

Abstract
Here we report the development of a C(sp3)–H cross-coupling platform enabled by the catalytic generation of chlorine radicals by nickel and photoredox catalysis. Aryl chlorides serve as both cross-coupling partners and the chlorine radical source for the α-oxy C(sp3)–H arylation of cyclic and acyclic ethers. Mechanistic studies suggest that photolysis of a Ni(III) aryl chloride intermediate, generated by photoredox-mediated single-electron oxidation, leads to elimination of a chlorine radical in what amounts to the sequential capture of two photons. Arylations of a benzylic C(sp3)–H bond of toluene and a completely unactivated C(sp3)–H bond of cyclohexane demonstrate the broad implications of this manifold for accomplishing numerous C(sp3)–H bond functionalizations under exceptionally mild conditions.
Free-radical halogenation of alkanes is one of the oldest and most well-studied reactions in organic chemistry.1,2 Though alkanes are notoriously unreactive compounds, halogen radicals, particularly chlorine radicals, can activate virtually any C(sp3)–H bond at room temperature by hydrogen atom abstraction.2 Interfacing the capability of chlorine atoms to activate C(sp3)–H bonds with catalytic functionalization reactions of the resulting alkyl radicals could allow for the development of a wide range of elusive bond constructions, enabling the functionalization of both complex molecules and feedstock hydrocarbons.
Over the past decade, nickel catalysis has emerged as a powerful platform for C(sp3)–C bond formation.3,4 Recently, it has been shown that Ni catalysts can intercept and functionalize alkyl radicals generated by distinct pathways, and that this approach can enable the development of C(sp3)–C bond-forming reactions utilizing starting materials otherwise unlikely to engage in cross coupling.3,5–7 In this context, we questioned whether hydrogen atom abstraction by chlorine radicals could be integrated with Ni-catalyzed alkyl cross coupling. Such a manifold would offer the possibility of achieving directing group-free C–H, C–X cross coupling with even the strongest C(sp3)–H bonds (Figure 1A).
Figure 1.
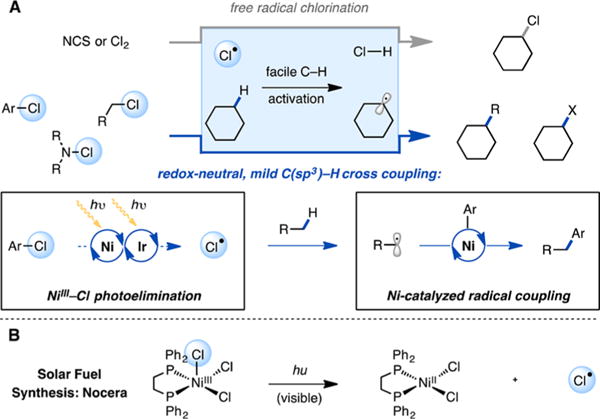
(A) Interfacing chlorine radical C(sp3)–H bond activation and cross coupling. (B) Chlorine radical generation via photolysis of high-valent nickel chloride complexes.
Successful realization of this goal, however, would require the identification of a mechanism for the catalytic generation of chlorine radicals under mild conditions from a stable precursor. Chlorine radicals are usually derived from highly reactive reagents such as chlorine gas or N-chlorosuccinimide that can competitively oxidize a Ni catalyst, chlorinate alkyl radical intermediates, and react with desirable functional groups present in organic substrates. Despite the positive attributes of free radical halogenation, these characteristic challenges would impede the development of a general C(sp3)–H, C–X cross-coupling platform.
We recognized that it should be possible to address these challenges by using the Ni cross-coupling catalyst to generate chlorine radicals from chemically inert chloride precursors. It has been shown that visible light excitation of high oxidation state transition metal halides can lead to halogen radical formation via dissociation from a charge-transfer excited state.8 Specifically, the Nocera laboratory recently reported the reversible, visible-light-promoted elimination of chlorine atoms from Ni(III) trichloride complexes (Figure 1B).9,10 We reasoned by analogy that photolysis of a Ni(III) aryl chloride species, generated by single-electron oxidation of a typical Ni(II) intermediate in cross coupling, might allow for the catalytic generation of chlorine atoms. Taken together with the ability of Ni(II) to accept alkyl radicals, it was hypothesized that photocatalytically generated chlorine atoms could participate in C–H abstraction to generate a substrate-derived alkyl radical poised to rebound to the Ni center in a manner compatible with Ni-catalyzed cross coupling. A photoredox catalyst was envisioned to promote the necessary single-electron oxidation and reduction of the Ni catalyst to facilitate the overall redox-neutral process. Importantly, whereas most photoredox catalysts do not have sufficient energy to oxidize chloride anion (E° = 2.03 V vs SCE in MeCN),11 this strategy offers a visible-light-driven mechanism for halogen radical formation enabled by the sequential capture of two photons.
As a model coupling reaction, we examined the direct C–H functionalization of ethers to generate benzylic ethers,12 a valuable pharmacophore in drug development. We recently reported Ni/photoredox-catalyzed functionalization of anilines via a photoredox-driven oxidation/α-C–H deprotonation sequence.6 Ethers, however, are not susceptible to this activation mechanism, given their high oxidation potential (tetrahydrofuran (THF) solvent oxidation onset potential E = 1.75 V vs SCE).13 We envisioned instead that oxidative addition of Ni(0) (1) into an aryl chloride would produce Ni(II) aryl chloride intermediate 2 (Figure 2). Concurrently, irradiation of iridium(III) photocatalyst 3 would produce a highly oxidizing long-lived triplet excited *Ir(III) state (τ0 = 2.3 μs, *E1/2 = 1.21 V vs SCE in MeCN) (4) which would be capable of oxidizing 2 (EP = 0.85 V vs SCE in THF, Figure S33) to produce fleeting Ni(III) intermediate 5.14 Photolysis of 5 would result in the generation of a chlorine atom and Ni(II) species 6, and the resulting chlorine radical would rapidly abstract a hydrogen atom from THF (H–Cl BDE = 102 kcal mol−1, THF BDE = 92 kcal mol−1). Rebound of the resulting carbon-centered radical to 6 would produce Ni(III) species 7. Subsequent reductive elimination would forge the desired C(sp3)–H cross-coupling product. Finally, single-electron-transfer (SET) reduction of the resulting Ni(I) intermediate 8 (EP = −1.17 V vs SCE in THF, Figure S32) by highly reducing Ir(II) species 9 (E1/2 = −1.37 V vs SCE in MeCN) would regenerate both the Ni(0) and Ir(III) catalysts.14
Figure 2.
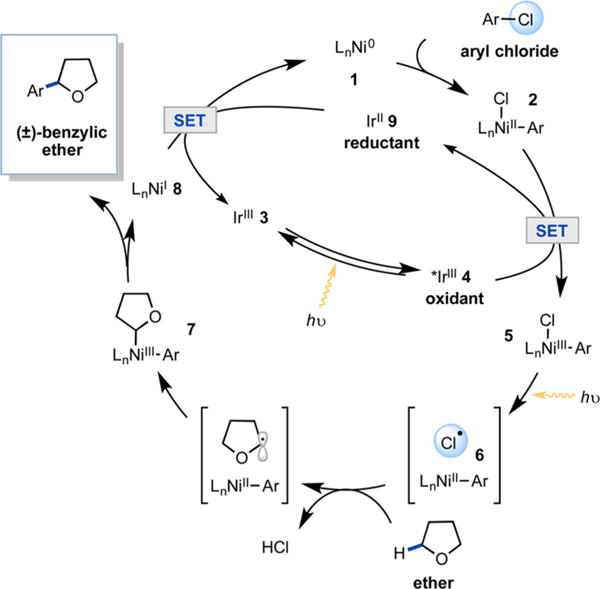
Proposed catalytic cycle. 3 = Ir[dF(CF3)ppy]2(dtbbpy)PF6; dF(CF3)ppy = 2 (2,4-difluorophenyl)-5-(trifluoromethyl)pyridine; dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine.
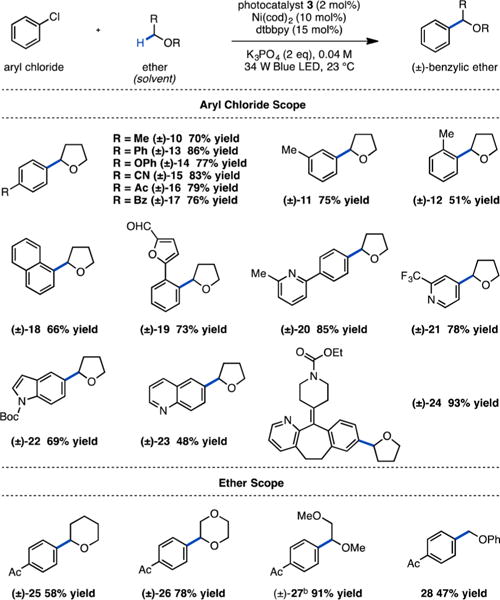
With this mechanistic hypothesis in hand, we examined the proposed α-oxy functionalization with aryl chlorides and THF as solvent. We were pleased to find that the combination of Ni(cod)2 (cod = 1,5-cyclooctadiene), dtbbpy, and Ir[dF(CF3)-ppy]2 (dtbbpy)PF6 (3) with 2 equiv of potassium phosphate under irradiation with blue LEDs enabled the cross coupling of numerous aryl chlorides with THF in good to excellent yields at room temperature (Table 1). Optimization studies revealed that in the absence of Ni catalyst, light, or photoredox catalyst, no product is formed. Furthermore, the reaction takes place in the absence of base, albeit with reduced reaction efficiency (Table S1). Notably, this transformation employs abundant, low cost aryl chlorides, which have seen little use in the emerging field of metallaphotoredox catalysis. A variety of electron-neutral (10–13), -rich (14), and -poor (15–17) chloroarenes as well as substrates bearing steric encumbrance (12, 18, 19) underwent efficient coupling to give the corresponding α-arylated products. Gratifyingly, aryl chlorides with diverse functionality including nitrile 15, ketones (16, 17), aldehyde 19, amides (22, 24), and alkene 24 were well tolerated, a showcase of the mild reaction conditions. Finally, medicinally relevant heteroaryl chlorides derived from pyridine (21), indole (22), and quinoline (23) and the complex aryl chloride loratadine (24) performed well in the reaction, demonstrating the potential applicability of this system to late-stage coupling.
Table 1.
Scope Studiesa
|
Yields are isolated yields. Reaction times varied between 36 and 75 h; see SI for individual substrates. bMixture of regioisomers.
We next evaluated ether coupling partners in the C–H functionalization reaction. We were pleased to find that, along with THF, other cyclic and acyclic ethers performed well under identical conditions, delivering both secondary (25, 26, 27) and primary (27, 28) C–H functionalization products. Interestingly, the reaction efficiency with different ether substrates trends with radical nucleophilicity, suggesting that polar effects may play an important role in these reactions. In addition, functionalization of 1,2-dimethoxyethane occurred at both primary and secondary carbons, giving the branched product preferentially (1.35:1 branched:linear) in a combined 91% yield, indicating moderate selectivity for the weaker C–H bond. Notably, this method produces benzylic ethers under extraordinarily mild conditions, a technology that could be of great utility for modulating small molecule hydrophobicity, an important consideration in drug design.15
We next turned our attention to mechanistic experiments designed to interrogate the halogen photoelimination and hydrogen atom abstraction hypothesis, beginning with an evaluation of the role of chloride. For the proposed mechanism, we anticipated that aryl iodides would not be competent coupling partners because the weak H–I bond (BDE = 71 kcal mol−1) renders an iodine radical incapable of abstracting a hydrogen atom from THF. Indeed, the yield of the coupling reaction showed a strong dependence on the halide identity: aryl iodide 29-I performed poorly, giving only trace THF product (10) (Figure 3A, entries 1–3). Nevertheless, the generation of even trace product 10 with 29-I suggests that an alternative mechanism could be operative. In seminal studies by Kochi and co-workers, it was shown that oxidative addition of aryl halides to Ni(0) proceeds via SET to generate an aryl radical anion intermediate that can collapse to generate a Ni(II) aryl halide or fragment toward a Ni(I) halide and aryl radical.16 To account for product formation with aryl iodide, we hypothesized that the aryl radical resulting from cage escape could abstract a hydrogen atom from THF (k = 4.8 × 106 M−1 s−1).17 If the THF radical intercepted a Ni(II) aryl halide complex, a false positive could in principle be observed in up to 50% yield. To test this, we evaluated reactions with aryl halides (30-Cl, -Br, and -I) and THF-d8 for deuterodehalogenated arene (31), a requisite product of the aryl radical abstraction pathway (Figure 3B). Notably, aryl iodide (30-I) delivered 66% yield of deuterodeha-logenated 31 while ≤3% yield of 31 was observed with 30-Cl and 30-Br. This result indicates that hydrogen atom abstraction by aryl radicals is significant only with aryl iodides and is fully consistent with chlorine radical generation in reactions of aryl chlorides.
Figure 3.
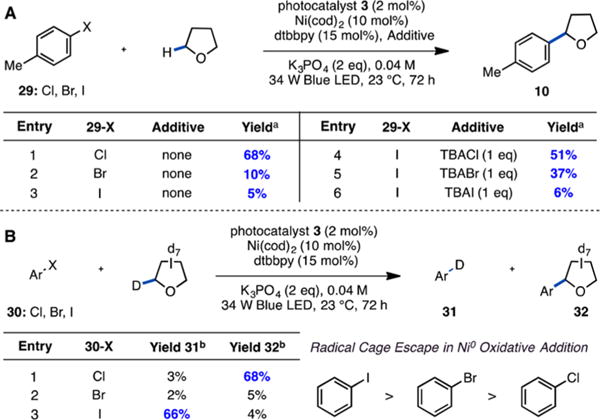
(A) Halide additive studies. See Table S6 for additional experiments. (B) Deuterium labeling experiments. Ar = 2-(4-halophenyl)-6-methyl-pyridine. aYields determined by GC-FID or 1H NMR using 1-fluoronaphthalene as an external standard. bYields determined by 2H NMR using DMF-d7 as an external standard.
It was hypothesized that the addition of chloride salts might resurrect the reaction of aryl iodides by halide exchange on Ni. Indeed, the reaction of 29-I containing 1 equiv of TBACl (TBA = tetrabutylammonium) showed a dramatic improvement in reaction efficiency, demonstrating the critical role of chloride (Figure 3A, entries 4–6). This observation indicates the potential to utilize a broad range of electrophiles that do not contain chloride such as aryl triflates (see Table S6). Monitoring the reaction of 29-I with TBACl over time it was observed that the generation of approximately equimolar aryl chloride (29-Cl) accompanied product formation (see Figures S3–S5). A Ni-catalyzed halogen exchange mechanism, through either a common catalytic intermediate or generation and subsequent consumption of aryl chloride, readily accommodates this observation.18 To our knowledge, no previously reported halogen exchange occurs under such mild conditions.19,20 Intriguingly, the reaction of 29-I with TBACl is complete after only 3 h, significantly faster than the reaction of 29-Cl. The improved reaction rate offers one pathway for further optimization (see Supporting Information (SI), section VII, for a discussion of mechanistic implications).
We next sought to gain insight into the role of photoredox catalyst 3. According to our mechanistic hypothesis, the excited photocatalyst serves as an oxidant to generate a transient Ni(III) complex from Ni(II) oxidative adduct 2. To test this, we synthesized Ni(II) complex 33 (Ar = 2-methylphenyl) (Figure 4A). Complex 33 was stoichiometrically competent in the presence of light and photocatalyst, and its first oxidation potential (EP = 0.85 V vs SCE in THF), as measured by cyclic voltammetry, indicates that *Ir(III) 4 should be a suitable oxidant. Indeed, Stern–Volmer quenching studies indicate that 33 is not only capable of quenching *Ir(III) 4 competitively, it is likely the species responsible for quenching in the reaction mixture (see SI for additional data, controls, and statistics).
Figure 4.
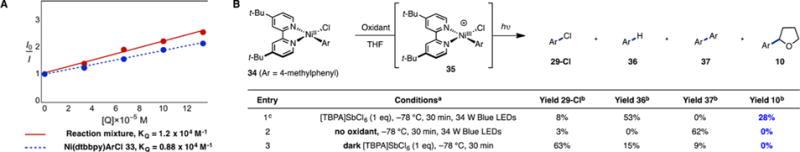
(A) Emission quenching of *Ir(III) 4. Ni complex 33 (Ar = 2-methylphenyl) gives a similar quenching rate to the reaction mixture consisting of Ni(dtbbpy)(cod) and 2-chlorotoluene. Additional data, controls, and statistics may be found in the SI. (B) Stoichiometric studies with Ni(II) complex 34 (Ar = 4-methylphenyl). See Table S13 for additional experiments. adtbbpy (1 equiv), K3PO4 (2 equiv), 2 mM THF. bYields determined by GC-FID using 1-fluoronaphthalene as an external standard. c1 mM THF.
In a stoichiometric sense, it should therefore be possible to use a non-photoredox oxidant to generate transient Ni(III) in order to separate the two roles of light in the oxidation and halogen elimination. Indeed, reaction of 34 (Ar = 4-methylphenyl) with 1 equiv of the single-electron oxidant [TBPA]SbCl6 (tris(4-bromophenyl)ammoniumyl hexachloroantimonate, E°′ = 1.16 V vs SCE in dichloromethane) under irradiation with 34 W blue LEDs gave 10 in 28% yield (Figure 4B; see Table S13 for additional data and controls).21 Importantly, in the absence of light or oxidant, 10 was not observed. These data demonstrate that, from the isolable Ni(II) aryl chloride complex, oxidant, and light are necessary. The mass balance of this key experiment can be accounted for by competitive reaction pathways such as formation of aryl chloride 29-Cl, likely the result of reductive elimination from 35 and consistent with halogen exchange experiments.22
We recognize this system operates in a distinct ligand field from the recently reported Ni(III) trihalide complexes.9,10 Though the structure of the proposed Ni(III) intermediate is unknown, preliminary DFT studies suggest that photoelimination even from a distorted square planar Ni(III) complex is feasible (see SI for computational studies). The calculated absorption spectrum for four coordinate [Ni(dtbbpy)(Ph)Cl]+ shows high-energy features (∼400–500 nm) that arise from Ni–Cl σ→σ* charge-transfer transitions (Figures S38 and S39). Taken together with the computed Ni–Cl bond dissociation free energy of 47 kcal mol−1, these states are expected to be dissociative.
Overall, these preliminary studies are consistent with a mechanism in which photolysis of a Ni(III) aryl chloride intermediate, generated by single-electron oxidation, leads to elimination of a chlorine radical capable of activating C(sp3)–H bonds by abstraction. Chlorine radicals are known to form stabilized adducts with aromatic functional groups.9,10 Therefore, we reasoned that if benzene were utilized as a solvent, we could promote the desired photoelimination and simultaneously prevent competitive solvent C(sp3)–H abstraction. We recognized that the ability to use an inert solvent and limiting C–H coupling partner would be highly desirable, facilitating the application of this manifold to a broad range of C–H coupling partners under general reaction conditions. Gratifyingly, with benzene as a solvent, the α-arylation reaction could be carried out with only 10 equiv of THF to give 16 in 71% yield, a 30-fold reduction in THF (Figure 5; see Table S3 for experiments with 1 and 5 equiv of THF). In addition, the benzylic C(sp3)–H arylation of toluene could be carried out to give 38 in 60% unoptimized yield. We were also pleased to find that employing benzene as solvent, C(sp3)–H arylation of the unactivated alkane cyclohexane could be accomplished to give 39 in 41% unoptimized yield. These demonstrations highlight the ability of this novel reaction platform to accomplish C(sp3)–H functionalization under mild conditions, enabled by catalytic access to the chemistry of chlorine radicals.
Figure 5.
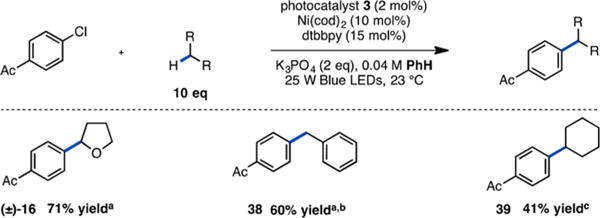
Preliminary demonstration of reaction generality. aYield determined by 1H NMR using 1-fluoronaphthalene as an external standard. bReaction was carried out using toluene as a solvent. cYield determined by GC-FID using 1-fluoronaphthalene as an external standard.
Supplementary Material
Acknowledgments
Financial support was provided by NIGMS (R01 GM100985). We thank Kevin Wu, Marherita Maiuri, Daniel Oblinsky, Andrew Proppe, Hilary Shi, and Robert Knowles for helpful discussions. We thank Máté Bezdek of the Chirik laboratory for the kind donation of FcBArF4 and Eric Webb for the preparation of 30-I. The authors thank G. Molander and co-workers for graciously offering to concurrently publish a related study.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b08397.
Procedures, experimental data, emission quenching data and statistics, computational data, and spectroscopic data, including Tables S1–S18 and Figures S1–S39 (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Fokin AA, Schreiner PR. Chem Rev. 2002;102:1551–1594. doi: 10.1021/cr000453m. [DOI] [PubMed] [Google Scholar]
- 2.Carey FA, Sundberg RJ. Advanced Organic Chemistry Part A: Structure and Mechanisms. Springer; New York: 2007. pp. 965–1063. [Google Scholar]
- 3.Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299–309. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ananikov VP. ACS Catal. 2015;5:1964–1971. [Google Scholar]
- 5.(a) Tellis JC, Primer DN, Molander GA. Science. 2014;345:433–436. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weix DJ. Acc Chem Res. 2015;48:1767–1775. doi: 10.1021/acs.accounts.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cornella J, Edwards JT, Qin T, Kawamura S, Wang J, Pan C, Gianatassio R, Schmidt M, Eastgate MD, Baran PS. J Am Chem Soc. 2016;138:2174–2177. doi: 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shaw MH, Twilton J, MacMillan DWC. J Org Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437–440. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Joe CL, Doyle AG. Angew Chem Int Ed. 2016;55:4040–4043. doi: 10.1002/anie.201511438. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ahneman DT, Doyle AG. Chem Sci. 2016 doi: 10.1039/c6sc02815b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC. Science. 2016;352:1304–1308. doi: 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esswein AJ, Nocera DG. Chem Rev. 2007;107:4022–4047. doi: 10.1021/cr050193e. [DOI] [PubMed] [Google Scholar]
- 9.Hwang SJ, Powers DC, Maher AG, Anderson BL, Hadt RG, Zheng SL, Chen S, Nocera DG. J Am Chem Soc. 2015;137:6472–6475. doi: 10.1021/jacs.5b03192. [DOI] [PubMed] [Google Scholar]
- 10.Hwang SJ, Anderson BL, Powers DC, Maher AG, Hadt RG, Nocera DG. Organometallics. 2015;34:4766–4774. [Google Scholar]
- 11.Isse AA, Lin CY, Coote ML, Gennaro A. J Phys Chem B. 2011;115:678–684. doi: 10.1021/jp109613t. [DOI] [PubMed] [Google Scholar]
- 12.(a) Liu D, Liu C, Li H, Lei A. Angew Chem Int Ed. 2013;52:4453–4456. doi: 10.1002/anie.201300459. [DOI] [PubMed] [Google Scholar]; (b) Qvortrup K, Rankic DA, MacMillan DWC. J Am Chem Soc. 2014;136:626–629. doi: 10.1021/ja411596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luca OR, Gustafson JL, Maddox SM, Fenwick AQ, Smith DC. Org Chem Front. 2015;2:823–848. [Google Scholar]
- 14.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712–5719. [Google Scholar]
- 15.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 16.Tsou TT, Kochi JK. J Am Chem Soc. 1979;101:6319–6332. [Google Scholar]
- 17.Scaiano JC, Stewart LC. J Am Chem Soc. 1983;105:3609–3614. [Google Scholar]
- 18.Tsou TT, Kochi JK. J Org Chem. 1980;45:1930–1937. [Google Scholar]
- 19.Sheppard TD. Org Biomol Chem. 2009;7:1043–1052. doi: 10.1039/b818155a. [DOI] [PubMed] [Google Scholar]
- 20.Preliminary studies suggest that Ni, the Ir catalyst, and light are all necessary for halogen exchange (Table S10).
- 21.Connelly NG, Geiger WE. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 22.Zheng B, Tang F, Luo J, Schultz JW, Rath NP, Mirica LM. J Am Chem Soc. 2014;136:6499–6504. doi: 10.1021/ja5024749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.