

Abstract
Current guidelines for hypercholesterolemia treatment emphasize lifestyle modification and lipid‐modifying therapy to reduce the risk for cardiovascular disease. Statins are the primary class of agents used for the treatment of hypercholesterolemia. Although statins are effective for many patients, they fail to achieve optimal reduction in lipids for some patients, including those who have or are at high risk for cardiovascular disease. The PCSK9 gene was identified in the past decade as a potential therapeutic target for the management of patients with hypercholesterolemia. Pharmacologic interventions to decrease PCSK9 levels are in development, with the most promising approach using monoclonal antibodies that bind to PCSK9 in the plasma. Two monoclonal antibodies, alirocumab and evolocumab, have recently been approved for the treatment of hypercholesterolemia, and a third one, bococizumab, is in phase 3 clinical development. All 3 agents achieve significant reductions in levels of low‐density lipoprotein cholesterol, as well as reductions in non‐high‐density lipoprotein cholesterol, apolipoprotein B, and lipoprotein(a). Long‐term outcome trials are under way to determine the sustained efficacy, safety, and tolerability of PCSK9 inhibitors and whether this novel class of agents decreases the risk for major cardiovascular events in patients on lipid‐modifying therapy. Available data suggest that PCSK9 inhibitors provide a robust reduction in atherogenic cholesterol levels with a good safety profile, especially for patients who fail to obtain an optimal clinical response to statin therapy, those who are statin intolerant or have contraindications to statin therapy, and those with familial hypercholesterolemia.
Keywords: alirocumab, bococizumab, evolocumab, hyperlipidemia, low‐density lipoprotein cholesterol, proprotein convertase subtilisin/kexin type 9
Elevated low‐density lipoprotein (LDL) is well recognized as the root cause of atherosclerosis,1, 2 with recent evidence confirming that non‐high‐density lipoprotein cholesterol (non‐HDL‐C), apolipoprotein B (apo B), and lipoprotein(a) [Lp(a)] levels are also significant risk factors for the occurrence of atherosclerotic cardiovascular disease (ASCVD) events.3, 4, 5 The evidence for a positive relationship between hypercholesterolemia and risk for ASCVD has prompted numerous guidelines and evidence‐based recommendations for interventions to reduce lipid levels. These recommendations have evolved over time to emphasize reductions in plasma LDL cholesterol (LDL‐C) and non‐HDL‐C levels. Although statins are the mainstay of treatment to accomplish this goal, many patients still have atherogenic cholesterol levels greater than the recommended values.6
A variety of factors have been implicated in this treatment gap, including barriers to access to health care, nonadherence to statins and lifestyle regimens, and high rates of discontinuation of statin therapy.6, 7 Patients who are intolerant to statins, as well as those who fail to adhere to the optimal dose of statins, are at significantly increased risk for ASCVD.8 Statin‐related muscle symptoms affect as many as 5% to 29% of patients in clinical practice8, 9 and increase the risk for treatment discontinuation or suboptimal adherence to therapy.6, 10 Additional gaps in care are evident among individuals with familial hypercholesterolemia (FH). It is estimated that approximately half of individuals with FH are not prescribed cholesterol‐lowering medications, which places them at a 13‐fold increased risk for a CV event.11
The discovery of proprotein convertase subtilisin/kexin type 9 (PCSK9) in 2003 has opened the door to potentially address some of the gaps in the treatment of hypercholesterolemia.12 Gain‐of‐function (GOF) mutations in the PCSK9 gene decrease the number of LDL receptors (LDL‐Rs) at the hepatocyte surface, causing phenotypical FH.13, 14 In contrast, loss‐of‐function (LOF) mutations in the PCSK9 gene in African Americans were associated with 28% to 40% lower levels of plasma LDL‐C15, 16 and risk for coronary heart disease (CHD) reduced by 88% during a 15‐year follow‐up interval in the ARIC study.16 White individuals with an LOF mutation had a 15% lower LDL‐C level than unaffected individuals, and this was associated with a 47% reduction in risk for CHD.16
This review summarizes recommendations for lipid management by the National Lipid Association (NLA)2, 17 and guidelines issued by the American College of Cardiology (ACC) and the American Heart Association (AHA).18 The current and emerging evidence regarding the role of PCSK9 inhibition as a novel drug therapy for the management of hypercholesterolemia is discussed in detail, including a review of agents under evaluation in randomized controlled trials (RCTs) and the implications of this new class of medications for the optimal management of hypercholesterolemia, especially for patients at high risk for ASCVD, those with FH, and those who have contraindications to or who cannot tolerate statin therapy.
Recent Lipid‐Lowering Guidelines and Role of LDL‐C in Lipid Management
Current guidelines regarding the optimal strategies for the management of hypercholesterolemia and the prevention of ASCVD emphasize first‐line therapy with statins for specific groups of patients who are likely to receive the greatest clinical benefit from statin therapy for hypercholesterolemia.2, 18 There is also an emphasis on decreasing the risk for ASCVD rather than on achieving targeted reductions in LDL‐C levels.18
American College of Cardiology/American Heart Association Guidelines
The 2013 ACC/AHA guidelines emphasize the importance of heart‐healthy lifestyle modifications, simplify medical therapy by recommending moderate‐ or high‐intensity statin therapy, and apply pooled‐cohort risk‐assessment equations to determine an individual's risk for ASCVD.18 Notably, the guidelines abandon the use of LDL‐C targets based on evidence, suggesting that such targets may result in undertreatment with evidence‐based statin therapy. Lipid measurements are recommended to determine patients’ adherence to statins, with LDL‐C monitoring as an important component of ensuring an appropriate response to statin therapy.
These guidelines, which are based exclusively on evidence from RCTs, identify 4 patient groups for whom the potential benefit of reduced risk for ASCVD outweighs the potential for adverse effects associated with statin therapy (Figure 1). According to the level of ASCVD risk, moderate‐ or high‐intensity statin therapies are recommended to reduce LDL‐C by 30% to 50% or by ≥50%, respectively. Moreover, these guidelines recommend that nonstatin lipid‐modifying therapy (LMT) may be considered for high‐risk patients who do not have the anticipated response to statin therapy, who cannot tolerate the recommended intensity of statin therapy, or who are completely statin intolerant. Recently, an ACC expert consensus document provided guidance for the use of non‐statin therapies to lower LDL‐C in patients with high‐risk of ASCVD as well as defined thresholds LDL‐C, in terms of percentage reduction and absolute values.19 This guidance is more in line with the NLA recommendations.
Figure 1.
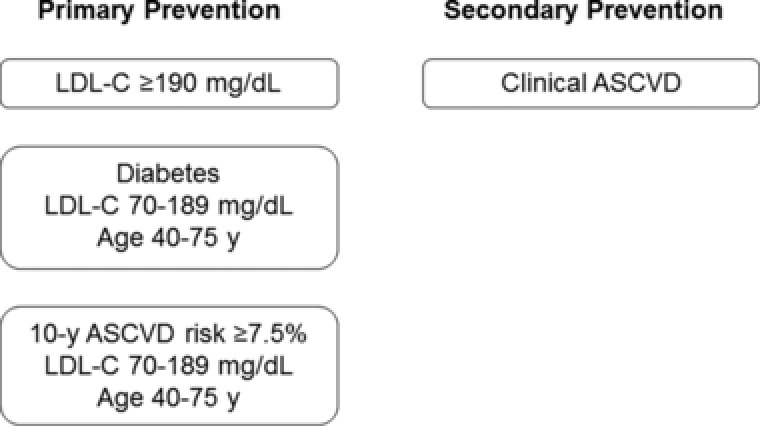
Statin benefit groups, as defined by the American College of Cardiology and the American Heart Association, for ASCVD risk reduction. ASCVD, atherosclerotic cardiovascular disease; LDL‐C, low‐density lipoprotein cholesterol.18
National Lipid Association Recommendations
The 2014 NLA recommendations reaffirm the importance of cholesterol goals and identify non‐HDL‐C and LDL‐C as coprimary targets for therapeutic interventions to achieve a sustained reduction in risk for ASCVD.2
The NLA expert panel concurred that statins are the primary pharmacologic agent to reduce the risk for ASCVD.2 Therapeutic goals are established for non‐HDL‐C and LDL‐C based on an individual's risk for ASCVD. Among individuals considered at low, moderate, or high risk for ASCVD, a treatment goal of <130 mg/dL is recommended for non‐HDL‐C and <100 mg/dL for LDL‐C, and goals for those at very high risk for ASCVD are <100 mg/dL and <70 mg/dL for non‐HDL‐C and LDL‐C, respectively. Alternative nonstatin drugs may be considered for patients who have contraindications or intolerance to statins.
Even though the ACC/AHA guidelines largely abandon the use of strict LDL‐C goals and recommend using LDL‐C lowering only as a measure of response to statin therapy, the optimal level of LDL‐C lowering for ASCVD risk reduction remains an important question. Boekholdt et al showed in a meta‐analysis that patients achieving LDL‐C <50 mg/dL had a significantly lower risk for major CV events than those who achieved LDL‐C between 75 and 100 mg/dL (adjusted hazard ratio, 0.81; 95%CI, 0.70 to 0.95).20 In a meta‐analysis of 27 randomized trials involving more than 174,000 patients comparing more intensive with less intensive statin treatment regimens and statin therapy with controls, the Cholesterol Treatment Trialists’ Collaboration demonstrated that a 39 mg/dL reduction in LDL‐C level leads to a 21% reduction in major vascular events at 1 year (rate ratio, 0.79; 95%CI, 0.77 to 0.81; P < .0001), with significant reductions in both women and men.21 Recently published results of IMPROVE‐IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial) also support the “lower‐is‐better” cholesterol premise. Adding ezetimibe to statin allowed patients to achieve a least squares mean (LSM) LDL‐C level of 55 mg/dL at 1 year (compared with 72 mg/dL for statin‐only patients) and was associated with a 6.4% relative risk reduction for major CV events at 7 years.22 Interestingly, this is the first trial that demonstrates a long‐term clinical benefit of adding a nonstatin treatment to statin therapy.
Gaps in the Treatment of Hypercholesterolemia
Although statins continue to be the gold standard of hypercholesterolemia therapy, many patients remain at high risk for CV disease despite treatment. In spite of modern lipid guideline recommendations and clinical trial evidence, statin therapy is often not titrated, with few patients receiving high‐intensity statins23 even after hospitalization for a CHD event.24 Additionally, according to a recent meta‐analysis of 8 randomized, controlled statin trials, more than 40% of patients on high‐dose statin therapy did not reach an LDL‐C target <70 mg/dL, and there was large interindividual variability in the reductions of LDL‐C, non‐HDL‐C, and apo B achieved with a fixed statin dose.20 Patients who fail to obtain an optimal clinical response to statin therapy include those with FH or with subtherapeutic response to statin treatment or those who are intolerant to or have contraindications to statin therapy.
Familial Hypercholesterolemia
Familial hypercholesterolemia is an autosomal codominant genetic disorder characterized by raised serum LDL‐C levels resulting from defects in hepatic uptake and degradation of LDL by the LDL‐R pathway.25 It is attributed primarily to mutations in the LDL‐R (60% to 90%), apo B (2% to 10%), and PCSK9 (˂5%) genes.25, 26, 27, 28 Individuals with FH are at increased risk for early‐onset CHD attributed to lifelong marked elevation in LDL‐C. Adults with heterozygous FH (HeFH) have total cholesterol (TC) levels between 310 and 580 mg/dL (8 to 15 mmol/L), with males likely to develop CHD before age 55 and women before age 60. Homozygous FH (HoFH) is a more severe and much rarer form of FH characterized by TC levels from 460 to 1160 mg/dL (12‐30 mmol/L), development of CHD, and aortic or supra‐aortic valve stenosis at very young ages, with death before age 20 or 30 if not treated.25, 29 Limited data are available to date on the prevalence of FH in an unselected sample of the general population; however, evidence suggests that there are 14 to 34 million individuals with FH worldwide.29 A recent analysis of HoFH, defined at the molecular level as homozygosity or compound heterozygosity for mutations in LDL‐R, apo B, or PCSK9 genes, determined the prevalence to be ∼1 in 300,000 inhabitants of the Netherlands.30
Despite the high risk for CHD, individuals with FH are underdiagnosed and undertreated, which can lead to poor outcomes.29 Notably, in a study of 69,000 Danish adults, the risk for CHD was strikingly high among individuals with definite or probable FH who did not receive medical therapy (adjusted odds ratio [OR], 13.2; 95%CI, 10.0 to 17.4) compared with non‐FH patients.11 The mainstay of treatment for FH has been diet, lifestyle modifications, and statins.25 Adults with FH should initiate medical therapy on diagnosis to the highest possible statin intensity tolerated. However, many patients with FH require concomitant treatment with nonstatin therapy, including ezetimibe, bile acid‐binding resin, LDL apheresis, or one of the new agents (PCSK9 inhibitors, lomitapide, or mipomersen).29, 31
Statin Intolerance and Subtherapeutic Response
Although statins are the most frequently prescribed pharmacologic agents for the treatment of hypercholesterolemia, issues relevant to intolerance, subtherapeutic response, and nonadherence are not uncommon barriers to long‐term statin therapy.
Statin Intolerance
Statin intolerance is generally defined as the inability to use statins or tolerate a full therapeutic dose because of significant adverse effects.10, 32, 33 Intolerance is most frequently attributed to myalgia or myopathy that can be associated with statins.33, 34
Although rates of statin‐induced myopathy in clinical trials are typically low (1.5% to 5.0%),34 they may be underestimated for a number of reasons, including exclusion of patients with a history of statin intolerance.34 A few large, community‐based studies indicate that occurrence of muscle symptoms may be between 10% and 20%.35 However, several studies have shown that more than half of the patients classified as statin intolerant can be rechallenged with an alternative statin.8, 36
Subtherapeutic Response to Statins
In terms of percentage LDL‐C reductions, interindividual response to statin therapy can vary from 5% to 70%, and many individuals do not meet treatment goals with the maximally tolerated statin intensity.32 A variety of factors, such as differences in drug absorption, drug transport, intrahepatic drug metabolism, drug metabolism in other organs, and mechanisms for drug excretion, are thought to play a role in subtherapeutic response to statins. Subtherapeutic response can also be due to variations in the level of target pathways that are not related to the pharmacokinetic features of the drug as well as, individual differences in cholesterol biosynthesis and lipoprotein metabolic pathways. Of note, it is assumed that the failure to achieve an optimal response to statin therapy is largely due to nonadherence with the treatment, also known as pseudoresistance.
PCSK9: Basic Physiology and Role as a Potential Therapeutic Target
The identification of the link between autosomal dominant hypercholesterolemia (ADH) and PCSK9 occurred in 2003 as a result of research conducted with 2 French families with TC levels in the 90th percentile37 and no genetic linkage to the LDL‐R or apo B genes.13, 38 Subsequently, 2 GOF mutations were identified at the PCSK9 locus and were found in 12.5% of the families with ADH.13 PCSK9 was ultimately found to encode a novel proprotein convertase in the subtilase subfamily that is involved in cholesterol homeostasis.13
These results established PCSK9 as the third gene associated with ADH in addition to genes encoding LDL‐R and apo B.26, 39 Increased production of PCSK9 is associated with a decrease in LDL‐R activity, which results in elevations in LDL‐C levels. Reductions in the production of PCSK9 were shown to exert the opposite effects on LDL‐R and LDL‐C,38 with LOF mutations associated with reduced LDL‐C levels and decreased risk for CHD.16, 26, 39
Physiological Function and Tissue Expression of PCSK9
The PCSK9 gene is located on chromosome 1p32.3 and is expressed primarily in the liver and small intestine, which play key roles in the synthesis and regulation of cholesterol.13, 14, 40 PCSK9 is also expressed in the kidney and cerebellum. The sterol‐responsive element binding protein 2 (SREBP‐2) regulates the transcription of HMG‐CoA reductase (the enzyme that statins inhibit) as well as both PCSK9 and LDL‐R and, consequently, the number of LDL‐R at the cell surface of hepatocytes, as shown in Figure 2. Synthesized in the endoplasmic reticulum as an ∼72‐kDa precursor, the PCSK9 protein undergoes autocatalytic cleavage (between the prodomain and catalytic domain), which is essential for its transport and secretion (Figure 3).14, 38 Circulating PCSK9 binds to LDL‐R on the hepatocyte cell membrane. This redirects LDL‐R from its normal pathway of recycling to the cell surface and instead directs LDL‐R to lysosomal degradation. Thus, circulating PCSK9 reduces the number of hepatic surface LDL‐Rs and increases plasma levels of LDL‐C. Proteolysis or target‐ and non‐target‐mediated pathways ultimately terminate the activity of the PCSK9 protein.12, 26, 38
Figure 2.
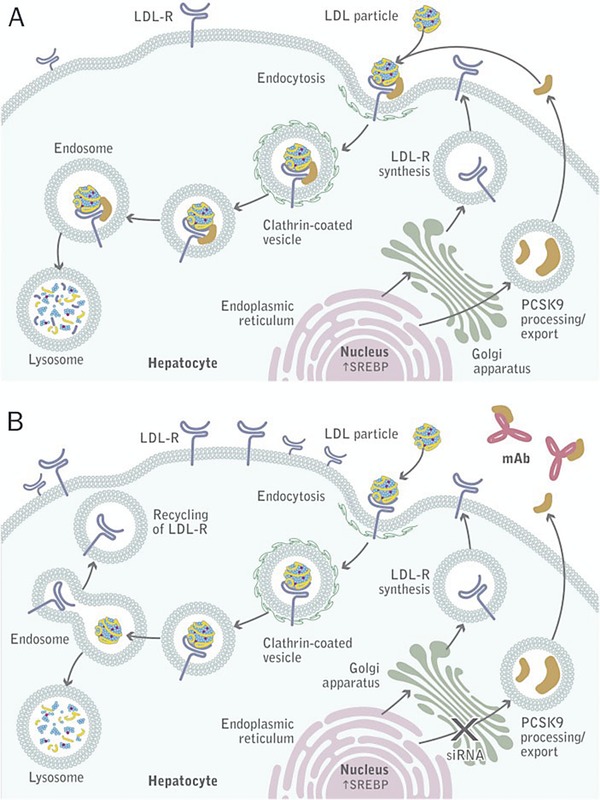
PCSK9 mode of action.26 (A) PCSK9 is synthesized in the hepatocyte and secreted into the plasma. On binding LDL‐R, PCSK9 in complex with LDL‐R is internalized through endocytosis and degraded in a lysosome. (B) When PCSK9 synthesis or binding is inhibited using interference RNA or a monoclonal antibody, respectively, LDL‐C bound by LDL‐R is internalized, but LDL‐R returns to the hepatocyte surface where it can bind another circulating LDL particle. LDL, low‐density lipoprotein; LDL‐C, LDL cholesterol; LDL‐R, LDL receptor; mAb, monoclonal antibody; PCSK9, proprotein convertase subtilisin/kexin type 9; siRNA, small interfering RNA; SREBP, sterol‐responsive element binding protein 2. Reproduced with permission.26 © 2012 The American Society for Biochemistry and Molecular Biology.
Figure 3.
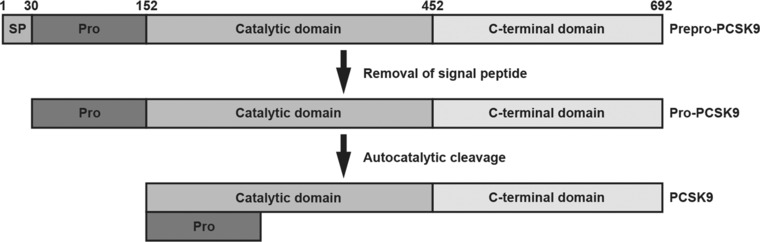
Protein domains of human PCSK9 include a signal sequence (amino acids 1‐30), prodomain (amino acids 31‐152), and catalytic domain (amino acids 153‐452), followed by a cysteine‐ and histidine‐rich C‐terminal region.14, 38 PCSK9, proprotein convertase subtilisin/kexin type 9; Pro, prodomain; SP, signal peptide. Reproduced with permission of Springer.37
Regulation of PCSK9 Expression
PCSK9 expression appears to be regulated by nutritional and hormonal status. Studies in vitro and in animals have shown that factors associated with the up‐regulation of PCSK9 include overexpression of SREBP‐2, cholesterol depletion, inflammation, insulin, and statin therapy.14 Notably, LMTs such as statins, fibrates, and ezetimibe increase levels of PCSK9. Decreased intracellular production of cholesterol by inhibition of HMG‐CoA reductase up‐regulates the expression of PCSK9 and LDL‐R, which may be 1 explanation for the increased levels of PCSK9 in response to statin therapy.38 This may explain the relatively small incremental reduction (6%) in LDL‐C with each doubling of the statin dose.41 Conversely, suppression of SREBP‐2, cholesterol feeding, berberine, glucagon, ethinylestradiol, chenodeoxycholic acid, and farnesoid X receptor agonists are all implicated in the down‐regulation of PCSK9.14
PCSK9 Inhibitors
The discovery of the role of PCSK9 in lipid metabolism has directed attention to the development of pharmacologic interventions to decrease PCSK9 levels or to inhibit PCSK9 by targeting either protein synthesis or binding to LDL‐R (Table 1). The strategies that target the binding include (1) monoclonal antibodies (mAbs) or other novel proteins that bind to PCSK9 in the plasma, thereby preventing it from binding to LDL‐R, (2) modified binding proteins such as adnectins, and (3) small‐molecule inhibitors. Notably, development of a small‐molecule inhibitor of PCSK9 that binds to LDL‐R might be challenging because of the relatively flat surface of PCSK9, which is devoid of the pockets necessary for small‐molecule binding.42 In addition, gene silencing of intracellular PCSK9 production by RNA interference disrupts the protein's synthesis.39 The development of mAbs has advanced tremendously in recent years. In 2015, alirocumab (Praluent, Sanofi‐Aventis US LLC, Bridgewater, New Jersey, and Regeneron Pharmaceuticals, Inc, Tarrytown, New York) and evolocumab (Repatha, Amgen, Inc, Thousand Oaks, California) were approved in the United States and in the European Union. Research on gene silencing approaches is currently being conducted in early clinical trials.38
Table 1.
Anti‐PCSK9 Therapeutic Approaches
| Mechanism of Action | Class | Agent | Company | Phase |
|---|---|---|---|---|
| PCSK9 binding | ||||
| Human monoclonal antibody | Alirocumab (REGN727/SAR236553) | Regeneron/Sanofi | Approved in USA and EU | |
| Human monoclonal antibody | Evolocumab (AMG145) | Amgen | Approved in USA and EU | |
| Humanized monoclonal antibody | Bococizumab (PF‐04950615) | Pfizer | 3 | |
| Human monoclonal antibody | LY3015014 | Eli Lilly | 2 | |
| Modified binding protein | Adnectin (BMS962476) | BMS/Adnexus | 1 | |
| Small‐molecule inhibitor | SX‐PCK9 | Serometrix | Preclinical | |
| PCSK9 synthesis | ||||
| RNA interference | ALN‐PCSsc | Alnylam/The Medicines Company | 1 | |
PCSK9, proprotein convertase subtilisin/kexin type 9.
In the United States, alirocumab and evolocumab are indicated as adjuncts to diet and maximally tolerated statin therapy for the treatment of adults with HeFH or clinical ASCVD who require additional lowering of LDL‐C.43, 44 Additionally, evolocumab is indicated as an adjunct to diet and other LDL‐lowering therapies (eg, statins, ezetimibe, LDL apheresis) for the treatment of patients with HoFH who require additional lowering of LDL‐C.44
Monoclonal Antibodies: The Science
The technology for the development of fully murine mAbs evolved during the 1970s and early 1980s and was based on fusion of immortal mouse myeloma cells and splenic cells from animals with stimulated immune systems.45, 46 Experience with early hybridoma technology revealed several factors that undermined the efficacy of long‐term, repeated administration of mAbs, including host immune response, pharmacokinetic limitations of mouse antibodies in humans,45 and safety issues.46 Importantly, when patients are treated with a mouse mAb, they can develop human antimouse antibodies that result in rapid mAb clearance, hypersensitivity, suboptimal penetration of the target site, and diminished efficacy due to cytotoxic effects.47
Efforts to overcome these barriers included the development of chimeric antibodies that contained mouse sequences in the variable region of the antibody and human sequences in the remainder of the antibody as a method of reducing the risk for immunogenicity in humans.45, 46 Recombinant DNA strategies allowed the development of more humanized antibodies that were less immunogenic, including chimeric and fully human antibodies (Figure 4).45, 47, 48
Figure 4.
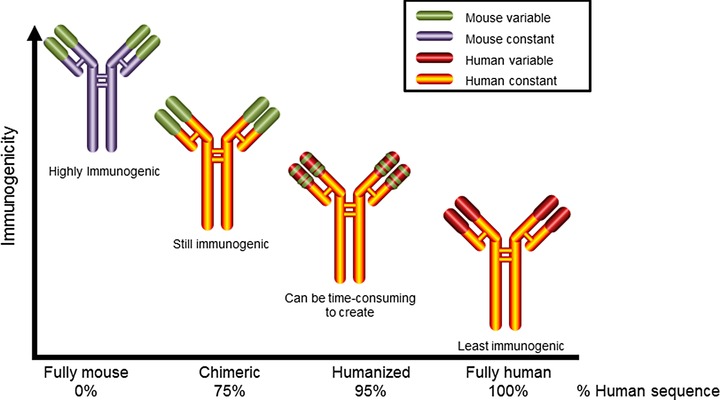
Evolution of therapeutic monoclonal antibodies. Fully mouse antibodies developed with early hybridoma technology were highly immunogenic. Development of recombinant DNA technologies resulted in more humanized and less immunogenic antibodies: chimeric, humanized, and fully human.45, 46, 48
Three functions allow mAbs to achieve their effects, including direct and indirect mechanisms. Direct effects are achieved when the antibody binds to a specific target such as cell‐surface receptors, membrane‐bound proteins, growth factors, or circulating proteins. Binding directly to the FAB domain (the antigen‐binding site) allows the mAb to promote or suppress a biological effect.45, 47 Alternatively, some mAbs work indirectly through an interaction with the Fc domain (constant domain) of the antibody. This causes cell‐surface receptors to produce immune‐mediated effector functions such as antibody‐dependent, cell‐mediated cytotoxicity, complement‐dependent cytotoxicity, or antibody‐dependent phagocytosis. An alternate indirect effect of mAbs involves delivery of the complement on multimeric immune complexes between the mAb and the target cell. This activates complement‐dependent cytotoxicity and results in cell death.45, 47 Specifically, the indirect effects of the antibody can be achieved through conjugation of mAbs with linkers such as drugs, toxins, radioisotopes, or cytokines, which results in targeted delivery of therapeutic or diagnostic agents.45 Both alirocumab and evolocumab are examples of antibodies with a direct mechanism of action: they target PCSK9 but not immune effectors.
Initially, mAbs were administered intravenously to ensure 100% bioavailability, accelerate systemic delivery, and ensure high final plasma concentrations.45, 47 Subcutaneous (SC) and intramuscular routes are now used, and, although associated with lower bioavailability (typically 20% to 60% for SC administration49), they offer the advantages of reduced cost, greater convenience to patients, who do not need to receive the infusion at special infusion centers, and lower risk for infusion reactions.45
Treatment with mAbs offers several advantages over other pharmacologic interventions (ie, small drug molecules), including a reduced risk for drug‐drug interactions because mAbs are not metabolized by the liver and kidneys and do not interact with cytochrome P450 and other transport proteins.45, 47 Monoclonal antibodies have high specificity for target antigens, can achieve high potency with less frequent dosing, and do not penetrate the central nervous system because of their size.45 Furthermore, mAbs are unlikely to block potassium channels, prolong the QT interval, and cause cardiac repolarization changes.45, 50
Overview of PCSK9‐Specific Monoclonal Antibody Clinical Trials
Identification of PCSK9 as a potential therapeutic target for the treatment of hypercholesterolemia has generated an extensive body of research to develop interventions to inhibit or reduce levels of PCSK9. Of particular interest are ongoing clinical research programs to evaluate the efficacy and safety of PCSK9‐specific mAbs, including alirocumab, evolocumab, and bococizumab (Figures 5, 6, 7, Tables 2 and 3).51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77
Figure 5.
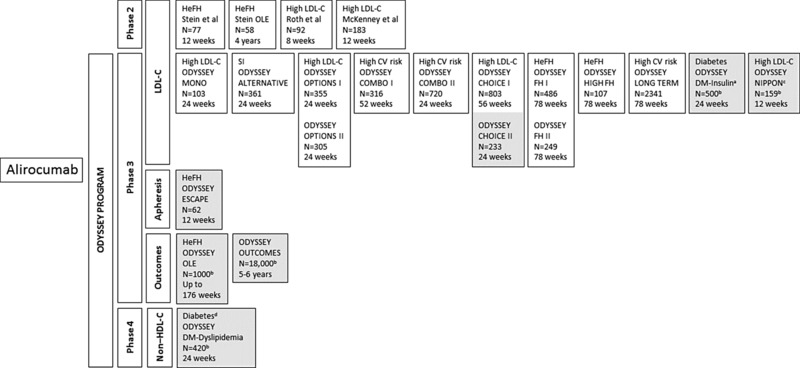
Overview of the ODYSSEY program for efficacy and safety of alirocumab (phase 2 and phase 3 clinical trials).52, 54, 58, 59, 65, 66, 67, 68, 73, 74, 76, 77, 78 Gray shading represents ongoing or planned studies. CV, cardiovascular; HeFH, heterozygous familial hypercholesterolemia; LDL‐C, low‐density lipoprotein cholesterol; SI, statin intolerance. aPatients with type 1 or type 2 diabetes who are treated with insulin. bEstimated enrollment. cJapanese population. dPatients with type 2 diabetes and mixed dyslipidemia.
Figure 6.
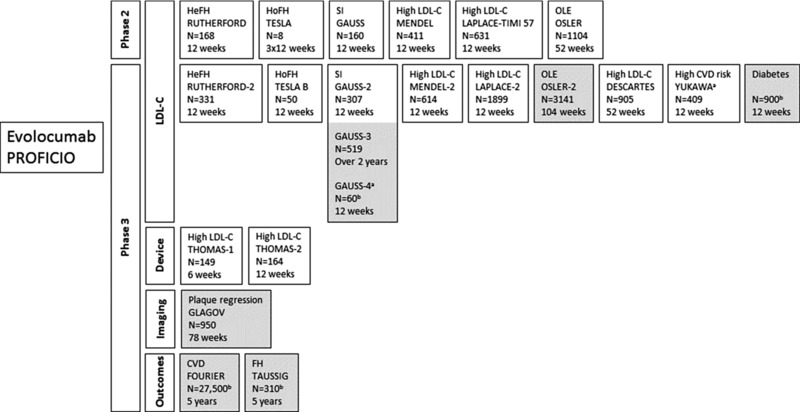
Overview of the PROFICIO program for efficacy and safety of evolocumab (phase 2 and phase 3 clinical trials).55, 56, 57, 60, 62, 63, 69, 70, 71, 75, 78, 84, 85 Gray shading represents ongoing or planned studies. CVD, cardiovascular disease; FH, familial hypercholesterolemia; HeFH, heterozygous FH; HoFH, homozygous FH; LDL‐C, low‐density lipoprotein cholesterol; OLE, open‐label extension; SI, statin intolerance. aJapanese population. bEstimated enrollment.
Figure 7.
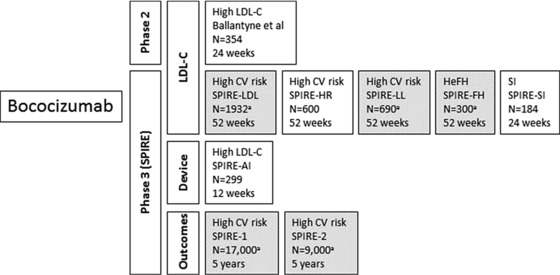
Overview of the SPIRE program for efficacy and safety of bococizumab (phase 2 and phase 3 clinical trials).72, 78 Gray shading represents ongoing or planned studies. CV, cardiovascular; HeFH, heterozygous familial hypercholesterolemia; LDL‐C, low‐density lipoprotein cholesterol; SI, statin intolerance. aEstimated enrollment.
Table 2.
Reduction of LDL‐C Reported to Date in Phase 2 and Phase 3 Clinical Trials of PCSK9‐Specific Antibodies
| Trial | Phase | Background Therapy | Treatment | Comparator | Time to Primary Endpoint | Dosing | LDL‐C LSM Change From Baseline to Primary Endpoint, % |
|---|---|---|---|---|---|---|---|
| Heterozygous familial hypercholesterolemia | |||||||
|
2 | Statin ± EZE | Aliroc | Placebo | 12 wk |
|
|
|
2 | LMT | Aliroc | OLE | 64 wk | 150 mg Q2W | |
|
3 | Max‐tolerated statin ± other LMT | Aliroc | Placebo | 24 wk | 75 mg/150 mg Q2Wa |
|
|
3 | Max tolerated statin ± other LMT | Aliroc | Placebo | 24 wk | 75 mg/150 mg Q2Wa |
|
|
3 | Max tolerated statin ± LMT | Aliroc | Placebo | 24 wk | 150 mg Q2W |
|
|
2 | Statin ± EZE | Evo | Placebo | 12 wk |
|
|
|
3 | Statin ± LMT | Evo | Placebo | 12 wk |
|
|
| Hypercholesterolemia and high cardiovascular risk | |||||||
|
3 | Max‐tolerated statin ± LMT | Aliroc | Placebo | 24 wk | 75 mg/150 mg Q2Wc |
|
|
3 | Max‐tolerated statin | Aliroc | Placebo + EZE | 24 wk | 75 mg/150 mg Q2Wc |
|
|
3 | Max‐tolerated statin or no statin | Aliroc | Placebo | 24 wk | 75 mg/150 mg Q2Wd
300 mg Q4W/150 mg Q2Wd |
|
| Additional patient populations | |||||||
|
2 | Diet + LMT | Evo | Open label | 12 wk |
|
|
|
3 | Diet + LMT | Evo | Placebo | 12 wk | 420 mg Q4W |
|
|
3 | LMT | Aliroc | EZE | 24 wk | 75 mg Q2W |
|
|
2 | Low‐dose statin or LMT | Evo | Placebo + EZE | 12 wk |
|
|
| Evo + EZE | Placebo + EZE | 12 wk | 420 mg Q4W |
|
|||
|
3 | Low‐dose statin ± LMT | Evo | Placebo + EZE | 12 wk |
|
|
|
3 | ATV 20 mg or 40 mg ± LMT | Aliroc + ATV 20 mg | EZE double statin | 24 wk | 75 mg/150 mg Q2Wf |
|
| Aliroc + ATV 40 mg | EZE double statin | 24 wk | 75 mg/150 mg Q2Wf |
|
|||
| Aliroc + ATV 40 mg | ROS 40 mg | 24 wk | 75 mg/150 mg Q2Wf |
|
|||
|
3 | ROS 10 or 20 mg ± LMT | Aliroc + ROS 10 mg | EZE double statin | 24 wk | 75 mg/150 mg Q2Wf |
|
| Aliroc + ROS 20 mg | EZE double statin | 24 wk | 75 mg/150 mg Q2Wf |
|
|||
|
2 | None | Aliroc + ATV 10 or 80 mg | Placebo + ATV 80 mg | 8 wk | Q2Wg |
|
|
2 | ATV 10, 20, or 40 mg | Aliroc | Placebo | 12 wk |
50 mg Q2W 100 mg Q2W 150 mg Q2W 200 mg Q4W 300 mg Q4W |
|
|
3 | None | Aliroc | Placebo + EZE | 24 wk | 75 mg/150 mg Q2Wc |
|
|
3 | None | Aliroc | Placebo | 24 wk | 75 mg/150 mg Q2Wd 150 mg Q4W/150 mg Q2Wd |
NR −56.4e |
|
2 | None | Evo | Placebo | 12 wk | 70 mg Q2W 105 mg Q2W 140 mg Q2W 280 mg Q4W 350 mg Q4W 420 mg Q4W |
|
|
2 | Statin ± EZE | Evo | Placebo | 12 wk | 70 mg Q2W 105 mg Q2W 140 mg Q2W 280 mg Q4W 350 mg Q4W 420 mg Q4W |
|
| 2 | ± Statin | SOC ± Evo | Open label | 52 wk | 420 mg Q4W | ||
| 3 | ± SOC | Evo | Open label | 12 wk |
140 mg Q2W or 420 mg QMj |
−64.0e | |
|
3 | Diet + none; diet + ATV | Evo + diet | Placebo | 52 wk | 420 mg Q4W |
|
| Diet + ATV + EZE | Evo + diet + ATV 10 mg | Placebo | 52 wk |
|
|||
| Evo + diet + ATV 80 mg | Placebo | 52 wk |
|
||||
| Evo + diet + ATV 80 mg + EZE | Placebo | 52 wk |
|
||||
|
3 | None | Evo | Placebo or EZE | 12 wk | 140 mg Q2W 420 mg QM |
|
|
3 | ATV 10 mg | Evo | Placebo ± EZE | 12 wk |
140 mg Q2W 420 mg QM |
|
| ATV 80 mg | Evo | Placebo ± EZE | 12 wk |
140 mg Q2W 420 mg QM |
|
||
| SIM 40 mg | Evo | Placebo | 12 wk |
140 mg Q2W 420 mg QM |
|
||
| ROS 5 mg | Evo | Placebo | 12 wk |
140 mg Q2W 420 mg QM |
|
||
| ROS 40 mg | Evo | Placebo | 12 wk |
140 mg Q2W 420 mg QM |
|
||
|
2 | Statin | Boco | Placebo | 12 wk |
50 mg Q2W 100 mg Q2W 150 mg Q2W Placebo Q2W 200 mg Q4W 300 mg Q4W Placebo Q4W |
|
|
3 | Max tolerated statin ± other LMT | Aliroc | Placebo | 24 wk | 150 mg Q2W |
|
Aliroc, alirocumab; ATV, atorvastatin; Boco, bococizumab; CV, cardiovascular; Evo, evolocumab; EZE, ezetimibe; FH, familial hypercholesterolemia; HeFH, heterozygous FH; HoFH, homozygous FH; LDL‐C, low‐density lipoprotein cholesterol; LMT, lipid‐modifying therapy; LSM, least squares mean; NR, not reported; OLE, open‐label extension; P, placebo; PCSK9, proprotein convertase subtilisin/kexin type 9; Q2W, every 2 weeks; Q4W, every 4 weeks; QM, once monthly; ROS, rosuvastatin; SI, statin intolerance; SIM, simvastatin; SOC, standard of care.
Potential dose increase to 150 mg Q2W at week 8 if LDL‐C >70 mg/dL.
Mean change from baseline.
Potential dose increase to 150 mg Q2W at week 12 if week 8 LDL‐C ≥70 mg/dL.
Potential dose increase to 150 mg Q2W at week 12 if LDL‐C <70 mg/dL or <100 mg/dL (dependent on CV risk) or if LDL‐C reduction <30% at week 8.
LSM change in LDL‐C vs placebo.
Potential dose increase to 150 mg Q2W at week 12 if week 8 LDL‐C ≥70 mg/dL or ≥100 mg/dL, dependent on risk level.
Alirocumab was supplied at a concentration of 150 mg/mL.
Mean change vs placebo.
OSLER‐1 enrolled patients with hypercholesterolemia, SI, HeFH, or high CV risk who participated in 1 of 5 phase 2 studies: MENDEL‐1, LAPLACE‐TIMI 57, GAUSS‐1, RUTHERFORD‐1, or YUKAWA‐1.
OSLER‐2 enrolled patients with hypercholesterolemia, SI, or HeFH who participated in 1 of 7 phase 3 studies: MENDEL‐2, LAPLACE‐2, GAUSS‐2, RUTHERFORD‐2, DESCARTES, THOMAS‐1, or THOMAS‐2.
Dose was dependent on the patient's choice.
Similar results were observed for the mean of weeks 10 and 12. Coprimary endpoints were the percentage change from baseline in LDL‐C level at the mean of weeks 10 and 12 and at week 12.
Table 3.
Change in Non‐LDL‐C Lipids Reported to Date in Phase 2 and Phase 3 Clinical Trials of PCSK9‐Specific Antibodies: Trials Reporting These Parameters to Date
| Change From Baseline to Primary Endpoint, % | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Trial | BG Therapy | Treatment | Comparator | Lp(a) (Median) | TC (LSM) | HDL‐C (LSM) | Non‐HDL‐C (LSM) | Apo B (LSM) | Apo A1 (LSM) | TG (Median) | VLDL‐C (LSM) | TC/HDL‐C Ratio (LSM) | Apo B/apo A1 Ratio (LSM) |
| Heterozygous familial hypercholesterolemia | |||||||||||||
| Stein 201268 N = 77 | Statin ± EZE | Aliroc | Placebo | −23.38 to −7.45 vs −3.91 | −43.55 to −18.10 vs −8.48 | +6.49 to +12.34 vs +2.20 | −57.90 to −26.84 vs −11.34 | −50.19 to −20.91 vs −6.39 | +1.69 to +8.82 vs −5.32 | −16.73 to −4.92 vs −10.55 | NR | NR | NR |
| Stein 201467 N = 58 | LMT | Aliroc | OLE | −24.5 | NR | +4.5 | NR | −45.5 | +7.6 | NR | NR | NR | NR |
| ODYSSEY FH I54 N = 486 | Max tolerated statin ± other LMT | Aliroc | Placebo | −25.2 vs −7.5a | NR | +8.8 vs +0.8 | −42.8 vs +9.6 | −41.1 vs +4.7 | +5.0 vs +0.3 | −9.6 vs +6.3a | NR | NR | NR |
| ODYSSEY FH II54 N = 249 | Max tolerated statin ± other LMT | Aliroc | Placebo | −30.3 vs −10.0a | NR | +6.0 vs −0.8 | −42.6 vs +3.1 | −42.8 vs −3.5 | +2.8 vs −1.6 | −10.4 vs +0.5a | NR | NR | NR |
| RUTHERFORD60 N = 168 | Statin ± EZE | Evo | Placebo | −27.4 to −19.1 vs +4.1a | −37.4 to −28.1 vs +2.9 | +9.1 to +10.1 vs +2.3 | −51.0 to −39.3 vs +2.5 | −43.3 to −31.9 vs +2.9 | +10.3 to +10.6 vs +8.6 | −10.5 to −5.6 vs +9.4a | −18.6 to −7.8 vs +17.0 | −42.0 to −33.7 vs +3.0 | −48.7 to −38.0 vs −4.1 |
| RUTHERFORD‐262 N = 331 | Statin ± LMT | Evo | Placebo | −22.9 to −21.6 vs +6.7 to +8.7 | NR | +5.4 to +8.1 vs −3.7 to −1.2 | −56.2 to −49.7 vs −1.4 to +5.3 | −49.8 to −44.8 vs −0.7 to +4.6 | +5.7 to +7.3 vs −1.4 to +1.8 | −16.1 to −5.1 vs +3.5 to +6.4 | NR | −46.0 to −38.3 vs +0.1 to +7.1 | −52.7 to −45.3 vs +1.5 to +4.2 |
| Hypercholesterolemia and high cardiovascular risk | |||||||||||||
| ODYSSEY COMBO I74 N = 316 | Max tolerated statin ± LMT | Aliroc | Placebo | −20.5 vs −5.9 | −27.9 vs −2.9 | +3.5 vs −3.8 | −39.1 vs −1.6 | −36.7 vs −0.9 | NR | −6.0 vs −5.4 | NR | NR | NR |
| ODYSSEY COMBO II73 N = 720 | Max‐ tolerated statin | Aliroc | Placebo + EZE | −27.8 vs −6.1 | −29.3 vs −14.6 | +8.6 vs +0.5 | −42.1 vs −19.2 | −40.7 vs −18.3 | +5.0 vs −1.3 | −13.0 vs −12.8 | NR | NR | NR |
| Additional patient populations | |||||||||||||
| TESLA69 N = 8 HoFH | Diet + LMT | Evo | Open label | −18.6 to −11.7 | NR | −1.4 to +4.7a | NR | −14.9 to −12.5a | +1.3 to +5.2a | −5.7 to +5.8a | NR | NR | NR |
| TESLA Part B61 N = 50 HoFH | Diet + LMT | Evo | Placebo | −9.4 vs +2.4a, b | −18.9 vs +7.8 | +4.0 vs +4.1 | −22.0 vs +8.1 | −19.2 vs +4.0 | NR | −1.4 vs −1.7 | +18.7 vs +62.6 | −21.6 vs +4.4 | −22.5 vs +5.3 |
| Additional patient populations (continued) | |||||||||||||
| ODYSSEY ALTERNATIVE59 N = 361 SI | LMT | Aliroc | EZE | −25.9 vs −7.3c | −31.8 vs −10.9c | +7.7 vs +6.8c, d | −40.2 vs −14.6c | −36.3 vs −11.2c | +4.8 vs +2.9c, d | −9.3 vs −3.6c, d | NR | NR | NR |
| GAUSS71 N = 160 SI | Low‐dose statin or LMT | Evo | Placebo + EZE | −25.9 to −20.3 vs −7.9a | −37.7 to −29.8 vs −10.7 | +5.5 to +7.4 vs −1.1 | −48.6 to −39.8 vs −15.0 | −42.1 to −33.6 vs −12.2 | +6.0 to +7.5 vs −1.4 | −19.3 to −14.2 vs −5.5 | −28.8 to −15.0 vs −13.2 | −40.6 to −32.0 vs −9.6 | −45.4 to −36.5 vs −11.4 |
| Evo + EZE | Placebo + EZE | −29.1 vs −7.9a | −44.3 vs −10.7 | +12.0 vs −1.1 | −59.8 vs −15.0 | −49.1 vs −12.2 | +8.3 vs −1.4 | −9.5 vs −5.5 | −37.8 vs −13.2 | −49.4 vs −9.6 | −52.0 vs −11.4 | ||
| GAUSS‐270 N = 307 SI | Low‐dose statin or LMT | Evo | Placebo + EZE | −27.0 to −22.1 vs −1.7 to +5.8a | NR | +5.3 to +6.5 vs +1.6 to +1.8 | −48.6 to −46.2 vs −16.5 to −13.2 | −45.8 to −43.1 vs −13.0 to −10.0 | +5.2 to +5.5 vs +1.1 to +3.3 | −3.9 to −2.5 vs −5.5 to +2.16 | −6.2 to −2.2 vs −5.5 to −2.3 | −40.4 to −38.6 vs −14.1 to −9.92 | −47.7 to −45.5 vs −13.1 to −11.4 |
| ODYSSEY OPTIONS I76 N = 355 Patients with subtherapeutic response to statins | ATV 20 mg or 40 mg ± LMT | Aliroc + ATV 20 mg | EZE double statin | −23.6 vs −10.6 (EZE) and −20.2 (ATV 40 mg) | NR | +4.8 vs −0.1 (EZE) and +1.9 (ATV 40 mg) | −36.7 vs −15.1 (EZE) and −6.3 (ATV 40 mg) | −33.7 vs −10.1 (EZE) and −4.4 (ATV 40 mg) | NR | −12.0 vs −3.3 (EZE) and −6.7 (ATV 40 mg)a | NR | NR | NR |
| Aliroc + ATV 40 mg | EZE double statin | −30.8 vs +0.2 (EZE) and −9.7 (ATV 80 mg) and −4.9 (ROS 40 mg) | NR | +7.7 vs +2.0 (EZE) and +4.7 (ATV 80 mg) and +5.7 (ROS 40 mg) | −47.6 vs −21.0 (EZE) and −6.5 (ATV 80 mg) and −17.4 (ROS 40 mg) | −41.9 vs −14.3 (EZE) and −3.5 (ATV 80 mg) and −10.9 (ROS 40 mg) | NR | −19.1 vs −13.9 (EZE) and −7.3 (ATV 80 mg) and −0.5 (ROS 40 mg)a | NR | NR | NR | ||
| ODYSSEY OPTIONS II77 N = 305 | ROS 10 mg or 20 mg ± LMT | Aliroc + ROS 10 mg | EZE double statin | −27.9 vs −4.3 (EZE) and −4.0 (ROS 20 mg)a | NR | +9.1 vs +4.0 (EZE) and +1.7 (ROS 20 mg) | −42.7 vs −13.4 (EZE) and −11.3 (ROS 20 mg) | −36.5 vs −9.7 (EZE) and −7.3 (ROS 20 mg) | NR | −11.2 vs −8.3 (EZE) and −1.8 (ROS 20 mg)a | NR | NR | NR |
| Aliroc + ROS 20 mg | EZE double statin | −22.7 vs −5.8 (EZE) and −5.2 (ROS 40 mg)a | NR | +7.2 vs −1.8 (EZE) and +1.5 (ROS 40 mg) | −31.4 vs −12.9 (EZE) and −11.2 (ROS 40 mg) | −28.3 vs −11.2 (EZE) and −9.8 (ROS 40 mg) | NR | −8.7 vs −11.1 (EZE) and −9.9 (ROS 40 mg)a | NR | NR | NR | ||
| Additional patient populations (continued) | |||||||||||||
| Roth 201265 N = 92 Hypercholesterolemia | None | Aliroc + ATV 10 mg | Placebo + ATV 80 mg | −34.7 vs −2.7 | −40.5 vs −16.6e | +2.6 vs −3.6 | −58.3 vs −22.3e | −54.4 vs −12.0e | +0.4 vs −5.2 | −4.0 vs −11.9 | NR | NR | NR |
| Aliroc + ATV 80 mg | Placebo + ATV 80 mg | −31.0 vs −2.7 | −47.2 vs ‐16.6e | +5.8 vs −3.6 | −63.9 vs −22.3e | −58.0 vs −12.0e | −2.2 vs −5.2 | −24.7 vs −11.9 | NR | NR | NR | ||
| McKenney 201258 N = 183 Hypercholesterolemia | ATV 10, 20, or 40 mg | Aliroc | Placebo | −28.6 to −7.9 vs 0.0 | −45.2 to −23.0 vs −1.6 | +4.1 to +8.5 vs −1.0 | −62.5 to −33.6 vs −2.2 | −56.1 to −27.3 vs +2.2 | +0.3 to +4.2 vs 0.0e | −18.9 to −5.5 vs +9.7 | NR | NR | NR |
| ODYSSEY MONO66 N = 103 Hypercholesterolemia; no background LMT | None | Aliroc | Placebo + EZE | −16.7 vs −12.3a , f | −29.6 vs −10.9 | +6.0 vs +1.6 | −40.6 vs −15.1 | −36.7 vs −11.0 | +4.7 vs −0.6 | −11.9 vs −10.8f | NR | NR | NR |
| MENDEL57 N = 411 Hypercholesterolemia | None | Evo | Placebo | −27.3 to −9.1 vs +2.0 to +9.2a | −34.0 to −26.7 vs −2.2 to +1.4 | +5.3 to +11.5 vs +1.1 to +5.7 | −48.0 to −37.9 vs −2.9 to −0.3 | −44.5 to −32.7 vs −0.3 to +0.2 | +1.2 to +9.0 vs −1.6 to +0.7 | −10.6 to −5.9 vs −4.2 to +1.4 | −26.0 to −10.3 vs −0.1 to +9.8 | −40.0 to −30.1 vs −3.4 to −1.2 | −48.1 to −32.8 vs +0.1 to +2.0 |
| LAPLACE‐TIMI 5785 N = 631 Hypercholesterolemia | Statin ± EZE | Evo | Placebo | NR | −42.5 to −26.2g | +1.6 to +8.1g | −61.4 to −37.8g | −56.4 to −34.4g | +0.31 to +4.8g | −33.7 to −13.4g | −44.3 to −21.1g | −47.7 to −27.7g | −53.4 to −33.8g |
| OSLER‐155 N = 1104 Hypercholesterolemia | ± Statin | Evo + SOC | Open label | −32.8 for no Evo/Evo vs −11.1 for no Evo/SOC and −8.7 for Evo/ SOC | −32.5 for no Evo/Evo vs −0.5 for no Evo/SOC and −1.5 Evo/SOCc | +8.5 for no Evo/Evo vs +3.5 for no Evo/SOC and +3.7 for Evo/SOCc | −45.9 for no Evo/Evo vs −1.2 no Evo/ SOC and −2.8 Evo/ SOCc | −42.1 for no Evo/Evo vs −4.2 for no Evo/ SOC and −3.6 for Evo/SOCc | +3.6 for no Evo/Evo vs +0.9 for no Evo/ SOC and +0.1 for Evo/ SOCc | −8.3 for no Evo/Evo vs +3.7 for no Evo/SOC and −1.4 for Evo/SOCe | −14.7 no Evo/Evo vs +11.6 no Evo/ SOC and −4.2 Evo/SOCe | −36.9 no Evo/Evo vs −2.3 no Evo/ SOC and −3.1 Evo/ SOCc | −43.7 no Evo/Evo vs −4.2 no Evo/SOC and −2.5 Evo/SOCc |
| SOC | Open label | −29.9 for Evo/Evo vs −11.1 for no Evo/SOC and −8.7 for Evo/ SOC | −33.0 for Evo/Evo vs −0.5 for no Evo/ SOC and −1.5 for Evo/ SOCc | +9.1 for Evo/Evo vs +3.5 for no Evo/SOC and +3.7 for Evo/SOCc | −46.5 vs −1.2 and −2.8c | −42.6 for Evo/Evo vs −4.2 for no Evo/SOC and −3.6 for Evo/SOCc | +4.9 for Evo/Evo vs +0.9 for no Evo/SOC and +0.1 for Evo/SOCc | −9.0 for Evo/Evo vs +3.7 for no Evo/SOC and −1.4 for Evo/SOCe | −18.8 for Evo/Evo vs +11.6 for no Evo/SOC and −4.2 for Evo/ SOCe | −37.5 for Evo/Evo vs −2.3 for no Evo/SOC and −3.1 for Evo/SOCc | −44.7 for Evo/Evo vs −4.2 for no Evo/SOC and −2.5 for Evo/SOCc | ||
| Additional patient populations (continued) | |||||||||||||
| OSLER‐1/OSLER‐275 , h N = 4465 Hypercholesterolemia | SOC | Evo | Open label | −25.5 vs 0.0 | −32.3 vs +3.8c | +8.7 vs +1.7c | −46.1 vs +5.9c | −41.7 vs +5.5c | +6.8 vs +2.6c | −9.1 vs +3.5 | NR | NR | NR |
| DESCARTES51 N = 905 Hypercholesterolemia | Diet + none; diet + ATV | Evo + diet | Placebo | −22.5 vs −12.8a | −30.9 vs +5.4 | +9.2 vs −2.1 | −44.9 vs +9.5 | −43.3 vs −0.4 | +3.1 vs −1.7 | −7.7 vs +15.4a | −2.1 vs +77.2 | −34.7 vs +9.7 | −44.1 vs +1.9c |
| Diet + ATV + EZE | Evo + diet + ATV 10 mg | Placebo | −32.8 vs −3.6a | −30.1 vs +5.3 | +4.8 vs −0.5 | −46.0 vs +8.5 | −44.8 vs +2.9 | +1.5 vs −0.9 | −1.0 vs +10.2a | +8.6 vs +28.7 | −32.1 vs +6.8 | −45.1 vs +4.4c | |
| Evo + diet + ATV 80 mg | Placebo | −30.1 vs −10.6a | −25.0 vs +8.1 | +5.4 vs +1.5 | −37.8 vs +11.7 | −39.2 vs +5.4 | +3.4 vs −0.1 | −0.9 vs +11.5a | +4.4 vs +35.5 | −28 vs +8.4 | −40.8 vs +6.1c | ||
| Evo + diet + ATV 80 mg + EZE | Placebo | −20.5 vs −1.1a | −27.0 vs +1.7 | +5.0 vs +1.0 | −38.9 vs +2.4 | −37 vs +0.8 | +0.9 vs −1.7 | −2.1 vs +1.6a | −3.9 vs +13.6 | −28.8 vs +2.2 | −36.2 vs +2.9c | ||
| MENDEL‐256 N = 614 Hypercholesterolemia | None | Evo | Placebo | −20.4 to −17.8 vs 0.0e | NR | +4.1 to +4.8 vs −5.3 to −1.2e | −50.1 to −49.7 vs −0.3 to +1.5 | −47.2 to −46.6 vs +0.6 to +1.8 | NR | −15.6 to −8.1 vs −1.9 to +2.0 | −16.3 to −9.5 vs −1.6 to 0.0e | NR | −48.5 to −48.3 vs +1.1 to +4.5 |
| EZE | −2.1 to 0.0e | NR | −2.8 to −1.5e | −16.5 to −14.9 | −14.0 to −13.2 | NR | −2.4 to 0.0 | −3.6 to −0.9e | NR | −14.3 to −12.7 | |||
| LAPLACE‐263 , i N = 1899 Hypercholesterolemia | ATV 10 mg | Evo | Placebo | −25.9 to −20.3 vs −0.4 to +7.3a | −37.2 to −36.5 vs +1.4 to +5.6 | +7.0 to +7.9 vs 0.0 to +0.2 | −53.4 to −52.5 vs +2.4 to +8.3 | −50.9 to −47.15 vs +0.21 to +7.89 | NR | −13.3 to −3.8 vs +8.3 to +14.4a | −11.7 to −6.2 vs +8.3 to +14.7 | NR | NR |
| EZE | +3.3 to +7.2a | −14.3 to −11.3 | −1.8 to −0.4 | −18.27 to −14.8 | −15.98 to −10.95 | NR | −0.4 to +4.9a | −4.6 to +3.5 | NR | NR | |||
| ATV 80 mg | Evo | Placebo | −24.7 to −24.6 vs −2.2 to +3.4a | −36.3 to −32.6 vs +6.0 to +9.34 | +7.4 to +9.1 vs +0.3 to +5.0 | −54.8 to −50.1 vs +10.0 to +11.8 | −49.77 to −46.47 vs +6.54 to +11.64 | NR | −10.1 to −1.1 vs +6.7 to +8.2a | −9.7 to −1.1 vs +6.7 to +8.5 | NR | NR | |
| EZE | +8.0 to +10.2a | −12.3 to −10.0 | +0.2 to +0.6 | −17.3 to −14.3 | −12.31 to −12.16 | NR | −7.4 to −3.1a | −7.9 to −6.0 | NR | NR | |||
| SIM 40 mg | Evo | Placebo | −38.06 to −29.23 vs −6.81 to −1.06a | −41.9 to −36.5 vs +0.4 to +0.7 | +6.4 to +10.9 vs −2.7 to +1.1 | −59.02 to −50.96 vs +1.89 to +5.66 | −55.95 to −49.16 vs +0.35 to +3.57 | NR | −14.7 to −13.7 vs +8.1 to +16.7a | −15.9 to −14.8 vs +7.6 to +21.0 | NR | NR | |
| Additional patient populations (continued) | |||||||||||||
| ROS 5 mg | Evo | Placebo | −25.09 to −20.85 vs +4.49 to +11.40a | −36.4 to −36.3 vs +3.1 to +6.3 | +6.1 to +7.2 vs −0.2 to +2.9 | −52.04 to −51.57 vs +5.85 to +7.92 | −50.15 to −48.58 vs +4.63 to +6.35 | NR | −6.9 to −4.5 vs +13.0 to +13.6a | −8.2 to −6.3 vs +12.5 to +13.8 | NR | NR | |
| ROS 40 mg | Evo | Placebo | −26.11 to −21.97 vs +10.21 to +10.38a | −33.3 to −29.8 vs +1.2 vs +4.3 | +4.7 to +5.6 vs −0.4 to +0.7 | −50.97 to −46.42 vs +3.35 to +8.61 | −45.61 to −43.71 vs +3.24 to +4.91 | NR | −10.5 to +5.6 vs +10.0 to +11.0a | −10.0 to −6.1 vs +8.6 to +10.1 | NR | NR | |
| Ballantyne 201572 N = 354 Hypercholesterolemia | Statin | Boco | Placebo | −10.7 to 0.0 vs 0.0 to +3.5 | −31.6 to −10.5 vs −2.4 to +1.2c | +2.7 to +7.1 vs −0.4 to +0.8c | −44.9 to −17.3 vs −2.3 to +2.8c | −37.4 to −13.6 vs −2.1 to +1.5c | +2.9 to +9.9 vs +1.8 to +2.1c | −18.6 to −7.6 vs −14.5 to +3.7 | NR | NR | NR |
| ODYSSEY LONG TERM64 N = 2341 HeFH, or high CV risk | Max‐tolerated statin ± other LMT | Aliroc | Placebo | −30.2 vs −3.9 | −38.8 vs −0.4 | +4.2 vs −0.7 | −53.1 vs +0.6 | −54.3 vs +1.2 | +4.2 vs +1.2 | −15.8 vs +1.4 | NR | NR | NR |
Aliroc, alirocumab; apo A1, apolipoprotein A1; apo B, apolipoprotein B; ATV, atorvastatin; BG, background; Boco, bococizumab; CV, cardiovascular; Evo, evolocumab; EZE, ezetimibe; HDL‐C, high‐density lipoprotein cholesterol; HeFH, heterozygous familial hypercholesterolemia; HoFH, homozygous familial hypercholesterolemia; LDL‐C, low‐density lipoprotein cholesterol; LMT, lipid‐modifying therapy; Lp(a), lipoprotein(a); LSM, least squares mean; NR, not reported; OLE, open‐label extension; ROS, rosuvastatin; SI, statin intolerance; SIM, simvastatin; SOC, standard of care; TC, total cholesterol; TG, triglycerides; VLDL‐C, very low‐density lipoprotein cholesterol.
LSM.
Multiplicity adjustments following the Hochberg procedure were used to control for overall significance at the 0.05 level of significance for the primary and secondary endpoints.
Mean change from baseline.
Hierarchical testing terminated at the endpoint of HDL‐C (baseline to week 24, intention‐to‐treat analysis), and this statistical comparison and all subsequent comparisons (TG and apo A1) were not considered statistically significant.
Median.
Combined estimate for adjusted mean (SE) percentage changes are shown.
Mean change vs placebo.
Change at 12 weeks in OSLER program (including OSLER‐1 and OSLER‐2).
Similar results were observed for the mean of weeks 10 and 12.
Alirocumab at varying doses (range 50 to 300 mg) combined with a statin, with or without ezetimibe, for the treatment of patients with primary hypercholesterolemia58, 65 or HeFH67, 68 was evaluated in phase 2 clinical trials (Figure 5, Table 2). The ODYSSEY program was established to further evaluate the efficacy, safety, and tolerability of alirocumab for the treatment of individuals with hypercholesterolemia, those at high risk for CV events, those with statin intolerance, those with HeFH, and those with diabetes. Since the inception of the program, 17 phase 3 trials enrolling more than 24,000 patients have been completed or are under way (Figure 5, Table 2). Results from these trials to date reveal favorable efficacy and safety of alirocumab compared with standard therapies or placebo.52, 54, 59, 64, 66, 73, 74, 76, 77 A phase 4 trial, ODYSSEY DM‐Dyslipidemia, will evaluate the efficacy of alirocumab in the reduction of non‐HDL‐C in patients with type 2 diabetes and mixed dyslipidemia.78 The FDA‐approved dose of alirocumab is 75 to 150 mg SC every 2 weeks (Q2W).43
The Program to Reduce LDL‐C and CV Outcomes Following Inhibition of PCSK9 in Different Populations (PROFICIO) is a multistudy clinical trial program evaluating the efficacy and safety of evolocumab (Figure 6). The research program includes 22 studies, 16 of which are phase 3 trials, and a projected enrollment of approximately 30,000 patients. Results from phase 2 and phase 3 studies demonstrate significant reductions in LDL‐C levels and a positive impact on secondary lipid parameters [eg, non‐HDL‐C, apo B, and Lp(a)] with a favorable safety profile.55, 56, 57, 61, 62, 63, 71, 75 The FDA‐approved dose of evolocumab is 140 mg SC Q2W or 420 mg SC once monthly (especially for HoFH).44
The bococizumab research program includes phase 2 and phase 3 trials (Figure 7).72, 79 In the SPIRE program, 3 phase 3 studies, SPIRE‐SI, SPIRE‐HR, and SPIRE‐AI, have recently been completed, and 5 other trials are currently planned or under way to continue efforts to evaluate the efficacy and safety of bococizumab, a humanized antibody for the treatment of hypercholesterolemia and HeFH.
Efficacy of PCSK9‐Specific Antibodies in Patient Populations
Heterozygous Familial Hypercholesterolemia. Patients with HeFH were studied in phase 2 and phase 3 trials evaluating the efficacy of alirocumab or evolocumab (Tables 2 and 3). Additionally, a total of 300 patients will be enrolled in SPIRE‐FH, a phase 3, 52‐week, randomized, double‐blind, parallel‐group study to determine the efficacy and safety of bococizumab in patients with HeFH at 12 weeks.
When alirocumab was administered with a daily statin with or without ezetimibe to patients with HeFH in a phase 2 trial, the LSM decrease in LDL‐C from baseline to week 12 ranged from 28.9% to 67.9% and was dependent on the alirocumab dose.68 In the follow‐up open‐label trial, results after 64 weeks of treatment with alirocumab revealed a 59.0% mean reduction in LDL‐C from baseline.67 ODYSSEY FH I, FH II, and HIGH FH enrolled 841 patients with HeFH on a maximally tolerated statin dose, with or without other LMTs (Figure 5, Table 2). In these 3 studies, the reduction in LDL‐C from baseline to week 24, the primary efficacy endpoint, was 45.7% to 48.8%.52, 54
The RUTHERFORD phase 2 trial evaluated the efficacy and safety of evolocumab in patients with HeFH.60 The LSM reduction in LDL‐C ranged from 42.7% to 55.2% from baseline to week 12. The LSM reduction in LDL‐C in the RUTHERFORD‐2 phase 3 trial was similar at week 12 and ranged from 55.7% to 61.3%.62
Interestingly, a phase 1 study of alirocumab demonstrated similar reductions in LDL‐C in patients with HeFH and those with nonfamilial hypercholesterolemia.80 Similarly, PCSK9‐specific mAbs led to a comparable reduction of LDL‐C in patients with HeFH (28.9% to 67.9%) and in other populations with hypercholesterolemia excluding patients with HoFH (33.3% to 73.2%; Table 2 and discussed below).
High CVD Risk
Evaluation of the efficacy of alirocumab in patients at high risk for CVD (established CVD or CHD risk equivalents and hypercholesterolemia) who received a maximally tolerated statin dose (ODYSSEY COMBO I phase 3 trial) demonstrated that the LSM reduction in LDL‐C was 48.2% for patients treated with alirocumab.74 The 104‐week ODYSSEY COMBO II study produced similar results, with an LDL‐C reduction of 50.6% after treatment with alirocumab.73 Alirocumab was also shown to maintain consistent LDL‐C reductions over 52 weeks. The ongoing ODYSSEY CHOICE I study included patients with inadequately controlled hypercholesterolemia, moderate (Systematic Coronary Risk Evaluation [SCORE] 10‐year risk of fatal CVD ≥1% and <5%) to very high CV risk (high CV risk: no CHD/CVD with SCORE ≥5%, chronic kidney disease, diabetes, or HeFH; very high CV risk: documented CHD/CVD), or muscle‐related statin intolerance.81 Treatment with alirocumab led to 58.7% and 52.4% LSM reductions in LDL‐C level compared with placebo at week 24 among patients receiving a background statin therapy and no statins, respectively.
In the recently completed SPIRE‐HR trial, patients (N = 600) with hypercholesterolemia or mixed dyslipidemia who are at increased risk for CV events were randomized to bococizumab and a statin or placebo plus statin to assess the percentage change from baseline in LDL‐C levels at week 12.78 Approximately 690 patients will be recruited to SPIRE‐LL, a 52‐week, double‐blind, parallel‐group study to assess the efficacy, safety, and tolerability of bococizumab in patients with primary hypercholesterolemia or mixed dyslipidemia who are at high or very high risk for a CV event. SPIRE‐LDL (N ≈ 1932) will assess the efficacy, safety, and tolerability of bococizumab vs placebo in patients who are also treated with a statin and who have a fasting LDL‐C level >70 mg/dL, triglycerides ≤400 mg/dL, and high or very high risk for a CV event. The primary endpoint will be percentage change from baseline in LDL‐C at week 12 in each of these trials.
Additional Patient Populations
Statin Intolerance
The ability of PCSK9‐specific antibodies to lower LDL‐C in patients intolerant to statins was assessed in the ODYSSEY ALTERNATIVE and in 2 trials in the PROFICIO program, GAUSS and GAUSS‐2. Additionally, more than 500 patients will participate in the GAUSS‐3 trial of evolocumab and 150 in the SPIRE‐SI study of bococizumab.
ODYSSEY ALTERNATIVE compared alirocumab with ezetimibe in patients with a history of muscle symptoms related to at least 2 previous statins.59 This was the only study design to include a placebo run‐in period and a statin rechallenge to confirm statin intolerance. The run‐in period was completed by 87.0% of patients, with 15.9% of patients receiving alirocumab discontinuing due to skeletal muscle–related symptoms. Alirocumab treatment led to a 45.0% reduction in LDL‐C level from baseline to week 24.
The GAUSS phase 2 randomized, double‐blind, placebo‐ and ezetimibe‐controlled trial evaluated the effect of evolocumab on LDL‐C in patients intolerant to at least 1 statin.71 Treatment with evolocumab resulted in dose‐dependent reductions in LDL‐C, with the LSM reduction from baseline at week 12 ranging from 40.8% to 63.0%, depending on the background therapy. Of 160 randomized patients, 5 (including 3 receiving evolocumab) discontinued the study because of muscle‐related symptoms.
The 12‐week, double‐blind, phase 3 GAUSS‐2 trial evaluated evolocumab compared with ezetimibe in patients with hypercholesterolemia who are unable to tolerate effective statin doses.70 The percentage of patients who discontinued participation in the study because of muscle‐related symptoms was similar among patients receiving evolocumab (5%) and ezetimibe (6%). Evolocumab achieved LDL‐C reduction from baseline to week 12 of 52.6% to 56.1%.
Subtherapeutic Response to Statins
Patients not able to reach an LDL‐C goal despite taking commonly used statin doses participated in the ODYSSEY OPTIONS I and II trials. The trials compared the efficacy and safety of alirocumab in addition to atorvastatin (OPTIONS I) or rosuvastatin (OPTIONS II) with double‐dose statin therapy (ie, atorvastatin 40 mg/day doubled to 80 mg/day in OPTIONS I, and rosuvastatin 10 mg/day doubled to 20 mg/day or rosuvastatin 20 mg/day doubled to 40 mg/day in OPTIONS II), a switch to a higher‐intensity statin, or the addition of ezetimibe.76, 77 Eligible patients had a history of CVD and LDL‐C ≥70 mg/dL or risk factors for CVD and LDL‐C ≥100 mg/dL. The intent‐to‐treat analysis in OPTIONS I evaluated the percentage change in LDL‐C levels at week 24, with alirocumab plus atorvastatin associated with a 44.1% to 54.0% LSM decrease in LDL‐C, depending on the background statin and the comparator.76 Results for OPTIONS II revealed an LSM decrease in LDL‐C of 50.6% to 36.3% for alirocumab plus rosuvastatin, depending on the rosuvastatin dose (10 mg or 20 mg).77
Homozygous Familial Hypercholesterolemia
The TESLA open‐label and TESLA Part B randomized, double‐blind, placebo‐controlled phase 2 and phase 3 trials, respectively, evaluated the efficacy and safety of evolocumab for the treatment of patients with HoFH. Evaluation of the primary endpoint of percentage change in LDL‐C from baseline to week 12 in TESLA Part B revealed an LSM decrease in LDL‐C of 23.1% in patients in the evolocumab arm. In contrast to HeFH, where the type of LDL‐R gene mutation does not affect the patient's response to treatment with PCSK9 inhibitors, with HoFH the treatment efficacy is dependent on the residual LDL‐R activity classified as receptor‐negative (<2% residual activity) or receptor‐defective (2% to 25% residual activity).82
Nonfamilial Hypercholesterolemia
In a 12‐week phase 2 trial of patients with hypercholesterolemia, alirocumab plus atorvastatin achieved significant reductions in LDL‐C compared with placebo, with the LSM decreases in LDL‐C ranging from 39.6% to 72.4%, depending on the alirocumab dose.58 The LSM percentage reduction in LDL‐C following 8 weeks of Q2W treatment with alirocumab plus atorvastatin was 73.2%.65 Another phase 2 study of alirocumab that enrolled 13 patients with 4 different PCSK9 gene GOF mutations showed a 62.5% LSM reduction from baseline in LDL‐C level at week 2.83 When alirocumab was used as a monotherapy in the phase 3 ODYSSEY MONO trial (n = 103), hypercholesterolemic patients with a 10‐year risk for fatal CV events of ≥1% and <5% achieved an LSM reduction of LDL‐C level of 47.2%.66 Fourteen of 52 patients treated with alirocumab were uptitrated at week 12 to alirocumab 150 mg Q2W because their LDL‐C level was ≥70 mg/dL at week 8. In the recently completed ODYSSEY CHOICE II study, patients with hypercholesterolemia who were not receiving statins achieved a 56.4% LSM reduction in LDL‐C level from baseline at week 24 compared with patients in the placebo group.81
The MENDEL and MENDEL‐2 studies evaluated the efficacy and safety of evolocumab used as a monotherapy in patients with hypercholesterolemia (Table 2). In DESCARTES, LAPLACE‐TIMI 57, and LAPLACE‐2, evolocumab was used in combination with statin therapy in patients with hypercholesterolemia or mixed dyslipidemia.63, 84, 85 OSLER‐1 and OSLER‐2 were open‐label studies that enrolled patients with hypercholesterolemia, high CV risk, statin intolerance, or HeFH who participated in 1 of 12 phase 2 or phase 3 studies of evolocumab.55, 75 The regimen in the OSLER studies involved treatment with evolocumab combined with standard of care (SOC) therapy. The MENDEL‐2 phase 3 trial compared biweekly evolocumab 140 mg and monthly evolocumab 420 mg with ezetimibe or placebo in patients with hypercholesterolemia.56 At the 12‐week evaluation, the mean decrease in LDL‐C levels was 57.0% for biweekly evolocumab and 56.1% for monthly evolocumab.
In DESCARTES, a phase 3 trial, patients with hypercholesterolemia received evolocumab for 52 weeks after a run‐in period of 4 to 12 weeks of background LMT.84 In the analysis, the LSM reduction in LDL‐C in the evolocumab patients at 52 weeks was 46.7% to 54.7%, depending on the background therapy.
Recently, results of a pooled analysis focusing on elderly patients who received evolocumab and enrolled in phase 2 and phase 3 trials and their open‐label extension studies became available.86 Patients ≥65 years receiving evolocumab achieved an LDL‐C reduction of 58.4% to 62.9% at the mean of weeks 10 and 12, and the LDL‐C level in patients ≥75 years was reduced by 59.9% to 68.6%.
Additionally, Navarese and colleagues analyzed 24 phase 2 and phase 3 RCTs (N = 10,159) comparing treatment using PCSK9‐specific mAbs with treatment with no PCSK9 antibody in adults with hypercholesterolemia.87 The meta‐analysis showed a mean reduction of 47.5% (95%CI 25.35 to 69.64; P < .001) in LDL‐C level in patients receiving PCSK9‐specific antibodies compared with those in the control group.
Finally, a 24‐week, phase 2, randomized, double‐blind, placebo‐controlled trial investigated the efficacy and safety of bococizumab in 354 patients with hypercholesterolemia on stable statin therapy.72 Patients receiving bococizumab had a significant reduction in LDL‐C from baseline at week 12 of 19.5% to 52.0% (depending on the dosing regimen).
Studies With Long‐Term Follow‐Up
Studies with a long‐term follow‐up evaluating the efficacy, safety, and tolerability of PCSK9 antibodies in clinical development are currently under way. The recently completed ODYSSEY LONG TERM study (N = 2341) was a phase 3 clinical trial with the objective of comparing the safety, tolerability, and efficacy of alirocumab with placebo in patients at high or very high risk for CVD, including those with HeFH, CHD, or risk equivalent and LDL‐C ≥70 mg/dL while treated with the maximally tolerated dose of statin with or without other LMT.64 The primary week 24 efficacy evaluation revealed a mean LDL‐C decrease from baseline of 61.0% for patients receiving alirocumab. The difference between alirocumab and placebo in the LDL‐C level reduction at week 24 was similar in patients with HeFH and non‐FH. Additionally, ODYSSEY FH I, FH II, and HIGH FH studies (described in the section Heterozygous Familial Hypercholesterolemia) assessed the long‐term (78 weeks) efficacy and safety of alirocumab in patients with HeFH.52, 54
Two ongoing trials of evolocumab will provide additional information about the long‐term safety and efficacy of this agent for diverse patient populations. The GLAGOV phase 3 trial will determine the effects of evolocumab administered Q4W for 78 weeks on atherosclerotic disease burden (percent atheroma volume assessed by intravascular ultrasonography) in patients with CHD, a clinical indication for coronary catheterization, and LDL‐C level ≥80 mg/dL or in patients with additional CV risk factors and LDL‐C ≥60 mg/dL and <80 mg/dL.79 Patients who have HoFH or PCSK9 mutations with an LDL‐C that exceeds the US National Cholesterol Program Adult Treatment Panel III target or who are currently undergoing apheresis and who have previously participated in an evolocumab trial will be eligible for participation in TAUSSIG. The TAUSSIG trial will evaluate the long‐term efficacy and safety of evolocumab given Q2W or Q4W on LDL‐C levels at 5 years.79
Outcomes Studies
In a post hoc safety analysis evaluating the occurrence of major cardiovascular events (MACE; composite endpoint of CHD death, nonfatal myocardial infarction [MI], fatal and nonfatal ischemic stroke, and unstable angina [UA] requiring hospitalization) in ODYSSEY LONG TERM, the rate of MACE was 48% lower in patients receiving alirocumab than in patients in the placebo group (95%CI 0.31 to 0.90; nominal P = .02).64 In addition, an open‐label extension study will assess the long‐term efficacy, safety, and immunogenicity of alirocumab in patients who participated in ODYSSEY FH I, FH II, HIGH FH, and LONG TERM studies.78
The effect of evolocumab on the rate of CV events was analyzed at 1 year in the open‐label extension OSLER‐1 and OSLER‐2 studies.75 An exploratory composite safety analysis published recently demonstrated that patients who received evolocumab in addition to SOC therapy had a significantly lower rate of all CV events than those who received SOC therapy alone, with Kaplan‐Meier estimates of 0.95% and 2.18%, respectively (hazard ratio 0.47; 95%CI 0.28 to 0.78; P = .003).
In the meta‐analysis of 24 RCTs, the difference in CV mortality between patients receiving PCSK9‐specific antibodies and those in the control group was not statistically significant (OR 0.50; 95%CI 0.23 to 1.10; P = .084).87 However, overall mortality was significantly lower with the use of PCSK9‐specific antibodies (OR 0.45; 95%CI 0.23 to 0.86; P = .015).
Additionally, randomized controlled outcomes trials are under way for each of the PCSK9 antibodies. The ODYSSEY OUTCOMES trial is under way to evaluate the occurrence of MACE in more than 18,000 patients with an acute coronary syndrome event 1 to 12 months before enrollment and LDL‐C ≥70 mg/dL who receive alirocumab.78 The FOURIER phase 3 trial will determine the effect of biweekly or monthly administration of evolocumab with a statin compared with placebo and a statin, with a primary endpoint of MACE defined as CVD death, nonfatal MI, UA requiring hospitalization, stroke, or coronary revascularization. Patients eligible for participation will include those with clinically confirmed CVD, a high risk for a recurrent CV event, and LDL‐C ≥70 mg/dL or non‐HDL‐C ≥100 mg/dL. Finally, SPIRE‐1 and SPIRE‐2 will evaluate the ability of bococizumab to reduce the occurrence of MACE, including CV death, MI, stroke, and UA requiring urgent revascularization at 5 years, in high‐risk patients who are receiving background LMT.
Inflammation and PCSK9 Inhibitors
Even though hypercholesterolemia is a well‐recognized factor contributing to atherosclerosis, chronic low‐grade inflammation is involved at all stages of atherosclerotic plaque development.2, 88 In fact, a number of anti‐inflammatory therapies have been tested in patients with ASCVD.89 Recently, Sahebkar and colleagues analyzed 7 studies of alirocumab or evolocumab and showed that PCSK9 inhibitors do not have a significant effect on the level of C‐reactive protein (hs‐CRP), a commonly used inflammatory marker.90 However, patients participating in the analyzed studies may have had normal baseline levels of hs‐CRP. The effect of PCSK9 inhibitors on atherosclerosis, beyond LDL‐C lowering, especially on plaque stability and growth, should be further elucidated.
Safety of PCSK9‐Specific Monoclonal Antibodies
Concerns regarding the treatment with PCSK9‐specific antibodies are related to the effects of low levels of PCSK9 in the plasma and, consequently, potentially low LDL‐C levels, as well as the safety, tolerability, and long‐term CV outcomes of the therapy.
Safety of Low Levels of PCSK9 and LDL‐C
It has been suggested that PCSK9 inhibition may have a deleterious effect on adrenal function as a result of impaired delivery of lipoprotein cholesterol to the adrenal glands to support adrenal steroidogenesis that is essential for regulation of stress responses, blood pressure, homeostasis of electrolytes, and secondary sexual traits.91 This concern is of particular relevance when LDL‐C is reduced to extremely low levels. Evaluation of a 54‐year‐old male patient who carried a heterozygous LOF mutation in PCSK9 provided the opportunity to determine the impact of PCSK9 deficiency on adrenal function. He had no detectable plasma PCSK9, an LDL‐C level of 24 mg/dL, and an HDL‐C of 66 mg/dL. Baseline adrenal function was normal, with normal levels of cortisol, aldosterone, androgens, and plasma adrenocorticotropic hormone. The cortisol response to adrenocorticotropic hormone stimulation was normal, suggesting that genetic deficiencies in PCSK9 were not associated with abnormal adrenal function. Moreover, the safety of PCSK9 inhibition is evident in family members of participants in the Dallas Heart Study who had a single mutation in PCSK9. One family of interest included the proband with an LDL‐C level of 49 mg/dL who had a 32‐year‐old daughter with an LDL‐C level of 14 mg/dL. Genetic evaluation of the daughter revealed she had a premature stop codon in the protein transcript inherited from her mother and a second mutation from her father that deleted an arginine at codon 97. These 2 mutations resulted in total deficiency of PCSK9. The daughter was reported to have normal intelligence, motor skills, kidney and liver function, and blood pressure.92, 93
Safety Summary for PCSK9 Monoclonal Antibodies
According to the meta‐analysis by Navarese and colleagues, the use of PCSK9‐specific mAbs is associated with no increase in serious adverse events (AEs).87 To date, phase 2 and phase 3 clinical trials for alirocumab and evolocumab have demonstrated that both agents are well tolerated. Another recent meta‐analysis by Lipinski and colleagues indicated that the rate of neurocognitive events in 17 phase 2 and phase 3 trials of alirocumab or evolocumab was significantly elevated in patients treated with a PCSK9‐specific mAB compared with placebo (OR 2.34; 95%CI 1.11 to 4.93; P = .02); however, this observation was mostly supported by results of the ODYSSEY LONG TERM and OSLER‐2 trials.94
Phase 2 trials for alirocumab58, 65, 68 reported that AEs were similar in all treatment groups. Injection site reactions and pruritus were among the most frequently occurring AEs,58, 68 and 7 of 56 patients developed antibodies to alirocumab at low titer levels.65 In the phase 3 ODYSSEY ALTERNATIVE trial that included patients with a history of statin intolerance, the rate of skeletal muscle–related treatment‐emergent AEs was significantly lower for patients receiving alirocumab than for patients receiving atorvastatin.59
To assess the safety profile of alirocumab, a pooled analysis of 3752 patients with hypercholesterolemia on stable background statin therapy who participated in 4 phase 2 studies and 5 phase 3 studies of up to 78 weeks in duration was conducted.95 Adverse events were comparable between the alirocumab and placebo groups with the exception of local injection site reaction, which was reported in 7.2% and 5.1% of patients, respectively. No significant elevation in liver or muscle enzymes was reported. Additionally, the analysis showed that the rate of neurologic or skeletal muscle–related treatment‐emergent AEs or neurocognitive disorders was similar for patients receiving alirocumab and those in the placebo group.
A similar analysis, including 14 phase 2 and phase 3 trials, showed that alirocumab was associated with a favorable safety profile when LDL‐C was reduced to very low levels.96 Of 3340 patients receiving alirocumab, 23.8% achieved LDL‐C <25 mg/dL on at least 2 consecutive visits, and 8.6% achieved LDL‐C <15 mg/dL. No safety signals were observed, and treatment‐emergent AEs were generally similar among all patients receiving alirocumab, those achieving very low LDL‐C levels, and those in the control group.
A pooled analysis of 12 phase 2 and phase 3 studies and 2 extension studies continuing the parent studies97 demonstrated that evolocumab has a safety profile similar to that of the control. The rates of AEs and serious AEs (including myalgia) were comparable between the evolocumab and control arms in the parent studies and year 1 of the extension studies. Nasopharyngitis was the most common AE documented in the parent studies and was reported by 5.9% of patients who received evolocumab and by 4.8% of control patients. Injection site reactions were reported by 3.3% of patients receiving evolocumab and 3.0% of patients in the control group in the parent studies and by 4.1% in the extension studies. No PCSK9 neutralizing antibodies were detected. Increases in creatine kinase and liver enzyme levels were infrequent and occurred with similar rates in patients receiving evolocumab and in those in the control group.
A similar safety profile, comparable for the tested PCSK9 antibody and placebo, was demonstrated for bococizumab, with nasopharyngitis and upper respiratory tract infections as the most frequently reported AEs.72
Long‐term studies are necessary to verify whether prolonged treatment with fully human PCSK9 antibodies could lead to the development of immune responses.79 Data on the long‐term effects of anti‐PCSK9 antibodies on the major CVD endpoints and any significant safety signals in the hepatic, gastrointestinal, or musculoskeletal systems are expected to be available by 2017.
Summary and Conclusions
Despite evidence‐based recommendations for the management of hypercholesterolemia, treatment gaps persist. In addition, current treatment options are not uniformly effective and/or tolerable for all patients, including those at high risk for ASCVD and individuals with HeFH and HoFH. PCSK9 inhibition represents an important advance in the clinical management of hypercholesterolemia, especially for patients who have a subtherapeutic response to a maximally tolerated statin dose or who are intolerant to current pharmacologic interventions. Emerging research on PCSK9 mAbs suggests that these agents offer an effective treatment with a good safety profile for individuals with contraindications to statins, those who are statin intolerant, and those who fail to reach non‐HDL‐C and LDL‐C goals as commonly seen in patients with higher baseline cholesterol levels, such as individuals with HeFH and HoFH.
Sources of Support
Support for this educational article was provided by Regeneron Pharmaceuticals, Inc, Tarrytown, New York, and Sanofi US, Bridgewater, New Jersey. The authors were responsible for all content and editorial decisions and received no honoraria related to the development of this publication.
Declaration of Conflicting Interests
Matthew K. Ito is employed by Sanofi US. At the time of original manuscript development, Dr Ito was Professor of Pharmacy Practice with the Oregon State University/Oregon Health and Science University, College of Pharmacy. During the last 2 years, Dr Ito has received a research grant from Kowa and consulting fees from Pfizer. Raul D. Santos has received honoraria related to consulting and/or speaker activities during the last 2 years from Amgen, Akcea, AstraZeneca, Biolab, Boehringer Ingelheim, Cerenis, Eli Lilly, Genzyme, Kowa, Merck, Pfizer, Praxis, Regeneron Pharmaceuticals, Sanofi, Torrent, and Unilever.
Acknowledgments
Writing and editorial support in the preparation of this publication was provided by MicroMass Communications, Inc, Cary, North Carolina, and funded by Regeneron Pharmaceuticals, Inc, Tarrytown, New York, and Sanofi US, Bridgewater, New Jersey.
References
- 1. Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832‐1844. [DOI] [PubMed] [Google Scholar]
- 2. Jacobson TA, Ito MK, Maki KC, et al. National Lipid Association recommendations for patient‐centered management of dyslipidemia: Part 1—full report. J Clin Lipidol. 2015;9(2):129‐169. [DOI] [PubMed] [Google Scholar]
- 3. Boekholdt SM, Arsenault BJ, Mora S, et al. Association of LDL cholesterol, non‐HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta‐analysis. JAMA. 2012;307(12):1302‐1309. [DOI] [PubMed] [Google Scholar]
- 4. Emerging Risk Factors Collaboration . Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clarke R, Peden JF, Hopewell JC, et al. PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518‐2528. [DOI] [PubMed] [Google Scholar]
- 6. Cohen JD, Brinton EA, Ito MK, Jacobson TA. Understanding Statin Use in America and Gaps in Patient Education (USAGE): an internet‐based survey of 10,138 current and former statin users. J Clin Lipidol. 2012;6(3):208‐215. [DOI] [PubMed] [Google Scholar]
- 7. Schultz AB, Chen CY, Burton WN, Edington DW. The burden and management of dyslipidemia: practical issues. Popul Health Manag. 2012;15(5):302‐308. [DOI] [PubMed] [Google Scholar]
- 8. Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings—a cohort study. Ann Intern Med. 2013;158(7):526‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stroes ES, Thompson PD, Corsini A, et al. European Atherosclerosis Society Consensus Panel. Statin‐associated muscle symptoms: impact on statin therapy—European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J. 2015;36(17):1012‐1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guyton JR, Bays HE, Grundy SM, Jacobson TA. An assessment by the Statin Intolerance Panel: 2014 update. J Clin Lipidol. 2014;8(3 suppl):S72‐S81. [DOI] [PubMed] [Google Scholar]
- 11. Benn M, Watts GF, Tybjaerg‐Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol‐lowering medication. J Clin Endocrinol Metab. 2012;97(11):3956‐3964. [DOI] [PubMed] [Google Scholar]
- 12. Farnier M. PCSK9: from discovery to therapeutic applications. Arch Cardiovasc Dis. 2014;107(1):58‐66. [DOI] [PubMed] [Google Scholar]
- 13. Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154‐156. [DOI] [PubMed] [Google Scholar]
- 14. Abifadel M, Elbitar S, El Khoury P, et al. Living the PCSK9 adventure: from the identification of a new gene in familial hypercholesterolemia towards a potential new class of anticholesterol drugs. Curr Atheroscler Rep. 2014;16(9):439. [DOI] [PubMed] [Google Scholar]
- 15. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37(2):161‐165. [DOI] [PubMed] [Google Scholar]
- 16. Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264‐1272. [DOI] [PubMed] [Google Scholar]
- 17. Jacobson TA, Maki KC, Orringer CE, et al. National Lipid Association recommendations for patient‐centered management of dyslipidemia: Part 2. J Clin Lipidol. 2015;9(6):S1‐S122,e121. [DOI] [PubMed] [Google Scholar]
- 18. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25):2889‐2934. [DOI] [PubMed] [Google Scholar]
- 19. Lloyd‐Jones DM, Morris PB, Ballantyne CM, et al. 2016. ACC expert consensus decision pathway on the role of non‐statin therapies for LDL‐cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology task force on clinical expert consensus documents [published online March 28, 2016]. JACC. 2016. doi:10.1016/j.jacc.2016.03.519. [DOI] [PubMed]
- 20. Boekholdt SM, Hovingh GK, Mora S, et al. Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta‐analysis of statin trials. J Am Coll Cardiol. 2014;64(5):485‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cholesterol Treatment Trialists' (CTT) Collaboration . Efficacy and safety of LDL‐lowering therapy among men and women: meta‐analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015;385(9976):1397‐1405. [DOI] [PubMed] [Google Scholar]
- 22. Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372(25):2387‐2397. [DOI] [PubMed] [Google Scholar]
- 23. Toth PP, Foody JM, Tomassini JE, et al. Therapeutic practice patterns related to statin potency and ezetimibe/simvastatin combination therapies in lowering LDL‐C in patients with high‐risk cardiovascular disease. J Clin Lipidol. 2014;8(1):107‐116. [DOI] [PubMed] [Google Scholar]
- 24. Rosenson RS, Kent ST, Brown TM, et al. Underutilization of high‐intensity statin therapy after hospitalization for coronary heart disease. J Am Coll Cardiol. 2015;65(3):270‐277. [DOI] [PubMed] [Google Scholar]
- 25. Bell DA, Hooper AJ, Watts GF, Burnett JR. Mipomersen and other therapies for the treatment of severe familial hypercholesterolemia. Vasc Health Risk Manag. 2012;8:651‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lambert G, Sjouke B, Choque B, Kastelein JJP, Hovingh GK. The PCSK9 decade. J Lipid Res. 2012;53(12):2515‐2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Singh S, Bittner V. Familial hypercholesterolemia—epidemiology, diagnosis, and screening. Curr Atheroscler Rep. 2015;17(2):482. [DOI] [PubMed] [Google Scholar]
- 28. Brautbar A, Leary E, Rasmussen K, Wilson DP, Steiner RD, Virani S. Genetics of familial hypercholesterolemia. Curr Atheroscler Rep. 2015;17(4):1‐17. [DOI] [PubMed] [Google Scholar]
- 29. Nordestgaard BG, Chapman MJ, Humphries SE, et al. European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478‐3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sjouke B, Kusters DM, Kindt I, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype‐phenotype relationship, and clinical outcome. Eur Heart J. 2015;36(9):560‐565. [DOI] [PubMed] [Google Scholar]
- 31. Rader DJ, Kastelein JJP. Lomitapide and mipomersen: two first‐in‐class drugs for reducing low‐density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129(9):1022‐1032. [DOI] [PubMed] [Google Scholar]
- 32. Reiner Z. Resistance and intolerance to statins. Nutr Metab Cardiovasc Dis. 2014;24(10):1057‐1066. [DOI] [PubMed] [Google Scholar]
- 33. Ahmad Z. Statin intolerance. Am J Cardiol. 2014;113(10):1765‐1771. [DOI] [PubMed] [Google Scholar]
- 34. Bitzur R, Cohen H, Kamari Y, Harats D. Intolerance to statins: mechanisms and management. Diabetes Care. 2013;36(Suppl 2):S325‐S330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Needham M, Mastaglia FL. Statin myotoxicity: a review of genetic susceptibility factors. Neuromuscul Disord. 2014;24(1):4‐15. [DOI] [PubMed] [Google Scholar]
- 36. Harris LJ, Thapa R, Brown M, et al. Clinical and laboratory phenotype of patients experiencing statin intolerance attributable to myalgia. J Clin Lipidol. 2011;5(4):299‐307. [DOI] [PubMed] [Google Scholar]
- 37. Marais AD, Kim JB, Wasserman SM, Lambert G. PCSK9 inhibition in LDL cholesterol reduction: genetics and therapeutic implications of very low plasma lipoprotein levels. Pharmacol Ther. 2015;145:58‐66. [DOI] [PubMed] [Google Scholar]
- 38. Stein EA, Swergold GD. Potential of proprotein convertase subtilisin/kexin type 9 based therapeutics. Curr Atheroscler Rep. 2013;15(3):310. [DOI] [PubMed] [Google Scholar]
- 39. Farnier M. PCSK9 inhibitors. Curr Opin Lipidol. 2013;24(3):251‐258. [DOI] [PubMed] [Google Scholar]
- 40. Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis‐regulated convertase 1 (NARC‐1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. 2003;100(3):928‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143‐3421. [PubMed] [Google Scholar]
- 42. Zhang Y, Eigenbrot C, Zhou L, et al. Identification of a small peptide that inhibits PCSK9 protein binding to the low density lipoprotein receptor. J Biol Chem. 2014;289(2):942‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sanofi‐Aventis US LLC and Regeneron Pharmaceuticals, Inc. Praluent [package insert]. Bridgewater, NJ, and Tarrytown, NY; 2015. [Google Scholar]
- 44. Amgen Inc. Repatha [package insert]. Thousand Oaks, CA; 2015. [Google Scholar]
- 45. Foltz IN, Karow M, Wasserman SM. Evolution and emergence of therapeutic monoclonal antibodies: what cardiologists need to know. Circulation. 2013;127(22):2222‐2230. [DOI] [PubMed] [Google Scholar]
- 46. Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9(10):767‐774. [DOI] [PubMed] [Google Scholar]
- 47. Catapano AL, Papadopoulos N. The safety of therapeutic monoclonal antibodies: implications for cardiovascular disease and targeting the PCSK9 pathway. Atherosclerosis. 2013;228(1):18‐28. [DOI] [PubMed] [Google Scholar]
- 48. Pucca MB, Bertolini TB, Barbosa JE, Galina SVR, Porto GS. Therapeutic monoclonal antibodies: scFv patents as a marker of a new class of potential biopharmaceuticals. Braz J Pharm Sci. 2011;47(1):31‐39. [Google Scholar]
- 49. Mould DR. Using pharmacometrics in the development of theraputic biological agents In: Ette EI, Williams PJ, eds. Pharmacometrics: The Science of Quantitative Pharmacology. Hoboken, NJ: John Wiley & Sons; 2007. [Google Scholar]
- 50. Ritter JM. Cardiac safety, drug‐induced QT prolongation and torsade de pointes (TdP). Br J Clin Pharmacol. 2012;73(3):331‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blom DJ, Hala T, Bolognese M, et al. A 52‐week placebo‐controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370(19):1809‐1819. [DOI] [PubMed] [Google Scholar]
- 52. Ginsberg HN, Rader DJ, Raal FJ, et al. ODYSSEY HIGH FH: efficacy and safety of alirocumab in patients with severe heterozygous familial hypercholesterolemia [abstract]. Circulation. 2014;130(23):2119. [Google Scholar]
- 53. Giugliano RP, Desai NR, Kohli P, et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE‐TIMI 57): a randomised, placebo‐controlled, dose‐ranging, phase 2 study. Lancet. 2012;380(9858):2007‐2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kastelein JJ, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. 2015;36(43):2996‐3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Koren MJ, Giugliano RP, Raal FJ, et al. Efficacy and safety of longer‐term administration of evolocumab (AMG 145) in patients with hypercholesterolemia: 52‐week results from the Open‐Label Study of Long‐Term Evaluation Against LDL‐C (OSLER) randomized trial. Circulation. 2014;129(2):234‐243. [DOI] [PubMed] [Google Scholar]
- 56. Koren MJ, Lundqvist P, Bolognese M, et al. Anti‐PCSK9 monotherapy for hypercholesterolemia: the MENDEL‐2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2531‐2540. [DOI] [PubMed] [Google Scholar]
- 57. Koren MJ, Scott R, Kim JB, et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomised, double‐blind, placebo‐controlled, phase 2 study. Lancet. 2012;380(9858):1995‐2006. [DOI] [PubMed] [Google Scholar]
- 58. McKenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand AC, Stein EA. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59(25):2344‐2353. [DOI] [PubMed] [Google Scholar]
- 59. Moriarty PM, Thompson PD, Cannon CP, et al. ODYSSEY ALTERNATIVE Investigators. Efficacy and safety of alirocumab vs ezetimibe in statin‐intolerant patients, with a statin rechallenge arm: the ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol. 2015;9(6):758‐769. [DOI] [PubMed] [Google Scholar]
- 60. Raal F, Scott R, Somaratne R, et al. Low‐density lipoprotein cholesterol‐lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL‐C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation. 2012;126(20):2408‐2417. [DOI] [PubMed] [Google Scholar]
- 61. Raal FJ, Honarpour N, Blom DJ, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double‐blind, placebo‐controlled trial. Lancet. 2015;385(9965):341‐350. [DOI] [PubMed] [Google Scholar]
- 62. Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD‐2): a randomised, double‐blind, placebo‐controlled trial. Lancet. 2015;385(9965):331‐340. [DOI] [PubMed] [Google Scholar]
- 63. Robinson JG, Nedergaard BS, Rogers WJ, et al. Effect of evolocumab or ezetimibe added to moderate‐ or high‐intensity statin therapy on LDL‐C lowering in patients with hypercholesterolemia: the LAPLACE‐2 randomized clinical trial. JAMA. 2014;311(18):1870‐1882. [DOI] [PubMed] [Google Scholar]
- 64. Robinson JG, Farnier M, Krempf M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1489‐1499. [DOI] [PubMed] [Google Scholar]
- 65. Roth EM, McKenney JM, Hanotin C, Asset G, Stein EA. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367(20):1891‐1900. [DOI] [PubMed] [Google Scholar]
- 66. Roth EM, Taskinen MR, Ginsberg HN, et al. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: results of a 24 week, double‐blind, randomized phase 3 trial. Int J Cardiol. 2014;176(1):55‐61. [DOI] [PubMed] [Google Scholar]
- 67. Stein EA, Bergeron J, Gaudet D, et al. One year open‐label treatment with alirocumab 150 mg every two weeks in heterozygous familial hypercholesterolemic patients [abstract]. J Am Coll Cardiol. 2014;63(12_S):A1371. [Google Scholar]
- 68. Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low‐density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380(9836):29‐36. [DOI] [PubMed] [Google Scholar]
- 69. Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128(19):2113‐2120. [DOI] [PubMed] [Google Scholar]
- 70. Stroes E, Colquhoun D, Sullivan D, et al. Anti‐PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS‐2 randomized, placebo‐controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2541‐2548. [DOI] [PubMed] [Google Scholar]
- 71. Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low‐density lipoprotein cholesterol levels in statin‐intolerant patients: the GAUSS randomized trial. JAMA. 2012;308(23):2497‐2506. [DOI] [PubMed] [Google Scholar]
- 72. Ballantyne CM, Neutel J, Cropp A, et al. Results of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, from a randomized, placebo‐controlled, dose‐ranging study in statin‐treated subjects with hypercholesterolemia. Am J Cardiol. 2015;115(9):1212‐1221. [DOI] [PubMed] [Google Scholar]
- 73. Cannon CP, Cariou B, Blom D, et al. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur Heart J. 2015;36(19):1186‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of the PCSK9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: the ODYSSEY COMBO I study. Am Heart J. 2015;169(6):906‐915. [DOI] [PubMed] [Google Scholar]
- 75. Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500‐1509. [DOI] [PubMed] [Google Scholar]
- 76. Bays H, Gaudet D, Weiss R, et al. Alirocumab as add‐on to atorvastatin versus other lipid treatment strategies: ODYSSEY OPTIONS I randomized trial. J Clin Endocrinol Metab. 2015;100(8):3140‐3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Farnier M, Jones P, Severance R, et al. Efficacy and safety of adding alirocumab to rosuvastatin versus adding ezetimibe or doubling the rosuvastatin dose in high cardiovascular‐risk patients: The ODYSSEY OPTIONS II randomized trial. Atherosclerosis. 2016;244:138‐146. [DOI] [PubMed] [Google Scholar]
- 78. Clinical Trials . www.clinicaltrials.gov. Accessed April 26, 2016.
- 79. Dadu RT, Ballantyne CM. Lipid lowering with PCSK9 inhibitors. Nat Rev Cardiol. 2014;11(10):563‐575. [DOI] [PubMed] [Google Scholar]
- 80. Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366(12):1108‐1118. [DOI] [PubMed] [Google Scholar]
- 81. Stroes E, Guyton JR, Farnier M, et al. Efficacy and safety of alirocumab 150 mg and 300 mg every 4 weeks in patients with poorly controlled hypercholesterolemia: the ODYSSEY CHOICE I and CHOICE II studies. Poster presented at: American College of Cardiology Congress, March 14‐16, 2015. San Diego, CA.
- 82. Santos RD, Watts GF. Familial hypercholesterolaemia: PCSK9 inhibitors are coming. Lancet. 2015;385(9965):307‐310. [DOI] [PubMed] [Google Scholar]
- 83. Hopkins PN, Defesche J, Fouchier SW, et al. Characterization of autosomal dominant hypercholesterolemia caused by PCSK9 gain of function mutations and its specific treatment with alirocumab, a PCSK9 monoclonal antibody. Circ Cardiovasc Genet. 2015;8(6):823‐831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Blom DJ, Hala T, Bolognese M, et al. A 52‐week placebo‐controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370(19):1809‐1819. [DOI] [PubMed] [Google Scholar]
- 85. Giugliano RP, Desai NR, Kohli P, et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE‐TIMI 57): a randomised, placebo‐controlled, dose‐ranging, phase 2 study. Lancet. 2012;380(9858):2007‐2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Koren M, Rosenson R, Khan B, et al. LDL cholesterol reduction in elderly patients with the PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of 1779 patients in phase 2, 3 and open label extension studies [abstract]. J Am Coll Cardiol. 2015;65(10S):A1366. [Google Scholar]
- 87. Navarese EP, Kolodziejczak M, Schulze V, et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta‐analysis. Ann Intern Med. 2015;163(1):40‐51. [DOI] [PubMed] [Google Scholar]
- 88. Golia E, Limongelli G, Natale F, et al. Inflammation and cardiovascular disease: from pathogenesis to therapeutic target. Curr Atheroscler Rep. 2014;16(9):435. [DOI] [PubMed] [Google Scholar]
- 89. Verweij SL, van der Valk FM, Stroes ES. Novel directions in inflammation as a therapeutic target in atherosclerosis. Curr Opin Lipidol. 2015;26(6):580‐585. [DOI] [PubMed] [Google Scholar]
- 90. Sahebkar A, Di Giosia P, Stamerra CA, et al. Effect of monoclonal antibodies to PCSK9 on high‐sensitivity C‐reactive protein levels: a meta‐analysis of 16 randomized controlled treatment arms. Br J Clin Pharmacol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cariou B, Benoit I, Le May C. Preserved adrenal function in fully PCSK9‐deficient subject. Int J Cardiol. 2014;176(2):499‐500. [DOI] [PubMed] [Google Scholar]
- 92. Cohen JC. Emerging LDL therapies: using human genetics to discover new therapeutic targets for plasma lipids. J Clin Lipidol. 2013;7(3 suppl):S1‐S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhao Z, Tuakli‐Wosornu Y, Lagace TA, et al. Molecular characterization of loss‐of‐function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79(3):514‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lipinski MJ, Benedetto U, Escarcega RO, et al. The impact of proprotein convertase subtilisin‐kexin type 9 serine protease inhibitors on lipid levels and outcomes in patients with primary hypercholesterolaemia: a network meta–analysis. Eur Heart J. 2016;37(6):536‐545. [DOI] [PubMed] [Google Scholar]
- 95. Jones PH, Bays H, Chaudhari U, et al. Pooled safety and adverse events in nine randomized, placebo‐controlled, phase 2 and 3 clinical trials of alirocumab [abstract]. J Am Coll Cardiol. 2015;65(10_S):A1363. [Google Scholar]
- 96. Robinson J, Farnier M, Chaudhari U, et al. Adverse events in patients with low‐density lipoprotein cholesterol levels <25 or <15 mg/dL on at least two consecutive visits in fourteen randomized, controlled, clinical trials of alirocumab [abstract]. J Am Coll Cardiol. 2015;65(10_S):A1350. [Google Scholar]
- 97. Toth PP, Sattar N, Genest J, et al. A comprehensive safety analysis of 6026 patients from phase 2 and 3 short and long term clinical trials with evolocumab (AMG 145) [abstract]. J Am Coll Cardiol. 2015;65(10_S):A1351. [Google Scholar]