

Abstract
1,4-Benzodiazepines are used in the treatment of anxiety disorders but have limited long term use due to adverse effects. HZ-166 (2) has been shown to have anxiolytic-like effects with reduced sedative/ataxic liabilities. A 1,3-oxazole KRM-II-81 (9) was discovered from a series of six bioisosteres with significantly improved pharmacokinetic and pharmacodynamic properties as compared to 2. Oxazole 9 was further characterized and exhibited improved anxiolytic-like effects in a mouse marble burying assay and a rat Vogel conflict test.
Graphical Abstract
INTRODUCTION
The γ-aminobutyric acid type A receptor (GABAAR), a heteropentameric chloride ion channel,1, 2 is the principle target for the major inhibitory neurotransmitter, γ-aminobutyric acid (GABA), within the central nervous system (CNS). Benzodiazepines (BZDs) are a general class of drugs known to bind to the GABAAR and are primarily used to treat anxiolytic and obsessive-compulsive disorders.3 The pharmacological action exerted by a BZD is dependent on the discrete subunits of the receptor complex. Classical BZDs, such as diazepam, bind non-selectively to α1-3,5βγ2 GABAAR at the well-documented BZD binding site located at the α+γ− interface.4, 5
A compelling clinical opportunity exists in the development of α2/α3 selective GABAAR ligands.6, 7 Based on the literature, these ligands are expected to result in superior treatments for seizures,8, 9 antihyperalgesia10, 11 and anxiety12–14 without causing sedation, amnesia and ataxia, or the propensity for addiction/dependence15, 16 or tolerance.17 The clinical value of classical BDZs is evidenced by the fact that some have been marketed for over 50 years without a suitable replacement. Continued research has seen a number of promising candidates8, 18–21 fail due to pharmacokinetic complications22 or a variety of adverse effects.19, 23, 24
Over the past decade a series of α2/α3 subtype selective imidazobenzodiazepines, using a privileged scaffold, have been designed, synthesized and studied. Some of the first compounds, ligands 1 – 3 (XHe-II-053, HZ-166 and JY-XHe-053, respectively; Chart 1) have been characterized previously for their selectivity at GABAAR containing α2/α3 subunits over α1.25 Previous investigations indicated that ligand 2 can engender anxiolytic-like effects in primates without sedative-like liabilities of non-selective positive allosteric modulators (e.g. diazepam).26 However, ligand 2, as well as ligands 1 and 3, suffered from poor exposure and high clearance in rodents due to hydrolysis of the ester to the carboxylic acid 4, which exhibited low penetration across the blood-brain-barrier. Therefore, the goal of the present study was to discover a lead-like molecule that would retain the anxiolytic-like properties of BZDs 1 – 3 with lower systemic clearance in rats and thus increase plasma and brain exposure. Our design strategies to prevent formation of the carboxylic acid metabolite included blocking a site of oxidation on the ester by replacing the –CH2 with a –CD2 (ligand 5, see Scheme S1) and replacing the ester altogether with an ester bioisostere.
Chart 1.

Imidazobenzodiazepines 1 – 5.
The newly synthesized ligands were assessed in vitro and in vivo to determine their metabolic stability, general anxiolytic-like effects, and possible adverse motor effects. As a result, a novel lead compound, KRM-II-81 (9), was identified with exceptional drug stability, pharmacokinetics, anxiolytic-like efficacy, and relatively benign motor-impairing characteristics.
CHEMISTRY
The ester bioisosteres 6 – 10 were prepared from compounds 1 – 3, as depicted in Scheme 1. The methyl and ethyl oxadiazoles 6 and 7 were synthesized by treating 2 with an amidoxime in the presence of sodium hydride. The alkyl group was limited to methyl and ethyl to resemble the ethyl ester functionality of 2. The 1,3-oxazoles 8 – 10 were synthesized via the aldehyde derived from 1 – 3 through a two-step reduction-oxidation protocol. The reduction of the ester in the presence of LiAlH4 afforded the alcohol which was subsequently oxidized with activated manganese dioxide. The oxazoles 8 (KRM-II-82), 9 and 10 (KRM-II-18B) were synthesized individually from the corresponding aldehydes using toluenesufonylmethyl isocyanide (TosMIC) in the presence of potassium carbonate.27
Scheme 1.

Synthesis of heterocyclic bioisosteres 6 – 10.
RESULTS and DISCUSSION
In Vitro Characterization
The GABAAR ligands depicted in Chart 1, Schemes 1 and 2 were triaged through a combination of in vitro potency, microsomal stability, and in vivo pharmacodynamics (PD) and motor side-effect liabilities to identify optimized compounds for further testing in electrophysiological and in vivo assays (Tables 1 and 2, see Supporting Information for methods).
Table 1.
In vitro data of compounds 1 – 10.
| α3 FLIPRa | Liver microsomal stabilityb (% Remaining, 0.5h, 37 0176C, 4 μM) | Cytotoxicityc (18 h) | |||
|---|---|---|---|---|---|
| EC50 (μM) | Human | Mouse | Rat | LD50 (μM) | |
| 1 | 0.146 | 35 | 6.3 | 36 | n.d. |
| 2 | 0.844 | 86 | 57 | 58 | > 100 |
| 3 | 0.029 | 6.1 | 6.1 | 0.5 | n.d. |
| 4 | 0.47 | 96 | 93 | 99 | n.d. |
| 5 | 1.17 | 95 | 63 | 77 | n.d. |
| 6 | 5.15 | 81 | 85 | 93 | > 100 |
| 7 | 3.02 | 91 | 92 | 97 | > 100 |
| 8 | 0.321 | 74 | 73 | 72 | 66.8 ± 9 |
| 9 | 0.937 | 91 | 90 | 90 | >100 |
| 10 | 0.011 | 78 | 78 | 68 | 61.5 ± 10 |
Membrane potential (fluorescence) changes were measured by FLIPR assay using a FLIPR Tetra instrument from Molecular Devices and stably transfected HEK293T cells expressing either α1β3γ2 (n = 2; all compounds EC50 > 20 μM) or α3β3γ2 (n = 1) GABAARs; details are listed in methods section.
Compounds (n = 1) were incubated with liver microsomes for 30 minutes at 37 °C at a final concentration of 4 μM. The percentage of compound remaining after incubation was determined by LC-MS/MS.
Compounds (n = 4) were incubated with HEK293T cells for 18 hours followed by the quantification of viable cells using CellTiter-Glo (Promega); n.d., not determined
Table 2.
Summary of exposure data
| Ligand | Total Plasma Concentrations (nM)a | Brain concentrations (T in min) (nM)b | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Time (min) | ||||||
| 15 | 30 | 60 | 120 | Conc. of dosed ligand | Conc. of 4 (as a metabolite) | |
|
| ||||||
| p.o. (10 mg/kg) | ||||||
| 2 | BQLc | BQLc | BQLc | BQLc | (120) BQLc | (120) 10d |
|
| ||||||
| 5 | BQLc | BQLc | BQLc | BQLc | (120) BQLc | (120) BQLc |
|
| ||||||
| 4 | 248 | 293 | 161 | 37.5 | (120) BQLc | (120) BQLc |
|
| ||||||
| 7 | n.d. | n.d. | 1720 | n.d. | (60) 3090 | n.d. |
|
| ||||||
| 9 | n.d. | n.d. | 3910 | n.d. | (60) 3640 | n.d. |
Sprague-Dawley rats (n = 3 per time point) were dosed p.o. (10 mg/kg).
Blood samples were collected at 15, 30, 60 and 120 minutes, extracted, and quantified by LC-MS/MS.
Brains were harvested after 60 or 120 minutes followed by extraction and quantification by LC-MS/MS;
Quantifiable limit < 12.2 nM;
The three samples averaged 10 nM; n.d. not determined.
Preferred potency for the α3β3γ2 subtype over the α1β3γ2 (all ligands EC50 > 20 μM) GABAAR in the FLIPR assay was displayed by all compounds; however, the oxadiazoles 6 (EC50 = 5.15 μM) and 7 (EC50 = 3.02 μM) were consistently less potent than their oxazole counterpart 9 (EC50 = 0.94 μM) at α3β3γ2 GABAAR.
Ligands containing a 2′-F in the pendant phenyl ring, 3 and 10 were especially potent at α3β3γ2 with EC50 values of 29 nM and 11 nM, respectively. The deuterated ethyl ester 5 had a similar activity in comparison to non-deuterated 2, while the acid 4 displayed an EC50 of 0.47 μM for the α3β3γ2 GABAAR. The lack of interaction with the α1β3γ2 GABAAR predicted low risk of motor impairment and sedative effects with these ligands.8, 9 In addition, these compounds are expected to be devoid of tolerance and dependence.15–17 Based on physicochemical properties, we anticipated that most molecules would have adequate blood-brain-barrier penetration, with the exception of the hydrophilic carboxylic acid 4. As expected, appreciable quantities of carboxylic acid 4 were not detected in the brain either by dosing 4 itself or monitoring for 4 as the metabolite of esters 2 and 5 (Table 2).
To assess metabolic stability all of the compounds were incubated for 30 minutes with human, mouse, or rat liver microsomes (Table 1). Consistent with our hypothesis of blocking a possible oxidation site in the ester, replacing the –CH2 of 2 with a –CD2 in 5 resulted in increased metabolic stability across all species. In addition, replacement of the C(3)-ester in 1 – 3 with a heterocycle, 6 – 10, resulted in improved metabolic stability for all related 2′-X ligands in liver microsomes. Cytotoxicity was evaluated by exposing human embryonic kidney cells (HEK293T) to the compounds for 18 hours. Ligand 2 and bioisosteres 6, 7, and 9 were non-toxic at a dose of 100 μM (Table 1). However, oxazoles 8 and 10 displayed LD50’s of 66.8 μM and 61.5 μM, respectively, while all other ligands had LD50 values higher than 100 μM.
In Vivo Exposure Characterization
Selected compounds were dosed in vivo in rats to determine their plasma and brain exposure profiles (Table 2). Both esters 2 and 5 had poor plasma and exposure when dosed either i.v. (1 mg/kg, see Table S1) or p.o. (Table 2). Unfortunately, the increased metabolic stability of 5 as compared to 2 in rat liver microsomes did not translate into increased in vivo exposure. However, we observed appreciable plasma levels 1 hour after dosing (10 mg/kg p.o.) of oxadiazole 7 and excellent levels of oxazole 9 (1720 nM and 3910 nM, respectively). Significant concentrations in the brain were observed for both compounds 60 minutes after administration.
Pharmacodynamic Assessment
Marble burying is a useful model of anxiety and has been used to evaluate potential anxiolytic compounds.28 Herein, we evaluated the anxiolytic-like effects of compounds 2, 6 – 10 in mice using a marble burying assay (Figure 1).28, 29
Figure 1.

Assessment of anxiolytic-like activity of 2 and 6 – 10 in the marble burying assay. Male, NIH Swiss Webster mice (n = 10) were injected i.p. with either vehicle or a test compound (10 or 30 mg/kg) 30 minutes prior to testing. Data were analyzed using ANOVA (Dunnett’s test: * P < 0.05 vs. vehicle). a Mild sedation-like behavior observed at 30 mg/kg.
At 10 mg/kg, only oxadiazole 7 produced a significant reduction in marble burying as compared to vehicle. At 30 mg/kg all compounds significantly decreased marble-burying. Mice treated with 30 mg/kg of 7, however, showed weak signs of sedation. To evaluate the potential for anti-anxiety versus motor side effects of these compounds, a motorsensory study using mice on a rotating rod (rotarod) was carried out (Figure 2) with the same mice as studied for marble-burying.
Figure 2.

Assessment of ataxic effects of 2 and 6 – 10 in the rotarod assay. Mice, as treated in the marble burying assay, were placed on a rotarod set at 4 r.p.m. and testing time was 2 min. Mice not falling off during the test were given a “Success” designation, while mice that fell once were assigned a “Partial” designation. Mice falling twice during the 2 minute time period failed the test
At a concentration of 10 mg/kg little change in motorsensory behavior was observed for all the compounds tested with the exception of compounds 6 and 10, which showed very minor impairment. At 30 mg/kg, the effective anxiolytic dose for most compounds, we observed a very weak increase in ataxia for all compounds except 9, which importantly did not significantly alter rotarod performance compared to vehicle and displayed zero failures.
From an in vivo perspective, the mouse marble burying assay allowed us to screen bioisosteres 6 – 10 for PD as well as potential anxiolytic-like effects while the rotarod assay permitted detection of motor side-effect liabilities. By triaging the molecules through a combination of in vitro potency, microsomal stability, and in vivo PD and motor side-effect liabilities we selected oxazole 9 for further characterization in electrophysiology, pharmacokinetic and in vivo rodent anxiolytic activity.
Electrophysiology
Oxazole 9 and for comparison ester 2 were assessed for electrophysiological efficacy profiles in recombinant α1β3γ2, α2β3γ2, α3β3γ2 and α5β3γ2 GABAA receptors expressed in Xenopus laevis oocytes as described previously.30 To minimize cell-to-cell variability, oxazole 9 was compared directly with ethyl ester 2 in the same cells at 100 nM, 1 μM, and 10 μM (Table 3).
Table 3.
Efficacy of 9 and 2 at αxβ3γ2 GABAAR subtypes as % of control current.
| Change of GABA EC3-6 current (100%), n=3–4 | ||||||
|---|---|---|---|---|---|---|
| 9 | 2 | |||||
| 100 nM | 1 μM | 10 μM | 100 nM | 1 μM | 10 μM | |
| α1 | n.d. | 155.2 ± 9.9 | 231.7 ± 13.75 | n.d. | 147.4 ± 11.43 | 178.4 ± 19.55 |
| α2 | 100 ± 11.85 | 200.6 ± 15.67 | 258.3 ± 18.07 | 111 ± 6.48 | 207.4 ± 19.72 | 244 ± 18 |
| α3 | 109.3 ± 6.33 | 187.5 ± 25.25 | 332 ± 48.14 | 119.1 ± 8.62 | 195.2 ± 32.82 | 305 ± 31 |
| α5 | n.d. | 157.5 ± 11 | 246.4 ± 6.15 | n.d. | 134 ± 9.4 | 158.5 ± 2.6 |
Dose-dependent modulation of GABA (EC3-6 concentration) by ligands 9 and 2 on Xenopus laevis oocytes expressing GABAAR subtypes α1-3,5β3γ2 was determined by two-electrode voltage clamp current recordings. Data represents mean ± SEM.
While oxazole 9 is slightly more efficacious than ethyl ester 2 in modulating α1β3γ2 receptors at 10 μM, it is of comparable efficacy at α2β3γ2 and α3β3γ2 receptors at all concentrations. Most importantly and indicated by the electrophysiological results above, oxazole 9 is less active at α1β3γ2 receptors than at α2β3γ2 and α3β3γ2 receptors at all concentrations rendering it an α1 sparing GABAAR ligand. Interestingly, it is significantly more efficacious than 2 at α5β3γ2 receptors (but only at the 10 μM dose), which very recently has been mechanistically-connected to possible anxiolytic activity.31
The oxazole 9, along with 2 and 5, were also assessed with the high-throughput Ion Works Barracuda (IWB) electrophysiology platform and compared to zolpidem and diazepam in HEK293 cells expressing human α1β3γ2, α2β3γ2, and α3β3γ2 GABAARs (see Supporting Information, Figure S1). The results of these concentration-dependent studies indicate that oxazole 9 modulates α2 (EC50 = 118 nM) and α3 (EC50 = 205 nM) containing GABAARs to a greater extent than α1-containing receptors (EC50 = 489 nM).
Characterization of Oxazole 9
Following a 1 mg/kg i.v. dose in rats, oxazole 9 exhibited a clearance of 21.7 ml/min/kg with a low Vdss of 1.4 L/kg and a good T½ of 1.4 hours. Following a 10 mg/kg i.p. dose in rats, the AUC was 16500 nM*hrs with a Cmax of 3090 nM occurring at 2.0 hours. Oxazole 9 had an i.p. T½ of 3.1 hours and very good bioavailability of 69%.
Understanding the relationship between in vivo CNS effects and the unbound concentration of ligand in the brain can be important when assessing dose-response relationships. Thus in a second experiment, the total plasma and brain concentrations were determined 4 hrs following a 10 mg/kg i.p. dose (Table 4). The fraction unbound in plasma and brain was then measured as 25.9% and 17.7%, respectively using in vitro equilibrium dialysis. Subsequently, the unbound concentration in each compartment was calculated by multiplying free fraction by total concentration to provide a Cu,plasma = 552 nM and a Cu,brain = 363 nM at 4 hours, which leads to a Kp,uu = 0.66 since Kp,uu = Cu,brain/Cu,plasma. A full unbound concentration-time profile in plasma and brain of unbound 9 is illustrated in the supporting information (Figure S2).
Table 4.
Total and unbound concentrations at 4 hours of 9 following a 10 mg/kg i.p. dose (n = 3).
| Total Concentration (nM) | Unbound Concentration (Cu) (nM)a | Free Fractionb Fu | |
|---|---|---|---|
| Plasma | 2130 | 552 | 0.259 |
| Brain | 2050 | 363 (Kp,uu = 0.66) | 0.177 |
Calculated from total concentration (nM) and fraction coefficient (Fu);
Determined with rat plasma protein and rat brain in vitro using equilibrium dialysis.
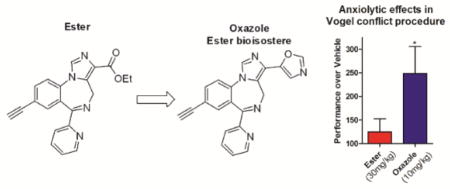
Next, oxazole 9 was evaluated further in the “Vogel conflict test” to detect anxiolytic-like behavioral effects. In this assay, water deprived rats were allowed to drink water during an unpunished and punished period in which licking was suppressed by a mild shock during punished periods. Anxiolytics cause significantly increased drinking during the punished period.32 Included in this assay was ester 2 for comparison and chlordiazepoxide (CDAP) as positive control32 (Figure 4).
Figure 4.

Vogel Conflict assessment of anxiolytic-like activity with male, Sprague-Dawley rats (n = 6–8). A. Evaluation of 2 and the anxiolytic CDAP given i.p. at 30 mg/kg and 20 mg/kg respectively; B. Dose response effects of 9 in comparison to CDAP. All compounds were administered 30 min prior to testing. Results were analyzed using ANOVA (Dunnett’s test: * P < 0.05; ** P < 0.01).
Ligand 2 was tested at 30 mg/kg, where it had been active in the marble burying assay (Figure 1). However, no discernable difference was observed from the vehicle in the Vogel assay. In contrast, CDAP induced a large and significant increase in drinking behavior during the punished period without significantly affecting drinking during the non-punished period. The 1,3-oxazole 9 produced no increase in response in comparison to the control when given a 3 mg/kg i.p. dose with a 30 min pretreatment. However, when dosed at 10 mg/kg, 9 increased the rate of response by 240% over the control (P < 0.05). Doses at 30 mg/kg and 60 mg/kg showed similar increases of about 220% and 200% over the control, respectively. In comparison, the marketed anxiolytic, CDAP, produced a 330% response over the control (P < 0.05) when dosed at 20 mg/kg. Neither 9 (up to 60 mg/kg) nor CDAP (20 mg/kg) affected the response rate in the unpunished session. Thus oxazole 9 possesses a significant anxiolytic signature at 10 mg/kg i.p. in the punished session without decreasing the response rate in the unpunished session. The lack of anxiolytic-like effects of 2 in rats using the Vogel conflict test is likely due to the instability of the ester function in rodents and further underscores the unique and improved drug-like properties of 9.
CONCLUSION
It has long been known that targeting specific GABAAR α subunits can result in desired pharmacological effects; however, due to the similar topology of the BzR binding site of the various α subunits,5, 33 designing selective ligands devoid of adverse effects, such as sedation, ataxia, amnesia, tolerance, and dependence, has been difficult. Results presented here indicate that oxazole 9 has potential as an anxiolytic devoid of these adverse effects. Consequently, oxazole 9 represents a unique and promising lead compound with attributes that include a desirable α2/α3-GABAAR selective profile, low molecular weight (MW = 351), superb lipophilicity (clogP = 2.3; calculated used the ChemAxon program), good pH=7 solubility (by light scattering), excellent total exposure in plasma and brain and favorable measured fraction unbound in plasma and brain (26% and 18% respectively), leading to in vivo activity in pre-clinical rodent models of anxiety. Preliminary studies have begun in models of epilepsy and neuropathic pain (data to be published) to further characterize 9.
EXPERIMENTAL SECTION
5-(8-Ethynyl-6-(pyridin-2-yl)-4H-benzo[f]-imidazo[1,5-a][1,4]diazepin-3-yl)oxazole (9)
Step 1
8-Ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carbaldehyde. Ethyl ester 2 (3 g, 8.4 mmol) was placed in an oven dried two neck round bottom flask and dry THF (300 mL) was added. The reaction mixture was cooled to 0 °C and the mixture allowed to stir after which LiAlH4 (320 mg, 8.42 mmol) was added to the reaction mixture at 0 °C. After 10 min the reaction mixture was stirred at rt with continued stirring for 45 min under an argon atmosphere. After 45 min at rt, analysis of the mixture by TLC (silica gel 1:9 MeOH/EtOAc) indicated the absence of starting ester 2. The reaction mixture was slowly quenched with a cold saturated aq solution of Na2SO4 (20 mL) at 0 °C and then the reaction mixture diluted with EtOAc (50 mL). After this, the mixture was filtered through a small pad of celite, followed by an EtOAc wash (30 mL) of the celite, and the filtrate mixture washed and extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with water, brine, and the solvent removed under reduced pressure to furnish the mixture of alcohols (imine alcohol 40% and reduced imine alcohol 60%, via analysis by 1H NMR spectroscopy) as a yellow solid. This mixture of alcohols was used directly in the next step. The mixture of 2′-pyridylalcohols was dissolved in dry DCM (300 mL) under an argon atmosphere, and activated MnO2 (10.9 g, 126 mmol) and Na2CO3 (2.7 g, 25.3 mmol) were added to the reaction mixture at 0 °C. The mixture was allowed to stir at rt overnight. After complete conversion of alcohol to aldehyde in the reaction mixture as indicated by TLC (silica gel), the reaction mixture was diluted with DCM (50 mL) and filtered through a small pad of celite. This pad of celite was washed with DCM (50 mL). The solvent was removed under reduced pressure and the residue that resulted purified by flash column chromatography (silica gel, EtOAc/DCM 2:1 with 1% each Et3N and CH3OH) to afford the pure aldehyde as a white solid (1.03 g, 39% over two steps). 1H NMR (300 MHz, CDCl3) δ 10.05 (s, 1H), 8.56 (d, J = 5.0 Hz, 1H), 8.08 (d, J = 7.5 Hz, 1H), 7.97 (s, 1H), 7.78 (ddd, J = 1.5, 6.0 Hz, 1H), 7.77 (dd, J = 1.5, 7.0 Hz, 1H), 7.55–7.57 (m, 2H), 7.38 (ddd, J = 1.5, 5.0 Hz, 1H), 6.00 (br s, 1H), 4.17 (br s, 1H), 3.16(s, 1H): 13C NMR (75 MHz, CDCl3) δ 186.9, 167.7, 156.2, 148.6, 137.7, 137.1, 136.7, 136.3, 135.4, 135.3, 135.0, 127.1, 124.9, 124.0, 122.8, 121.5, 81.5, 79.7, 44.4; HRMS (LCMS-IT-TOF) Calc. for C19H13N4O (M + H)+ 313.1080, found 313.1084.
Step 2
5-(8-Ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepin-3-yl)oxazole (KRM-II-81, 9). The toluenesulfonylmethyl isocyanide (TosMIC, 750 mg, 3.84 mmol) was placed in a dry two neck round bottom flask and dry MeOH (100 mL) added under an argon atmosphere. At rt, solid K2CO3 (1.33 g, 9.6 mmol) and the aldehyde from the preceding step (1.0 g, 3.2 mmol) were added to the reaction mixture and the mixture heated to reflux for 3 to 4 h. After completion of the reaction on analysis by TLC (silica gel, 1:10 MeOH and EtOAc), which was indicated by the absence of aldehyde starting material and complete conversion to the 1,3-oxazole (lower Rf,), the reaction mixture was quenched with cold water. After the mixture was quenched, 1/3 of the solvent was removed under reduced pressure and the product extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with water, brine and dried (Na2SO4). The solvent was then removed under reduced pressure and the residue purified by flash chromatography (silica gel) to give the pure oxazole 9 as white solid (821 mg, 73%). 1H NMR (300 MHz, CDCl3) δ 8.62 (d, J = 4.2 Hz, 1H), 8.12 (s, 1H), 8.06 (d, J = 7.8 Hz, 1H), 7.96 (s, 1H), 7.85 (ddd, J = 1.8, 6.0 Hz, 1H), 7.79 (dd, J = 1.8, 6.6 Hz, 1H), 7.62 (d, J = 8.4 Hz, 1H), 7.55 (d, J = 1.5 Hz, 1H), 7.53 (s, 1H), 7.41 (ddd, J = 1.5, 4.8 Hz, 1H), 5.78 (d, J = 12.9 Hz, 1H), 4.31 (d, J = 12.9 Hz, 1H), 3.71 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 167.9, 156.7, 149.9, 149.0, 146.6, 137.0, 136.4, 135.8, 135.5, 135.3, 129.8, 127.5, 127.0, 124.9, 124.0, 122.8, 122.7, 121.0, 81.8.7, 79.5, 45.3; HRMS (LCMS-IT-TOF) Calc. for C21H14N5O (M + H)+ 352.1188, found 352.1193.
Supplementary Material
Figure 3.

Total plasma concentration-time profile of 9. Male Sprague-Dawley rats (n = 3 per time point) were dosed i.v. (1 mg/kg) or i.p. (10 mg/kg) and plasma concentrations measured at time points noted.
Acknowledgments
Funding Sources
We would like to thank the NIH (MH096463, NS076517) for generous financial support (JMC), and the Austrian Science Fund for supporting ME with project FWF P 27746.
We are grateful for the expert experimental work of Xia Li, Denise Morrow, and Scott Gleason in contributing behavioral pharmacology data for this manuscript. Maria Cuadrado and Camino de los Llanos Martinez are gratefully acknowledged for their help in generating the FLIPR data. We would also like to acknowledge the Lilly Open Innovation Drug Discovery Program (OIDD) for its role in the initiation of this collaboration. In addition, the authors acknowledge support from the Milwaukee Institute for Drug Discovery and University of Wisconsin-Milwaukee’s Shimadzu Laboratory for Advanced and Applied Analytical Chemistry.
ABBREVIATIONS
- BQL
below quantification limit
- BZD(s)
benzodiazepine(s)
- CDAP
chlordiazepoxide
- CNS
central nervous system
- EC50
half maximal effective concentration
- Et3N
trimethylamine
- FLIPR
Fluorescence Imaging Plate Reader
- GABA
gamma-aminobutyric acid
- GABAAR
gamma-aminobutyric acid type A receptor
- HRMS
high-resolution mass spectrometry
- PD
pharmacodynamics
- TosMIC
toluenesulfonylmethyl isocyanide
Footnotes
ASSOCIATED CONTENT
Supporting Information. The electrophysiology using the Ion-Works Barracuda high-throughput platform and the unbound concentrations time-response curve can be found in the Supporting Information. Additional tables, figures, methods and experimental are also included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cromer BA, Morton CJ, Parker MW. Anxiety over GABAA receptor structure relieved by AChBP. Trends Biochem Sci. 2002;27:280–287. doi: 10.1016/s0968-0004(02)02092-3. [DOI] [PubMed] [Google Scholar]
- 2.Sieghart W, Sperk G. Subunit composition, distribution and function of GABAA receptor subtypes. Curr Top Med Chem. 2002;2:795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin DS, Polkinghorn C. Evidence-based pharmacotherapy of generalized anxiety disorder. Int J Neuropsychopharmacol. 2005;8:293–302. doi: 10.1017/S1461145704004870. [DOI] [PubMed] [Google Scholar]
- 4.Sieghart W, Ernst M. Heterogeneity of GABAA receptors: revived interest in the development of subtype-selective drugs. Curr Med Chem: Cent Nerv Syst Agents. 2005;5:217–242. [Google Scholar]
- 5.Clayton T, Chen JL, Ernst M, Richter L, Cromer BA, Morton CJ, Ng H, Kaczorowski CC, Helmstetter FJ, Furtmuller R, Ecker G, Parker MW, Sieghart W, Cook JM. An updated unified pharmacophore model of the benzodiazepine binding site on γ-aminobutyric acida receptors: Correlation with comparative models. Curr Med Chem. 2007;14:2755–2775. doi: 10.2174/092986707782360097. [DOI] [PubMed] [Google Scholar]
- 6.Atack JR. GABAA receptor subtype-selective modulators. I. α2/α3-selective agonists as non-sedating anxiolytics. Curr Top Med Chem. 2011;11:1176–1202. doi: 10.2174/156802611795371350. [DOI] [PubMed] [Google Scholar]
- 7.Da Settimo F, Taliani S, Trincavelli ML, Montali M, Martini C. GABAA/Bz receptor subtypes as targets for selective drugs. Curr Med Chem. 2007;14:2680–2701. doi: 10.2174/092986707782023190. [DOI] [PubMed] [Google Scholar]
- 8.McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, Farrar S, Myers J, Cook G, Ferris P, Garrett L, Bristow L, Marshall G, Macaulay A, Brown N, Howell O, Moore KW, Carling RW, Street LJ, Castro JL, Ragan CI, Dawson GR, Whiting PJ. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nat Neurosci. 2000;3:587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- 9.Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, Fritschy JM, Martin JR, Bluethmann H, Mohler H. Benzodiazepine actions mediated by specific γ-aminobutyric acidA receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- 10.Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, Ahmadi S, Brockhaus J, Sergejeva M, Hess A, Brune K, Fritschy JM, Rudolph U, Mohler H, Zeilhofer HU. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. 2008;451:330–334. doi: 10.1038/nature06493. [DOI] [PubMed] [Google Scholar]
- 11.Di Lio A, Benke D, Besson M, Desmeules J, Daali Y, Wang Z-j, Edwankar R, Cook JM, Zeilhofer HU. HZ166, a novel GABAA receptor subtype-selective benzodiazepine site ligand, is antihyperalgesic in mouse models of inflammatory and neuropathic pain. Neuropharmacology. 2011;60:626–632. doi: 10.1016/j.neuropharm.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Low K, Crestani F, Keist R, Benke D, Brunig I, Benson JA, Fritschy JM, Rulicke T, Bluethmann H, Mohler H, Rudolph U. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290:131–134. doi: 10.1126/science.290.5489.131. [DOI] [PubMed] [Google Scholar]
- 13.Morris HV, Dawson GR, Reynolds DS, Atack JR, Stephens DN. Both α2 and α3 GABAA receptor subtypes mediate the anxiolytic properties of benzodiazepine site ligands in the conditioned emotional response paradigm. Eur J Neurosci. 2006;23:2498–2504. doi: 10.1111/j.1460-9568.2006.04775.x. [DOI] [PubMed] [Google Scholar]
- 14.Dias R, Sheppard WFA, Fradley RL, Garrett EM, Stanley JL, Tye SJ, Goodacre S, Lincoln RJ, Cook SM, Conley R, Hallett D, Humphries AC, Thompson SA, Wafford KA, Street LJ, Castro JL, Whiting PJ, Rosahl TW, Atack JR, McKernan RM, Dawson GR, Reynolds DS. Evidence for a significant role of α3-containing GABAA receptors in mediating the anxiolytic effects of benzodiazepines. J Neurosci. 2005;25:10682–10688. doi: 10.1523/JNEUROSCI.1166-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan KR, Brown M, Labouebe G, Yvon C, Creton C, Fritschy JM, Rudolph U, Luscher C. Neural bases for addictive properties of benzodiazepines. Nature. 2010;463:769–775. doi: 10.1038/nature08758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Licata S, Platt D, Cook J, Van Linn M, Rowlett J. Contribution of alpha1 subunit-containing gamma-aminobutyric acid A (GABAA) receptors to motor-impairing effects of benzodiazepines in squirrel monkeys. Psychopharmacology (Berl) 2009;203:539–546. doi: 10.1007/s00213-008-1401-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Rijnsoever C, Tauber M, Choulli MK, Keist R, Rudolph U, Mohler H, Fritschy JM, Crestani F. Requirement of α5-GABAA receptors for the development of tolerance to the sedative action of diazepam in mice. J Neurosci. 2004;24:6785–6790. doi: 10.1523/JNEUROSCI.1067-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atack JR, Hallett DJ, Tye S, Wafford KA, Ryan C, Sanabria-Bohorquez SM, Eng W-s, Gibson RE, Burns HD, Dawson GR, Carling RW, Street LJ, Pike A, De Lepeleire I, Van Laere K, Bormans G, De Hoon JN, Van Hecken A, McKernan RM, Murphy MG, Hargreaves RJ. Preclinical and clinical pharmacology of TPA023B, a GABAA receptor α2/α3 subtype-selective partial agonist. J Psychopharmacol. 2011;25:329–344. doi: 10.1177/0269881109354928. [DOI] [PubMed] [Google Scholar]
- 19.Atack JR, Wafford KA, Street LJ, Dawson GR, Tye S, Van Laere K, Bormans G, Sanabria-Bohorquez SM, De Lepeleire I, de Hoon JN, Van Hecken A, Burns HD, McKernan RM, Murphy MG, Hargreaves RJ. MRK-409 (MK-0343), a GABAA receptor subtype-selective partial agonist, is a non-sedating anxiolytic in preclinical species but causes sedation in humans. J Psychopharmacol. 2011;25:314–328. doi: 10.1177/0269881109354927. [DOI] [PubMed] [Google Scholar]
- 20.Mirza NR, Larsen JS, Mathiasen C, Jacobsen TA, Munro G, Erichsen HK, Nielsen AN, Troelsen KB, Nielsen EO, Ahring PK. NS11394 [3′-[5-(1-Hydroxy-1-methyl-ethyl)-benzoimidazol-1-yl]-biphenyl-2-carbonitrile], a unique subtype-selective GABAA receptor positive allosteric modulator: In vitro actions, pharmacokinetic properties and in vivo anxiolytic Efficacy. J Pharmacol Exp Ther. 2008;327:954–968. doi: 10.1124/jpet.108.138859. [DOI] [PubMed] [Google Scholar]
- 21.Zuiker RGJA, Chen X, Osterberg O, Mirza NR, Muglia P, de Kam M, Klaassen ES, van Gerven JMA. NS11821, a partial subtype-selective GABAA agonist, elicits selective effects on the central nervous system in randomized controlled trial with healthy subjects. J Psychopharmacol. 2016;30:253–262. doi: 10.1177/0269881115620435. [DOI] [PubMed] [Google Scholar]
- 22.Scott-Stevens P, Atack JR, Sohal B, Worboys P. Rodent pharmacokinetics and receptor occupancy of the GABAA receptor subtype selective benzodiazepine site ligand L-838417. Biopharm Drug Dispos. 2005;26:13–20. doi: 10.1002/bdd.423. [DOI] [PubMed] [Google Scholar]
- 23.Mohler H. The rise of a new GABA pharmacology. Neuropharmacology. 2011;60:1042–1049. doi: 10.1016/j.neuropharm.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 24.Hofmann M, Kordas KS, Gravius A, Bolcskei K, Parsons CG, Dekundy A, Danysz W, Dezsi L, Wittko-Schneider IM, Saghy K, Gyertyan I, Horvath C. Assessment of the effects of NS11394 and L-838417, α2/3 subunit-selective GABAA receptor-positive allosteric modulators, in tests of pain, anxiety, memory and motor function. Behav Pharmacol. 2012;23:790–801. doi: 10.1097/FBP.0b013e32835a7c7e. [DOI] [PubMed] [Google Scholar]
- 25.Cook JM, Zhou H, Huang S, Sarma PVVS, Zhang C. Stereospecific anxiolytic and anticonvulsant agents with reduced muscle-relaxant, sedative-hypnotic and ataxic effects. 7,618,958 B2. US. 2009 Nov 17;
- 26.Fischer BD, Licata SC, Edwankar RV, Wang Z-j, Huang S, He X, Yu J, Zhou H, Jr, EMJ, Cook JM, Furtmuller R, Ramerstorfer J, Sieghart W, Roth BL, Majumder S, Rowlett JK. Anxiolytic-like effects of 8-acetylene imidazobenzodiazepines in a rhesus monkey conflict procedure. Neuropharmacology. 2010;59:612–618. doi: 10.1016/j.neuropharm.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Webb MR, Donald C, Taylor RJK. A general route to the Streptomyces-derived inthomycin family: the first synthesis of (+)-inthomycin B. Tetrahedron Lett. 2006;47:549–552. [Google Scholar]
- 28.Shimazaki T, Iijima M, Chaki S. Anxiolytic-like activity of MGS0039, a potent group II metabotropic glutamate receptor antagonist, in a marble-burying behavior test. Eur J Pharmacol. 2004;501:121–125. doi: 10.1016/j.ejphar.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 29.Li X, Morrow D, Witkin JM. Decreases in nestlet shredding of mice by serotonin uptake inhibitors: Comparison with marble burying. Life Sci. 2006;78:1933–1939. doi: 10.1016/j.lfs.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Ramerstorfer J, Furtmuller R, Vogel E, Huck S, Sieghart W. The point mutation gamma 2F77I changes the potency and efficacy of benzodiazepine site ligands in different GABAA receptor subtypes. Eur J Pharmacol. 2010;636:18–27. doi: 10.1016/j.ejphar.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Behlke LM, Foster RA, Liu J, Benke D, Benham RS, Nathanson A, Yee BK, Zeilhofer HU, Engin E, Rudolph U. A pharmacogenetic ‘restriction-of-function’ approach reveals evidence for anxiolytic-like actions mediated by α5-containing GABAA receptors in mice. Neuropsychopharmacology. 2016;41:2492–2501. doi: 10.1038/npp.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Millan MJ, Brocco M. The Vogel conflict test: procedural aspects, gamma-aminobutyric acid, glutamate and monoamines. Eur J Pharmacol. 2003;463:67–96. doi: 10.1016/s0014-2999(03)01275-5. [DOI] [PubMed] [Google Scholar]
- 33.Clayton T, Poe MM, Rallapalli S, Biawat P, Savic MM, Rowlett JK, Gallos G, Emala CW, Kaczorowski CC, Stafford DC, Arnold LA, Cook JM. A review of the updated pharmacophore for the alpha 5 GABA(A) benzodiazepine receptor model. Intl J Med Chem. 2015 doi: 10.1155/2015/430248. Article ID 430248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.