of the binding pose obtained for 4PIM (4, dN‐Fe=9.33±1.90 Å, RMSD=6.97±2.14 Å), whereas more stable binding poses are observed for the thiazolotriazole derivative (6, dN‐Fe=3.58±1.08 Å, RMSD=1.09±0.55 Å) and the imidazothiazole derivative (7, dN‐Fe=2.66±0.29 Å, RMSD=1.43±0.98 Å). In the case of the thiazolotriazole derivative (6), the slightly high average distance between the coordinating nitrogen and the iron‐heme (dN‐Fe=3.58±1.08 Å) is due to large drifts of the ligand during the first 9 ns of the simulation (Figure 7, dN‐Fe 1ns–9ns=4.57±0.84 Å). Then, its binding pose becomes stable with an average value of dN‐Fe 9ns–20ns=2.78±0.28 Å. Noteworthy, these early drifts are also observed, though to a less extent, in the simulation of the experimental pose of 6 bound to 4PK5 (dN‐Fe 1ns–9ns=3.11±0.82 Å; dN‐Fe 9ns–20ns=2.77±0.21 Å).
Figure 7.
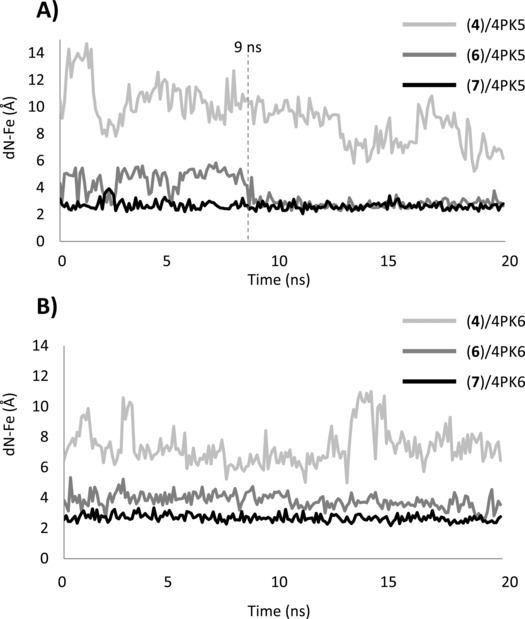
Variation of the distance between the iron‐heme (dN‐Fe) and the coordinating nitrogen atom of 4PIM (4), thiazolotriazole derivative (6) and imidazothiazole derivative (7) along the MD trajectories of 4PK5 (A) and 4PK6 (B).
The binding mode of 6 resulting from 4PK5 trajectories underlines the engagement of Tyr126, Phe163, Phe164 and Phe226 through hydrophobic and π‐stacking interactions (Table 2, occupancies 91 %–59 %, 83 %–89 %, 31 %–63 %, 56 %) by the p‐toluene group, and the presence of favourable hydrophobic contacts with Val130, Leu234, Ala264, Phe226, Ile354 and Leu384 (Table 2, occupancies 63 %, 71 %–48 %, 66 %, 56 %, 75 %, 78 %–41 %). Of note, the stability of the hydrogen bond interaction between the oxygen of the benzodioxane moiety and Arg231 is clear in the MD trajectory of the ligand‐bound complex resulting from self‐docking studies (Table 2, occupancy 29 %), while it seems unstable in the trajectory of the crystal structure wherein it is replaced by a π‐cation interaction (Table 2, occupancy 22 %). Likewise, the binding mode of the imidazothiazole derivative (7) from the 4PK5 trajectory shows a stable anchoring of the coordinating nitrogen to the iron‐heme along the entire simulation (dN‐Fe=2.66±0.29 Å). The occurrence of hydrophobic and π‐stacking interactions is observed between the p‐toluene group and Tyr126, Phe163 and Phe164 (Table 2, occupancies 98 %, 94 %, 40 %). The m‐chlorine benzyl side chain interacts with Arg231 and Phe226 in pocket B, making π‐cation and π‐stacking interactions (Table 2, occupancies 57 % and 70 %), respectively. Favourable van der Waals contacts are also found between compound 7 and the side chains of Val130, Leu234, Ala264, and Ile354 (Table 2, occupancies 26 %, 88 %, 83 %, 55 %).
Results of 4PK6 simulations reveal unstable binding poses for 4PIM (4, dN‐Fe=7.36±1.22 Å, RMSD=4.32±1.08 Å) and the thiazolotriazole derivative (6, dN‐Fe=3.81±0.47 Å, RMSD=2.13±0.27 Å), with both ligands showing high distances between the coordinating nitrogen atom and the iron‐heme (dN‐Fe) along the 20 ns trajectories. Conversely, stable binding modes are observed for the imidazothiazole derivative in the ligand‐bound crystal structure (dN‐Fe=2.46±0.41 Å, RMSD=0.57±0.17 Å) and in the binding mode resulting from self‐docking studies (dN‐Fe=2.68±0.24 Å, RMSD=0.94±0.42 Å).
Showing a similar pattern of interactions to 4PK5 simulation, in 4PK6 trajectories the imidazothiazole derivative (7) engages Tyr126, Phe163 and Phe164 with hydrophobic and π‐stacking interactions in pocket A (Table 2, occupancies 99 %–99 %, 97 %–98 %, 38 %–40 %), Arg231 and Phe226 with π‐cation and π‐stacking interactions in pocket B (Table 2, occupancies 62 %–22 % and 74 %–52 %), and Val130, Leu234, Ala264 and Ile354 with favourable van der Waals contacts (Table 2, occupancies 52 %–43 %, 88 %–82 %, 95 %–94 %, 32 %–94 %).
3.3. Discussion
Being acquainted with bioactive conformation and binding mode of a ligand to its biological target is the cornerstone of any SBDD strategy and H2L approach. This can be achieved using experimental methods such as crystallographic experiments and/or computational approaches including docking studies and molecular dynamic (MD) simulations. Compliant to this statement, the early disclosure of the crystal structure of IDO1 bound to 4PIM (4, pdb code: 2D0T) enabled the design and development of potent and selective inhibitors of the enzyme. Successful examples include NLG919 (1) and INCB024360 (2), that are currently advancing in clinical trials as single therapy for solid tumor.5
Recently, two additional crystal structures of IDO1 have been disclosed in complex with a thiazolotriazole derivative (6, pdb code: 4PK5) and imidazothiazole derivative (7, pdb code: 4PK6), respectively. These structures show different structural features with respect to the early ligand bound crystal structure of IDO1, including the absence of CHES ligands as observed in 2D0T, different size of the catalytic pocket, and different conformations of Phe226 and Arg231 which also result in diverse shapes of the ligand binding cleft (Figure 2). In order to understand the added value and/or redundancy that these features may bring in the design and development of second generation IDO1 inhibitors, we challenged the ability of each structure of IDO1 to reproduce the experimental binding modes of co‐crystallized inhibitors using self‐docking and cross‐docking studies with and without the IFD procedure, and combined with MD simulations.
Results of self‐docking studies show that all structures are able to recover the correct binding poses of co‐crystallized inhibitors among the top ten ranked solutions which are also stable in MD simulations with the only exception of 4PIM (4) docked into 2D0T (Table 1, row 2). Conversely, poor solutions are generally obtained when the IFD procedure is adopted (see supplementary materials, Table S1–S10) evidencing the fallacy of the IFD approach in finding appropriate conformational rearrangements of binding site residues that are able to resume correct binding poses of IDO1 co‐crystallized ligands.
Moreover, although the good stability of ligand‐bound crystal complexes in MD trajectories (Table 1, rows 1, 6, 10) suggests that force field, atomic charge parameterization and length of simulations can be deemed satisfactory, a case mounts for the structural features of 2D0T. Indeed, this crystal structure shows 4PIM (4) bound to IDO1 with two CHES molecules (5) into the catalytic site (Figure 2A). Keeping CHES molecules (5) into the binding site of IDO1 during self‐docking studies (Table 1, row 3) allows improving the results. Specifically, a binding pose of 4PIM (4) is found that shows a very low RMSD‐xray value (<0.5 Å) and is very stable along the entire MD simulation (RMSD=0.19±0.06 Å). Hence, the presence of CHES molecules (5) into the binding site of 2D0T may contribute to the binding mode of 4PIM (4) as observed into the relative crystal structure of the enzyme. Noteworthy, very recent crystallographic studies of 4PIM (4) bound to mutant forms of IDO1 have shown again the presence of CHES molecules into the binding cleft (Figure 8, pdb codes: 4U72, 4U74), further confirming the relevance of this structural feature for the binding mode of 4 to the enzyme.
Figure 8.
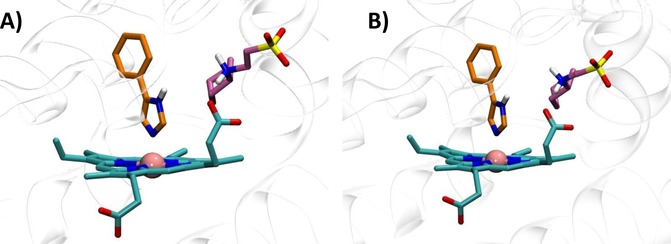
Features of the catalytic cleft of IDO1 in 4U72 (A) and 4U74 (B).
As far as cross‐docking studies are concerned, a trend towards satisfactory results is observed when using structures with a large size of the binding cleft such as 4PK5 (SASA=669 Å2) and 4PK6 (SASA=631 Å2). Indeed, these structures provide better results in cross‐docking studies than 2D0T, which is endowed with the smallest binding site volume (SASA=327 Å2). Accordingly, the use of 2D0T in cross‐docking studies yields poor solutions for the thiazolotriazole derivative (6, RMSD‐xray=5.09 Å) and the imidazothiazole derivative (7, RMSD‐xray=5.73 Å), being remotely located from the experimental binding modes. This structure of IDO1 could thus not anticipate the structural features observed in 4PK5 and 4PK6.
Structures 4PK5 and 4PK6 show also diverse shapes of the ligand binding cleft as a result of diverse conformations of Phe226 and Arg231 (Figure 2B–C). These conformations remain rather stable along the MD trajectories, as evidenced by the low root mean square fluctuations (RMSF) of the relative side chains (Table 3). Although in 4PK6 the conformations of Arg231 and Phe226 do not remarkably influence cross‐docking results, the stability of the obtained binding poses of 4PIM (4) and the thiazolotriazole derivative (6) is poor in MD simulations, as evidenced by high average RMSD values. In the case of 4PIM (4) this may also be ascribed to the lack of CHES molecules in the catalytic cleft, since a poor stability of the binding pose of this ligand is also observed in 4PK5 and in self‐docking studies using 2D0T without CHES molecules. In the case of cross‐docking of thiazolotriazole derivative (6) in 4PK6, the conformations of Phe226 and Arg231 provide steric bumps that hamper the correct positioning of the ligand into the catalytic cleft of IDO1. Conversely, the extended conformation of Arg231 in 4PK5 seems to provide the best results in terms of cross‐docking studies of the imidazothiazole derivative (7). In particular, the bioactive conformation of 7 bound to IDO1 as observed in the MD simulation of 4PK5 is very similar to that observed in the MD simulation of 4PK6, though a slightly different pattern of interactions is also found (Table 2, Figure 9). This latter is due to the conformational plasticity of Phe226 that directly engages the m‐chlorine benzyl side chain of 7 with a stable π‐π interaction in 4PK5 (Figure 9A), while it indirectly promotes a π‐cation interaction with Arg231 in the MD simulation of 4PK6 (Figure 9B). Overall, this result suggests that the use of 4PK5 is able to predict the experimental bioactive conformation and binding mode of 7 in cross‐docking studies combined with MD simulations, whereas this does not hold true for 4PK6. As a consequence, 4PK5 may represent an unbiased structure of IDO1 for SBDD strategy and H2L approach, enabling a wider exploration of the chemical space than 4PK6 and 2D0T structures.
Table 3.
Root mean fluctuations (RMSF) of Arg231 and Phe226 in the catalytic cleft of IDO1 during MD simulations.
| Complex | RMSF (Å) | |
|---|---|---|
| R231 | F226 | |
| (4)/2D0T*xray | 0.61 | 0.58 |
| (4)/2D0T*self | 0.61 | 0.62 |
| (6)/4PK5xray | 0.62 | 0.58 |
| (6)/4PK5self | 1.04 | 0.71 |
| (7)/4PK6xray | 0.75 | 0.60 |
| (7)/4PK5cross | 0.68 | 0.60 |
| (7)/4PK6self | 0.68 | 1.16 |
*2D0T including CHES (5) molecules.
Figure 9.
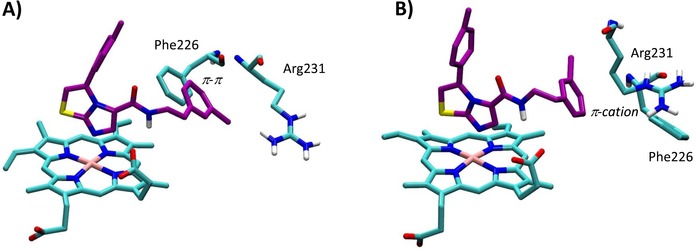
Bioactive conformation and binding mode of imidazothiazole derivative (7) into 4PK5 (A) and 4PK6 (B) along 20 ns MD simulations.
4. Conclusions
IDO1 plays an essential role as key regulator of immunosuppressive pathways in the tumor immuno‐editing process. Embracing this notion, a large number of IDO1 inhibitors have been disclosed as potential anticancer agents in the last decade. The design of many of these compounds has been inspired by the availability of the early crystal structure of the enzyme in complex with 4PIM (4). Nevertheless, only few of these inhibitors have nowadays entered in clinical trials. As a consequence, the quest of novel potent and selective inhibitors of the enzyme is still active, and further fostered by the recent availability of additional crystal structures of IDO1. In this work, we have analyzed the extent of value and/or redundancy that these new structures bring to the design and development of novel IDO1 inhibitors. Using self‐ and cross‐docking studies combined with MD simulations, we have shown that 4PK5 and 4PK6 unveil unprecedented structural features of the enzyme that could not be anticipated using 2D0T. Furthermore, the early 2D0T structure is somehow biased by the presence of CHES molecules that bind to the catalytic site and affect the binding mode of 4PIM (4) to the enzyme. Finally, this study pinpoints 4PK5 structure as of interest for the application of SBDD strategy and H2L development approach to design novel potent and selective modulators of IDO1.
Supporting Information
Tables S1–S9 report top ten ranked solutions of self‐docking and cross‐docking studies in 2D0T, 2D0T including CHES (5), 4PK5 and 4PK6.
Conflict of Interest
None declared.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the European Research Council (ERC‐2013‐AdG 338954‐DIDO) and the Italian Ministry of University and Research (PRIN 2012S47X27).
F. A. Greco, A. Bournique, A. Coletti, C. Custodi, D. Dolciami, A. Carotti, A. Macchiarulo, Mol. Inf. 2016, 35, 449.
References
- 1.
- 1a. Lancellotti S., Novarese L., Cristofaro R. De, Curr. Med. Chem. 2011, 18(15), 2205–2214; [DOI] [PubMed] [Google Scholar]
- 1b. Macchiarulo A., Camaioni E., Nuti R., Pellicciari R., Amino Acids 2009, 37(2), 219–229. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Ball H. J., Yuasa H. J., Austin C. J., Weiser S., Hunt N. H., Int. J. Biochem. Cell. Biol. 2009, 41(3), 467–471; [DOI] [PubMed] [Google Scholar]
- 2b. Pantouris G., Serys M., Yuasa H. J., Ball H. J., Mowat C. G., Amino Acids 2014, 46(9), 2155–2163. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Austin C. J., Rendina L. M., Drug Discov. Today 2015, 20(5), 609–617; [DOI] [PubMed] [Google Scholar]
- 3b. Prendergast G. C., Smith C., Thomas S., Mandik-Nayak L., Laury-Kleintop L., Metz R., Muller A. J., Cancer Immunol. Immunother. 2014, 63(7), 721–735; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Mellor A. L., Munn D. H., Nat. Rev. Immunol. 2004, 4(10), 762–774. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Röhrig U. F., Majjigapu S. R., Vogel P., Zoete V., Michielin O., J. Med. Chem. 2015, DOI: 10.1021/acs.jmedchem.5b00326; [DOI] [PubMed] [Google Scholar]
- 4b. Jiang T., Sun Y., Yin Z., Feng S., Sun L., Li Z., Future Med. Chem. 2015, 7(2), 185–201; [DOI] [PubMed] [Google Scholar]
- 4c. Dolušić E., Frédérick R., Expert Opin. Ther. Pat. 2013, 23(10), 1367–1381; [DOI] [PubMed] [Google Scholar]
- 4d. Delfourne E., Mini Rev. Med. Chem. 2012, 12(10), 988–996; [DOI] [PubMed] [Google Scholar]
- 4e. Di Pucchio T., Danese S., De Cristofaro R., Rutella S., Expert Opin. Ther. Pat. 2010, 20(2), 229–250. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Soliman H. H., Jackson E., Neuger T., Dees E. C., Harvey R. D., Han H., Ismail-Khan R., Minton S., Vahanian N. N., Link C., Sullivan D. M., Antonia S., Oncotarget 2014, 5(18), 8136–8146; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Vacchelli E., Aranda F., Eggermont A., Sautès-Fridman C., Tartour E., Kennedy E. P., Platten M., Zitvogel L., Kroemer G., Galluzzi L., Oncoimmunology 2014, 3(10):e957994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lob S., Konigsrainer A., Schafer R., Rammensee H. G., Opelz G., Terness P., Blood 2008, 111(4), 2152–2154. [DOI] [PubMed] [Google Scholar]
- 7. Sugimoto H., Oda S., Otsuki T., Hino T., Yoshida T., Shiro Y., Proc. Natl. Acad. Sci. USA 2006, 103(8), 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Macchiarulo A., Nuti R., Bellocchi D., Camaioni E., Pellicciari R., Biochim. Biophys. Acta 2007, 1774(8), 1058–1068. [DOI] [PubMed] [Google Scholar]
- 9. Orabona C., Pallotta M. T., Volpi C., Fallarino F., Vacca C., Bianchi R., Belladonna M. L., Fioretti M. C., Grohmann U., Puccetti P., Proc. Natl. Acad. Sci. USA 2008, 105(52), 20828–20833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Kumar S., Jaller D., Patel B., LaLonde J. M., DuHadaway J. B., Malachowski W. P., Prendergast G. C., Muller A. J., J. Med. Chem. 2008, 51(16), 4968–4977; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Yue E. W., Douty B., Wayland B., Bower M., Liu X., Leffet L., Wang Q., Bowman K. J., Hansbury M. J., Liu C., Wei M., Li Y., Wynn R., Burn T. C., Koblish H. K., Fridman J. S., Metcalf B., Scherle P. A., Combs A. P., J. Med. Chem. 2009, 52(23), 7364–7367; [DOI] [PubMed] [Google Scholar]
- 10c. John S., Thangapandian S., Sakkiah S., Lee K. W., Eur. J. Med. Chem. 2010, 45(9), 4004–4012; [DOI] [PubMed] [Google Scholar]
- 10d. Röhrig U. F., Awad L., Grosdidier A., Larrieu P., Stroobant V., Colau D., Cerundolo V., Simpson A. J., Vogel P., Van den Eynde B. J., Zoete V., Michielin O., J. Med. Chem. 2010, 53(3), 1172–1189; [DOI] [PubMed] [Google Scholar]
- 10e. Dolušić E., Larrieu P., Blanc S., Sapunaric F., Pouyez J., Moineaux L., Colette D., Stroobant V., Pilotte L., Colau D., Ferain T., Fraser G., Galleni M., Frère J. M., Masereel B., Van den Eynde B., Wouters J., Frédérick R., Eur. J. Med. Chem., 2011, 46(7), 3058–3065; [DOI] [PubMed] [Google Scholar]
- 10f. Smith J. R., Evans K. J., Wright A., Willows R. D., Jamie J. F., Griffith R., Bioorg. Med. Chem. 2012, 20(3), 1354–1363; [DOI] [PubMed] [Google Scholar]
- 10g. Röhrig U. F., Majjigapu S. R., Grosdidier A., Bron S., Stroobant V., Pilotte L., Colau D., Vogel P., Van den Eynde B. J., Zoete V., Michielin O., J. Med. Chem. 2012, 55(11), 5270–5290. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Gaspari P., Banerjee T., Malachowski W. P., Muller A. J., Prendergast G. C., DuHadaway J., Bennett S., Donovan A. M., J. Med. Chem. 2006, 49(2), 684–692; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Huang Q., Zheng M., Yang S., Kuang C., Yu C., Yang Q., Eur. J. Med. Chem. 2011, 46(11), 5680–5687. [DOI] [PubMed] [Google Scholar]
- 12. Tojo S., Kohno T., Tanaka T., Kamioka S., Ota Y., Ishii T., Kamimoto K., Asano S., Isobe Y., ACS Med. Chem. Lett. 2014, 5(10), 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., Nucleic Acids Res. 2000, 28, 235–242 (www.rcsb.org). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a.Maestro, version 10.1, Schrödinger, LLC, New York, NY, 2015;
- 14b.Schrödinger Suite 2015-1 Protein Preparation Wizard;
- 14c.Epik version 3.1, Schrödinger, LLC, New York, NY, 2015;
- 14d.Impact version 6.6, Schrödinger, LLC, New York, NY, 2015;
- 14e.Prime version 3.9, Schrödinger, LLC, New York, NY, 2015.
- 15. Rörhig U. F., Majjigapu S. R., Grosdidier A., Bron S., Stroobant V., Pilotte L., Colau D., Vogel P., Van den Eynde B. J., Zoete V., Michielin O., J. Med. Chem. 2012, 55(11), 5270–5290. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a.Glide, version 6.6, Schrödinger, LLC, New York, NY, 2015;
- 16b. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S., J. Med. Chem. 2004, 47, 1739–1749; [DOI] [PubMed] [Google Scholar]
- 16c. Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L., J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 17.LigPrep, version 3.3, Schrödinger, LLC, New York, NY, 2015.
- 18. Harvey M. J., Giupponi G., De Fabritis G., J. Chem. Theory Comput. 2009, 5(6), 1632–1639. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a.Jaguar, version 8.7, Schrödinger, LLC, New York, NY, 2015;
- 19b. Autenrieth F., Tajkhorshid E., Baudry J., Luthey-Schulten Z., J. Comp. Chem. 2004, 25(13), 1613–1622. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., A. D. Mackerell Jr , J. Comput. Chem. 2010, 31, 671–690; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Yu W., He X., Vanommeslaeghe K., A. D. Mackerell Jr , J. Comput. Chem. 2012, 33, 2451–2468; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20c. Vanommeslaeghe K., A. D. MacKerell Jr , J. Chem. Inf. Model. 2012, 52, 3144–3154; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20d. Vanommeslaeghe K., Raman E. P., A. D. MacKerell Jr , J. Chem. Inf. Model. 2012, 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Humphrey W., Dalke A., Schulten K., J. Mol. Graph. 1996, 14, 33–38 (http://www.ks.uiuc.edu/Research/vmd/). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary