

Abstract
Rationale
Epidemiologic evidence indicates that exposures to fine particulate matter air pollution (PM2.5) contribute to global burden of disease, primarily as a result of increased risk of cardiovascular morbidity and mortality. However, mechanisms by which PM2.5 exposure induces cardiovascular injury remain unclear. PM2.5-induced endothelial dysfunction and systemic inflammation have been implicated, but direct evidence is lacking.
Objective
To examine whether acute exposure to PM2.5 is associated with endothelial injury and systemic inflammation.
Methods and Results
Blood was collected from healthy, non-smoking, young adults over three study periods that included episodes of elevated PM2.5 levels. Microparticles and immune cells in blood were measured by flow cytometry, and plasma cytokine/growth factors were measured using multiplexing laser beads. PM2.5 exposure was associated with elevated levels of endothelial microparticles (annexin V+/CD41−/CD31+) including subtypes expressing arterial-, venous-, and lung-specific markers, but not microparticles expressing CD62+. These changes were accompanied by suppressed circulating levels of pro-angiogenic growth factors (EGF, sCD40L, PDGF, RANTES, GROα, and VEGF), and an increase in the levels of anti-angiogenic (TNFα, IP-10) and proinflammatory cytokines (MCP-1, MIP-1α/β, IL-6, and IL-1β), and markers of endothelial adhesion (sICAM-1 and sVCAM-1). PM2.5 exposure also was associated with an inflammatory response characterized by elevated levels of circulating CD14+, CD16+, CD4+, and CD8+, but not CD19+ cells.
Conclusions
Episodic PM2.5 exposures are associated with increased endothelial cell apoptosis, an anti-angiogenic plasma profile, and elevated levels of circulating monocytes, and T, but not B, lymphocytes. These changes could contribute to the pathogenic sequelae of atherogenesis and acute coronary events.
Keywords: Endothelial microparticles, endothelial dysfunction, inflammation, air pollution, cardiovascular disease, particulate matter, vascular disease
Subject Terms: Epidemiology, Cardiovascular Disease, Vascular Disease, Inflammation, Biomarkers
INTRODUCTION
Exposure to fine particulate matter air pollution (≤2.5 μm in aerodynamic diameter, PM2.5) increases the risk of developing cardiovascular disease (CVD) and premature cardiovascular mortality.1,2 Prospective cohort studies3–6 indicate that chronic exposure to PM2.5 may contribute to the initiation and progression of atherosclerosis, hypertension, and type 2 diabetes,7 and is associated with increased risk of adverse cardiovascular outcomes,5,6,8,9 resulting in reduced life expectancy.10 In addition, short-term exposures to PM2.5 are associated with increased daily mortality,11 acute coronary events12 and ischemic stroke,13 especially in individuals with pre-existing atherosclerotic disease.14–16 A recent assessment of factors that contribute to global burden of disease estimates that ambient and household air pollution are among the top ten contributors to premature mortality worldwide—largely because of the estimated effect of PM2.5 on ischemic heart disease.17
Despite extensive investigations, it is unclear how inhaled PM2.5, a pulmonary insult, can initiate adverse cardiovascular responses. Studies investigating the underlying processes have implicated the generation or exacerbation of a dysfunctional endothelium due to systemic inflammation.1 The endothelium plays a key role in regulating blood pressure, atherogenesis, and thrombosis, and therefore endothelial dysfunction could, in part, contribute to cardiovascular morbidity and mortality associated with PM2.5 exposure. However, previous data showing associations between ambient PM2.5 with endothelial dysfunction18,19 or systemic inflammation,20–22 are from individuals with moderate to high CVD risk,19,22 diabetes,18,20 or ischemic heart disease.21 As a result, it remains unclear whether endothelial dysfunction reflects direct PM2.5-induced injury or is secondary to disease exacerbation due to PM2.5 inhalation. Because endothelial dysfunction and systemic inflammation are strongly associated with atherosclerosis23,24 and diabetes,25,26 an increase in the severity of these diseases would affect both the immune system and the endothelial function, even if these processes were not direct targets of PM2.5. Therefore, it is difficult to conclude that inhaling PM2.5 leads to systemic inflammation or endothelial dysfunction or to determine how these changes may be related to the pathogenesis of atherosclerotic lesion formation and acute coronary events.
Hence, to determine whether PM2.5 directly affects the endothelium, we examined biomarkers of subtle endothelial injury and mild systemic inflammation in a cohort of young healthy individuals with very low CVD risk. It has been previously reported that acute inhalation of PM2.5 does not induce frank endothelial dysfunction in healthy individuals,18 therefore, to identify subclinical, endothelial injury, we measured circulating levels of endothelium-derived microparticles. Microparticles are submicron vesicular structures that are shed from activated or apoptotic cells.27–29 Elevated circulating levels of such microparticles reflect endothelial damage.30,31 To evaluate systemic inflammatory responses, we measured changes in immune cells, cytokines, growth factors, and vascular adhesion molecules. We found robust evidence of endothelial injury and systemic inflammation upon exposure to increased PM2.5 levels even in young healthy individuals. These findings lend support to the view that PM2.5 causes endothelial injury, potentially by establishing a mild inflammatory state. Our results are also consistent with the possibility that aberrant immune responses and endothelial injury may be early causes of endothelial dysfunction and CVD attributable to PM2.5 exposure in susceptible individuals.
METHODS
Study population
Research subjects included three groups of 24 persons (Table 1) who were recruited for each of three consecutive winter/spring study periods, including January – March 2013, January – March 2014, and December 2014 – April 2015. All study participants were healthy, young adults recruited from Provo, Utah. All subjects were non-smokers without exposure to second-hand smoke at home, work, or school. Air pollution episodes in Utah Valley occur under predictable conditions that include a combination of snow cover, relatively high barometric pressure, and stagnant atmospheric conditions. For each of the study periods, subjects were further divided into two “draw groups” of 12. At prearranged times (between 11:00 am and 12:00 noon on Tuesdays or Thursdays) and during times of variable air pollution, the subjects in each “draw group” had their blood drawn (see Figure 1) and completed a questionnaire regarding current health status, level of exercise, beginning date of last menstrual period and supplement use.
Table 1.
Summary of research subject characteristics*
| All | 2013 | 2014 | 2015 | |
|---|---|---|---|---|
| Number of subjects | 72 | 24 | 24 | 24 |
| Number of blood draws | 384 | 144 | 120 | 120 |
| Age, yrs, mean (SD) | 23 (2) | 23 (2) | 23 (2) | 23 (2) |
| Race (% white/other) | 88/13 | 92/8 | 83/17 | 88/12 |
| % Male | 65 | 100 | 46 | 50 |
| BMI, mean (SD) | 24 (3) | 24 (3) | 23 (3) | 24 (3) |
| Systolic Blood Pressure (SD) | 126 (14) | 130 (12) | 130 (12) | 118 (16) |
| Diastolic Blood Pressure (SD) | 74 (8) | 70 (9) | 76 (6) | 76 (9) |
Subject characteristics data, including biometric data, were collected at time of enrollment or at time of initial blood draw.
Figure 1.

PM2.5 concentrations and blood-draw dates plotted over study periods. Line plots indicate ambient PM2.5 concentrations for various sites and methods where: “1-d FRM” indicates daily concentrations based on the Federal Reference Method and “24-h Real time” indicates 24-hour average concentrations based on continuous monitors from the Department of Environmental Quality sites (Lindon, North Provo, and Spanish Fork). “FOB 24-h Real time” indicates 24-hr concentrations from monitor located adjacent to blood-draw building and “Blood draw room 24-h” indicates 24-hr concentrations from monitor inside the blood-draw room. Dots indicate the times of and PM2.5 concentrations at blood draws for each 12-subject blood-draw group.
In the first study period, only males were enrolled, while in the other periods, both genders were enrolled. For the second and third study periods, subjects were provided with dietary supplements and asked to consume two 640 mg soft gel tablets in the morning and evening with food. Consumption of these supplements began 3 weeks prior to the first blood draw and continued until the final blood draw. Subjects were randomly (stratified by gender and group) assigned capsules of omega-3 fatty acids (a total daily consumption of 2,560 mg, within the upper range of recommended dose) or identically sized placebo capsules of soybean oil (Pro Omega softgel/lemon capsules and Placebo Soybean Oil softgel/lemon, Nordic Naturals, Inc., Watsonville, California). Informed consent was obtained from all research subjects. The study was approved by the Institutional Review Board for Human Subjects at Brigham Young University and procedures followed were in accordance with institutional guidelines.
Blood collection and processing
For the analysis of microparticles and immune cell populations, 8 ml of blood was collected in sodium citrate-containing cell preparation tubes (CPT Vacutainer; Becton Dickinson). These tubes were centrifuged at 1700xg for 30 min at room temperature and shipped overnight to the University of Louisville for analysis. Upon arrival, the tubes were centrifuged again and the upper layer containing mononuclear cells and plasma was collected. This material was diluted with an equal volume of PBS and centrifuged at 500xg for 10 min. Aliquots of the supernatant were used for analysis of microparticles while the cell pellet was washed once with PBS and centrifuged. The final cell pellet was resuspended in a volume of 300 μl PBS+2% FCS, 150 μl of which was used for analysis of immune cell populations.
For analysis of platelet-monocyte aggregates, 3ml of blood was collected in an acid-citrate dextrose tube (ACD Vacutainer; Becton-Dickinson), and then 1ml aliquots were diluted with 3ml PBS and fixed with 1.3ml of 4% paraformaldehyde for 30 min on ice. Red blood cells were lysed by addition of 24 ml of water, the samples centrifuged at 400xg for 10min and the cell pellet resuspended in 1 ml Tyrode’s buffer and shipped to University of Louisville as above.
Inflammatory cytokines were measured in frozen plasma aliquots by analytical services at Eve Technologies (Calgary, Alberta, Canada). An array of 42 human cytokines (Human Cytokine Array/Chemokine Array 42-Plex, Discovery Assay®) and an array that included soluble ICAM-1 and soluble VCAM-1 (Human Neurodegenerative Disease Array 2-Plex, Discovery Assay®) were analyzed using multiplexing laser bead technology.
Flow cytometry
To measure microparticles, plasma aliquots were centrifuged for 2 min at 10,000xg to pellet residual cells and debris. The supernatant was collected and then centrifuged for 45 min at 17,000xg. The resulting microparticle pellet was resuspended in PBS containing 1% FCS and 2.5mM Ca+2 and incubated with Fc block for 10 min at 4°C. Then a cocktail of fluorescently conjugated antibodies including, Pacific blue-Annexin V (Life Technologies), APC-anti-CD34 (Becton Dickinson), PECy7-anti-CD41 (Becton Dickinson), PE-anti-CD31 (eBioscience), PECy5.5-anti-CD62E (Becton Dickinson), FITC-anti-EphB4 (R&D Systems), and APC-anti CD143 (Biolegend) was added. In addition, one antibody (anti-Ephrin B2; Santa Cruz) was labeled in the laboratory (Zenon Alexa 488 labeling kit; Life Technologies) and also added to the staining cocktail. After 30 min at room temperature, 25,000 volumetric counting beads were added and 10,000 events collected on an LSR II flow cytometer. An identical sample with no antibodies was used as a gating control. Microparticle numbers were quantified in gated populations <1μm in size and positive for Annexin V staining using the FlowJo software package and normalized to the number of counting beads (volume) collected. Specific populations were defined by phenotype as documented in Table 2 and illustrated in Supplemental Figure I in the Online Data Supplement.
Table 2.
Description and summary statistics of microparticles and immune cells and regression coefficients for PM2.5 from the subject-mean adjusted regression models.
| Outcome variables | Phenotype | No. of obs. | Mean* | Std. Dev. | Coefficient (x 10) (Std. Error) | P-value | R2 |
|---|---|---|---|---|---|---|---|
| Microparticles | |||||||
| MP, EPC | CD34+/CD31+ | 332 | 22.03 | 21.68 | −0.09 (0.34) | 0.796 | 0.00 |
| MP, Platelet | CD41+ | 332 | 37.37 | 34.94 | −1.33 (0.55) | 0.017 | 0.02 |
| MP, Endothelial | CD31+/CD41− | 332 | 6.76 | 10.14 | 1.00 (0.16) | <0.001 | 0.11 |
| MP, Lung Endothelial | CD31+/CD41−/CD143+ | 331 | 2.82 | 4.48 | 0.42 (0.07) | <0.001 | 0.10 |
| MP, Non-lung Endothelial | CD31+/CD41−/CD143− | 331 | 3.88 | 6.40 | 0.56 (0.10) | <0.001 | 0.09 |
| MP, Venous Endothelial | CD31+/CD41−/EphB4+ | 329 | 2.55 | 5.54 | 0.48 (0.09) | <0.001 | 0.09 |
| MP, Lung Venous Endothelial | CD31+/CD41−/EphB4+/CD143+ | 329 | 2.06 | 4.57 | 0.39 (0.07) | <0.001 | 0.08 |
| MP, Arterial Endothelial | CD31+/CD41−/EphrinB2+ | 331 | 3.68 | 5.03 | 0.37 (0.07) | <0.001 | 0.07 |
| MP, Lung Arterial Endothelial | CD31+/CD41−/EphrinB2+/CD143+ | 331 | 3.27 | 4.56 | 0.31 (0.07) | <0.001 | 0.06 |
| MP, Activated Endothelial | CD62+ | 332 | 17.91 | 16.11 | −0.63 (0.26) | 0.014 | 0.02 |
| MP, Lung Activated Endothelial | CD62+/CD143+ | 332 | 3.93 | 4.25 | 0.005 (0.06) | 0.943 | 0.00 |
| MP, Venous Activated Endothelial | CD62+/EphB4+ | 332 | 4.40 | 6.67 | −0.02 (0.10) | 0.876 | 0.00 |
| MP, Lung Venous Activated Endothelial | CD62+/EphB4+/CD143+ | 329 | 3.57 | 3.80 | −0.002 (0.06) | 0.980 | 0.00 |
| MP, Arterial Activated Endothelial | CD62+/EphrinB2+ | 330 | 5.17 | 5.03 | 0.03 (0.08) | 0.702 | 0.00 |
| MP, Lung Arterial Activated Endothelial | CD62+/EphrinB2+/CD143+ | 330 | 4.52 | 4.47 | 0.05 (0.07) | 0.518 | 0.00 |
| Immune cells | |||||||
| Monocytes | CD14+ | 365 | 22,503 | 15,535 | 863.99 (185.95) | <0.001 | 0.06 |
| Natural killer cells | CD16+ | 365 | 17,530 | 16,784 | 660.22 (182.24) | <0.001 | 0.03 |
| Helper T cells | CD4+ | 365 | 72,604 | 42,633 | 2151.75 (504.36) | <0.001 | 0.05 |
| Killer T cells | CD8+ | 365 | 39,259 | 24,421 | 1038.21 (323.52) | 0.001 | 0.03 |
| B cells | CD19+ | 365 | 18,242 | 22,527 | −310.72 (304.33) | 0.308 | 0.00 |
| Platelet-monocyte aggregates | CD45+/CD41+ | 368 | 4.71 | 5.62 | 0.20 (0.09) | 0.020 | 0.01 |
Per volume of the analytical tube. All microparticle subpopulations were <1 μm and AnnexinV+
To quantify immune cell populations, resuspended cell pellets from above were incubated with Fc block (Miltneyi) for 10min at 4°C, followed by the addition of fluorescently tagged antibodies (eBioscience) recognizing CD14 (650NC), CD16 (FITC), CD19 (Alexa 700), CD8 (APC-e780), and CD4 (PECy7) for 30 min at 4°C. The cells were washed once, resuspended in 350ul PBS +2% FCS and 50,000 volumetric counting beads (Accucount Particles; Spherotech) were added prior to analysis. Samples were collected for 2 min on an LSR II flow cytometer (Becton Dickinson). Cell numbers were quantified using the FlowJo software package and normalized to the number of counting beads (volume) collected.
To quantify platelet-monocyte aggregates, cell pellets from above were washed once in Tyrode’s solution and then incubated with Fc block (Miltenyi) for 10 min on ice. The samples were then incubated with an FITC-conjugated anti-CD41 antibody (eBioscience) and an APC-conjugated anti-CD45 (eBioscience) antibody for 30 min on ice, washed, resuspended in Tyrode’s and 10,000 events collected on the LSR II flow cytometer. A sample with isotype control antibodies was used as a control. The percent of double positive events was determined using FloJo software.
Measurement of PM2.5 air pollution
Daily ambient concentrations of PM2.5 in Utah Valley were collected from three monitoring sites: Lindon (located at the northern part of the valley), North Provo (approximately centrally located), and Spanish Fork (located at southern end of valley). At the Lindon and North Provo sites, hourly concentrations of PM2.5, based on continuous ambient monitors, were also collected and used to estimate average PM2.5 concentrations for the 24-h period from noon to noon for each day—the approximate 24-h time period before each blood draw. These data were obtained from the Utah Department of Environmental Quality (Salt Lake City, Utah). During the last two study periods, supplemental continuous ambient monitoring of PM2.5 (using a Thermo Scientific™ tapered element oscillating microbalance TEOM™ monitor) was conducted outside of the building where the blood draws were conducted and indoor monitoring (using a Thermo Scientific™ portable DataRAM4™ monitor) was conducted inside the blood-draw room. Average PM2.5 concentrations for the 24-h period from noon to noon for each day were calculated. Monitored PM2.5 concentrations were nearly identical for all ambient monitors (Figure 1). The indoor measured concentrations of PM2.5 were much lower. PM2.5 concentrations from the North Provo monitor were used as the primary exposure measure.
Statistical analysis
Associations with PM2.5 exposures and microparticles and markers of inflammation were evaluated by estimating two similar regression approaches: fixed effects regression models controlling for subject-specific differences and a subject-mean adjusted regression that accounts for subject-specific differences by subtracting out subject-level means. The fixed effects model controls for subject-level differences by estimating subject-specific fixed effects as part of the model, whereas the subject-mean adjusted model controls for subject-level differences by first subtracting out the subject-level means, and regressing deviations from these means on PM2.5. (See expanded statistical methods in the Online Data Supplement.)
Plots of subject-specific differences over pollution concentrations along with regression plots were generated. To explore the sensitivity of the results, models were estimated that excluded observations from participants who reported any acute illness at time of the blood draw, models that controlled for time exercised on the day of the draw and the day before the draw, models that controlled for menstruation (as indicated by blood-draw date being less than six days since beginning date of last menstrual period), and models that excluded observations for days with PM2.5 concentrations greater than 100 μg/m3. Additionally, rather than using PM2.5 concentrations 24 h prior to the blood draws, models that used PM2.5 concentrations 12 h and 48 h prior to the blood draws, respectively, were estimated. Finally, using data for the final two winter/spring time periods, models that included interaction terms for sex and PM2.5, and fish oil (versus placebo) and PM2.5 were estimated to test for effect modification by sex and by fish oil supplement use.
For analysis of the multiple inflammatory cytokines and adhesion molecules, we estimated the fixed effects and subject-means adjusted models. The percent change (and 95% CIs) for each analyte per 10 μg/m3 increase in PM2.5 relative to its mean value was calculated and plotted in order based on t-values. This approach comprehensively evaluates all measured analytes, allows for direct comparisons and evaluation of the strength of the statistical associations, and mitigates concerns regarding multiple testing and selective reporting. All statistical analyses were conducted using SAS, version 9.4 (SAS Institute, Inc., Cary, North Carolina).
RESULTS
A summary of selected characteristics of research subjects is provided in Table 1. PM2.5 concentrations and timing of blood draws for the three study periods are illustrated in Figure 1.
PM2.5, microparticles and immune cells
Table 2 presents descriptions and summary statistics of microparticle and immune cell measurements and subject-mean adjusted regression results. Supplemental Table I in the Online Data Supplement compares regression results for the fixed-effects and subject-mean regressions. Delayed shipment of blood due to weather, broken blood collection tubes during centrifugation, and instrument malfunction during analysis resulted in some missing data. PM2.5 concentrations were significantly (p < 0.001) associated with elevated endothelial microparticles and all endothelial subgroups (venous, arterial, lung, non-lung, lung arterial, and lung venous). The associations were significant even after using a Bonferroni correction for multiple testing (p < 0.05/15). Microparticles derived from endothelial progenitor cells (EPC), platelets and activated endothelial cells (CD62+) were not positively associated with PM2.5. PM2.5 concentrations were also significantly associated with elevated immune cell levels, including monocytes, natural killer cells, helper T cells, and killer T cells. PM2.5 concentrations were not associated with B cells, but were weakly associated with platelet-monocyte aggregates.
The associations between PM2.5 and subject-mean adjusted values for endothelial microparticles and monocytes are illustrated in Figure 2, panels A and B. Associations for lung, non-lung, venous, lung venous, arterial, and lung arterial endothelial microparticles are similar to that illustrated in Figure 2, panel A. Indeed, relative effects of PM2.5 exposures were similar across all endothelial subgroups. Expressed as a percent increase relative to the mean level of microparticles, a 10 μg/m3 increase in PM2.5 was associated with a 15% increase in all endothelial microparticles and a 15%, 14%, 10%, 9%, 19% and 19% increase in lung, non-lung, arterial, lung arterial, venous and lung venous endothelial microparticles, respectively. With regards to the immune cell responses, a 10 μg/m3 increase in PM2.5 was associated with a 4%, 4%, 3%, and 3% increase in levels of monocytes, natural killer cells, helper T cells, and killer T cells, respectively.
Figure 2.

Subject-mean adjusted values for endothelial microparticles (panel A), monocytes (panel B), TNFα (panel C), and sICAM-1(panel D) plotted over PM2.5 concentrations, with fitted regression lines, 95% confidence limits, and 95% prediction limits.
PM2.5, inflammatory cytokines, and adhesion molecules
Figure 3 presents the estimated associations between PM2.5 and all 42 measured growth factors and cytokines and 2 soluble adhesion molecules. The estimated associations are presented as percent change (and 95% CIs) in each analyte per 10 μg/m3 increase in PM2.5 relative to the mean. Estimated associations are ordered from left to right based on t-values. PM2.5 was associated with changes in several circulating growth factors and cytokines involved in systemic inflammation including, TNFα, MCP-1, MIP-1α and MIP-1β, IP-10, IL-8, and IL-6, and IL-1β, as well as the soluble adhesion proteins sICAM-1 and sVCAM-1.
Figure 3.

Associations between elevated PM2.5 exposures biomarkers of inflammation. The biomarkers include all 42 measured cytokines and 2 adhesion molecules. The results are presented as percent change (and 95% CIs) in each analyte per 10 μg/m3 increase in PM2.5 relative to the mean. Estimates are derived from the subject-mean adjusted regressions and are ordered from left to right based on t-values—resulting in the most statistically significant positive associations being on the left and the most statistically significant negative associations being on the right.
For TNFα, MCP-1, IL-8, MIP-1α, MIP-1β, and IP-10 the associations were highly statistically significant with a 10 μg/m3 incremental increase in PM2.5 associated with a 1.25 (SE=0.19, p < 0.0001), 5.22 (SE=1.09, p < 0.0001), 2.96 (SE=0.70, p < 0.0001), 0.86 (SE=0.21, p < 0.0001), 1.65 (SE=0.52, p = 0.002), and 4.05 (SE=1.34, p = 0.003) pg/ml increase in each cytokine, respectively. The associations with TNFα, MCP-1, IL-8, MIP-1α, and sICAM-1 were statistically significant even when using the Bonferroni correction for multiple testing of 44 analytes (p < 0.05/44). For IL-6, IL-10, and IL-1β the associations were also observed but they were marginally statistically significant with a 10 μg/m3 increase in PM2.5 associated with a 0.09 (SE=0.05, p = 0.05), 0.11 (SE=0.06, p = 0.05), and 0.29 (SE=0.15, p = 0.06) pg/ml increase in each cytokine, respectively. Elevated PM2.5 exposures were associated with significant reductions in the growth factors EGF (epidermal growth factor) and PDGF (platelet-derived growth factor), as well as sCD40L (CD40 ligand), GROα (growth-regulated protein alpha), and RANTES (regulated on activation, normal T cell expressed and secreted) (Figure 3).
Elevated PM2.5 exposures were also significantly associated with the two soluble adhesion molecules, sICAM-1 and sVCAM-1, with a 10 μg/m3 increase in PM2.5 associated with a 628.43 (SE=125.19, p < 0.0001) and 2,288.44 (SE=1,030.08, p = 0.03) pg/ml increase, respectively. The associations between subject-mean adjusted values for TNFα and sICAM-1 with PM2.5 concentrations are presented in Figure 2, panels C and D. Highly significant positive associations are observed, but this illustration also demonstrates that most of the variability remains unexplained by PM2.5.
Sensitivity analysis
Overall, the results were not highly sensitive to alternative modeling choices. Similar results were obtained for the fixed-effects and subject-mean adjusted regression models as well as models that treated all out-of-range cytokine/growth factor observations as missing observations (See Supplemental Tables I, II, and III in the Online Data Supplement). Also, similar results were obtained from models excluding draws from participants who reported illness, models controlling for concurrent and previous days’ time exercising; models controlling for menstruation, models that exclude days with PM2.5 concentrations greater than 100 μg/m3, and models that used PM2.5 concentrations 12 h and 48 h prior to blood draws (rather than 24 h). A formal comparison of effect estimates for these regression models for selected variables is presented in Supplemental Figure II in the Online Data Supplement. Furthermore, for the final two study periods, models that included sex and PM2.5, and fish oil (versus placebo) and PM2.5, interaction terms were estimated to test for effect modification. Overall there was no consistent evidence of effect modification by either sex or use of fish oil supplements.
DISCUSSION
The major findings of this study, as stylistically illustrated in Figure 4, are that episodic exposure to PM2.5 was associated with an increase in circulating microparticles indicative of endothelial apoptosis, and an inflammatory, anti-angiogenic blood profile associated with selective increases in T, rather than B, lymphocytes. Taken together, these findings reveal a characteristic signature of systemic injury inflicted by PM2.5 exposure and could provide new insights into potential mechanisms by which inhalation of PM2.5 increases CVD risk and severity, leading to premature cardiovascular mortality. Because plasmatic changes were observed in a young, healthy population with low CVD risk burden, these findings suggest that even in the absence of pre-existing disease, and un-confounded by significant disease progression, inhalation of PM2.5 induces endothelial injury and inflammation. Therefore, these changes are likely to be early signs of systemic injury, which if sustained, could contribute to the development or exacerbation of atherosclerotic disease as well as the precipitation of acute cardiovascular events in susceptible individuals.
Figure 4.
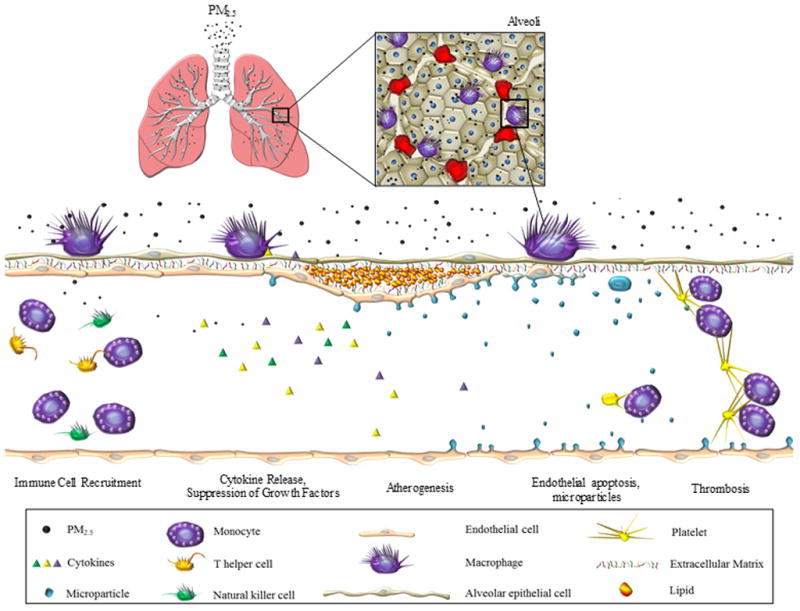
Schematic of the proposed mechanism by which PM2.5 induces vascular injury. Inhalation and deposition of PM2.5 in the lungs triggers inflammatory responses that lead to the release of inflammatory cytokines (TNFα, MCP-1 and IL-8); the recruitment of immune cells (CD14+ monocytes, CD16+ natural killer cells, CD4+ and CD8+ T cells); suppression of growth/angiogenic factors (EGF, CD40L, PDGF-AA, RANTES, GROα); and an increase in anti-angiogenic factors (TNFα and IP-10), resulting in endothelial cell apoptosis and the generation of endothelial microparticles in blood. These events are associated with an increase in circulating levels of soluble adhesion molecules (sICAM-1 and sVCAM-1) and platelet-monocyte aggregates. Collectively, these responses contribute to the pathogenic sequelae of atherogenesis and may increase thrombotic potential increasing risk of acute cardiovascular events.
Although previous epidemiological evidence suggests that exposure to PM2.5 contributes to the development of cardiovascular disease,1–6,9,12,14–16 the mechanistic basis of this injury is not fully understood. One potential pathway is the release of pro-inflammatory mediators by alveolar macrophages or epithelial cells upon exposure to airborne particles. Indeed, exposure to concentrated PM results in the release of pro-inflammatory cytokines such as IL-6 or TNFα from alveolar32 or peritoneal33 macrophages. Similarly, human airway cells34 or bronchial epithelial cells35 release TNFα, IL-6 or IL-8 upon incubation of ambient air particles. Our observation showing that circulating levels of TNFα, IL-8, and IL-6 are increased upon PM2.5 inhalation is consistent with the possibility that the release of cytokines in the lung could trigger and sustain a state of mild systemic inflammation.
Systemic inflammation due to PM2.5 inhalation, could by itself contribute to both an increase in the thrombotic propensity of the blood as well as pro-atherogenic changes in the vessel wall. Our observation of an association between increased platelet-monocyte aggregate levels in the peripheral blood and PM2.5 exposure (Table 2) suggests that PM2.5 exposure does indeed establish an early pro-coagulation state, even in healthy adults, which might be an important contributor to CVD risk associated with PM2.5 exposure. While in humans it is difficult to directly measure pro-atherogenic changes in the vessel wall and atherosclerotic disease progression in young healthy humans, we did find clear signs of endothelial injury, reflected by an increase in circulating levels of endothelial microparticles, derived mostly from the venous endothelium. Notably, this location is just upstream of the coronary circulation putting these particles potentially at higher levels near susceptible coronary artery plaques in high-risk individuals. Thus, increased levels of endothelial microparticles in disease-free individuals is likely suggestive of injury that precedes the development of frank disease. Nonetheless, the mechanisms by which PM2.5 triggers the release of microparticles from the lung remain unclear. Exposure to inflammatory cytokines such as TNFα could stimulate the release of microparticles by activating the endothelium36 and increased levels of activated (CD62+) endothelial microparticles are associated with states of high inflammation that accompany acute cardiovascular events.37 However, we found no association between CD62+ microparticles and PM2.5 exposure, suggesting that most endothelial microparticles released upon exposure to PM2.5 are derived from apoptosis, not cytokine-induced activation, of the endothelium. A similar increase in the plasma levels of apoptotic, not activated, endothelial ACE+ microparticles has been observed also in smokers with normal spirometry,38 suggesting that pathologic changes associated with smoking and PM2.5 exposure share overlapping mechanisms that involve early lung destruction, followed by subsequent cardiovascular injury.
Although elevated levels of specific cytokines such as TNFα cannot be linked to the generation of activated endothelial microparticles, the collective pattern of changes in plasma cytokines is indicative of an anti-angiogenic state. All the cytokines and growth factors that were suppressed with PM2.5 exposure—EGF, CD40L, PDGF-AA, GROα, RANTES and VEGF—have a key property in common. They are all potent angiogenic factors. The growth factor VEGF is required for in vivo angiogenesis and the growth of macrovascular endothelial cells,39 whereas EGF increases the growth and proliferation of microvascular endothelial cells, particularly in the presence of PDGF.40,41 EGF is also a strong trophic factor that prevents endothelial cells against TNFα-induced apoptosis42 and blockage of EGFR signaling in vivo induces endothelial apoptosis.43 RANTES is also a pro-angiogenic factor. It promotes endothelial cell migration, spreading and neo-vessel formation, and RANTES-mediated angiogenesis depends at least partly on VEGF.44,45 The chemokine, GROα is essential for thrombin-induced angiogenesis and it increases VEGF production by endothelial cells.46 Down regulation or inhibition of GROα markedly decreases VEGF expression and the angiogenic potential of endothelial cells.47 Likewise, the interaction of sCD40L with CD40 on endothelial cells has been shown to increase the expression of VEGF and stimulate angiogenesis.48,49 Overall, the association of PM2.5 with lower circulating levels of sCD40L, FGF, PDGF, GROα, RANTES and VEGF suggests that loss of trophic, angiogenic factors could account for the increase in apoptosis of endothelial cells upon PM2.5 exposure.
The anti-angiogenic state established by the loss of angiogenic growth factors appears to be further exacerbated and reinforced by a corresponding increase in anti-angiogenic cytokines such as TNFα and IP-10. IP-10 is secreted by activated T cells, monocytes and endothelial cells and elevated levels of this cytokine has been linked to inflammatory disorders such as asthma.50 It inhibits the development of new vasculature and causes the regression of newly formed vessels51 and is strongly induced upon stimulation of monocytes with TNF-α.52 Elevation in the circulating levels of TNFα and other cytokines such as MCP-1, IL-8, and MIP1α/β are indicative of a pro-inflammatory state associated with increased chemotaxis and atherogenesis. High circulating levels of MCP-153 and IL-854 are associated with increased risk of all-cause mortality in CVD patients. Interestingly, circulating levels of MIP-1β and TNFα correlate significantly with plaque levels of these cytokines,55 suggesting that PM2.5 associated increases in the plasma levels of these cytokine may be reflective of increased vascular inflammation.
The pattern of changes in plasma cytokine levels associated with PM2.5 exposure may also be linked with the observed increases in circulating lymphocytes. Cytokines such as MCP-156 and IP-1057 exert potent chemotactic activity towards monocytes and T lymphocytes, whereas IL-9 stimulates the proliferation of activated T cells. Hence, an increase in these cytokines may be linked to the increased levels of monocytes and T cells. Notably, no change in the levels of B cells was observed, suggesting a lack of an adaptive immune response. Because PM2.5 lacks proteins or other T-cell dependent antigens, as well as non-T cell dependent antigens such as foreign polysaccharides or DNA, it may be insufficient to induce a humoral response. Nevertheless, selective increase in T, but not B, cell populations may be indicative of increased cytokine production and/or the formation of auto-antigens generated by injured or dying endothelial cells.
Previous studies have shown that PM2.5 exposure of just a few hours or days is associated with both fatal and non-fatal ischemic stroke as well as acute coronary syndrome events.12–16 This evidence suggests that there are systemic responses to even short-term PM2.5 exposure that play a role in triggering acute cardiovascular events.1 Our observation that episodic increases in PM2.5 are associated with an increase in endothelial microparticles, lymphocytes and cytokines suggests a potential mechanism for the acute effects of PM2.5. While cause-and-effect relationships are not easily disentangled, our results are consistent with a scenario in which inhalation of PM2.5 triggers pro-inflammatory and anti-angiogenic cytokine profile which in turn induces endothelial cell apoptosis, leading to increased levels of circulating microparticles. These microparticles carry metalloproteases,58 which can induce endothelial cell apoptosis28 or contribute to the disruption or erosion of unstable plaques in susceptible individuals. Notably, metalloproteases are responsible for cleavage and generation of soluble adhesion molecules including sICAM-1 and sVCAM-1 from the endothelium, both of which were increased in association with PM2.5 exposure. Thus, an increase in microparticles upon PM2.5 exposure could disseminate pro-inflammatory mediators and spread inflammation beyond the lung to establish and sustain low-grade inflammation and aggravate the atherothrombotic process.29
The characteristic pattern of changes in cytokines, chemokines, endothelial microparticles, and blood lymphocytes reported here may be diagnostic of exposure to PM2.5 and may be useful in distinguishing PM2.5-induced injury from that induced by inhalation of other toxins such as tobacco smoke. While direct comparisons are not yet available, the pattern of changes we observed upon exposure to PM2.5 seems to differ from that observed with smoking. Although like PM2.5 exposure, smoking is also associated with endothelial apoptosis and a decrease in plasma levels of CD40L,59 EGF,60 and GROα61 it is generally associated with increased blood levels of VEGF,62 which due to stimulatory effect of nicotine on VEGF63 or because of hypoxia induced by CO in cigarette smoke. Also, unlike PM2.5 exposure, smoking is not usually associated with an increase in plasma TNFα or MCP-1 levels.64 Hence, while further research is clearly needed, direct future comparisons between the pattern of changes with PM2.5 and other inhaled toxic substances may help discern, and thereby more reliably attribute, cardiovascular injury due to PM2.5 exposure.
Strengths of this study are its size and design. The number of available observations over the three-year period, multiple observations per subject, and the ability to control for subject-specific differences using fixed-effects models or models using subject-mean adjusted variables provided adequate power to test the primary hypotheses. This approach allows for matching and control for subject-specific differences such as age, gender, race, genetics, health, etc. In this study, there was minimal potential for confounding by active smoking or exposure to secondhand cigarette smoke. Another primary strength of our study is that we evaluated PM2.5 associations under real-world conditions with relevant exposures. Despite these strengths, the study also has limitations. Although pollution episodes were predictable, we could only control the timing of the blood draws. We were generally successful at getting observations during pollution episodes but, in the third year study period, no substantive pollution episode occurred.
In conclusion, episodic PM2.5 air pollution exposures were associated with increased levels of endothelial microparticles and systemic increase in anti-angiogenic cytokines and a suppression of angiogenic growth factors in young healthy adults. The effects are statistically significant and the pattern of results is coherent and consistent. Although these findings need validation with additional research, they suggest that inhalation of PM2.5 can instigate adverse cardiovascular responses through changes in cytokine and growth factor levels leading to endothelial injury and increased abundance of monocytes and T cell in the peripheral blood; responses that could potentially initiate and promote atherosclerotic lesions and trigger acute cardiovascular and cerebrovascular events. Nevertheless, we found that air pollution exposure explained only a small amount of the variability in endothelial microparticles and markers of inflammation, suggesting that exposure to air pollution is one of multiple factors that influences cardiovascular health.
Supplementary Material
Novelty and Significance.
What Is Known?
Exposure to fine particulate air pollution (PM2.5) is associated with increased risk of cardiovascular disease and mortality and contributes substantially to global burden of disease.
Although PM2.5-induced systemic inflammation and endothelial dysfunction have been implicated, it remains unclear how inhaled PM2.5, a pulmonary insult, can induce cardiovascular injury and exacerbate cardiovascular disease.
What New Information Does This Article Contribute?
In healthy, non-smoking, young adults, episodic exposure to PM2.5 was associated with elevated circulating endothelial microparticles, indicative of endothelial cell apoptosis and endothelial injury.
PM2.5 exposure was associated with inflammatory responses including an increase in immune cells, a systemic increase in anti-angiogenic cytokines, and a suppression of pro-angiogenic growth factors.
Circulating levels of soluble adhesion molecules and platelet-monocyte aggregates were also elevated with PM2.5 exposure.
Extensive epidemiological evidence indicates that exposure to ambient PM2.5 contributes to cardiovascular disease and mortality, but it is unclear how exposure to PM2.5 causes cardiovascular injury. We collected blood from panels of healthy, non-smoking young adults who were environmentally exposed to episodes of elevated PM2.5 levels. Exposure to this pollution was positively associated with markers of endothelial injury and systemic inflammation. The evidence suggests that inhalation and pulmonary deposition of PM2.5 triggers inflammatory responses characterized by an increase in anti-angiogenic cytokines and suppression of pro-angiogenic growth factors, which could result in increased endothelial cell death and the generation of endothelial microparticles. In combination with observed elevated levels of soluble adhesion molecules and platelet-monocyte aggregates, these responses could contribute to atherogenesis, and thrombosis, and thereby increase the risk of acute cardiovascular events.
Acknowledgments
SOURCES OF FUNDING
Supported by grants from the National Institutes of Health (NIH ES019217; GM103492).
Nonstandard Abbreviations and Acronyms
- CD40L
CD40 ligand
- EGF
Epidermal growth factor
- EPC
Endothelial progenitor cells
- GROα
Growth-regulated protein alpha
- ICAM-1
Intercellular adhesion molecule 1
- IL-6
Interleukin 6
- IL-8
Interleukin 8
- IL-1β
Interleukin 1β
- IP-10
Interferon gamma-induced protein 10
- MCP-1
Monocyte chemoattractant protein 1
- MIP-1α
Macrophage inflammatory protein 1α
- MIP-1β
Macrophage inflammatory protein 1β
- MP
Microparticles
- PDGF
Platelet-derived growth factor
- PM2.5
Fine particulate matter < 2.5 μm in aerodynamic diameter
- RANTES
Regulated on activation, normal T cell expressed and secreted
- sICAM-1
soluble Intercellular adhesion molecule 1
- sVCAM-1
soluble Vascular cellular adhesion molecule 1
- TNFα
Tumor necrosis factor α
- VCAM-1
Vascular cellular adhesion molecule
Footnotes
DISCLOSURES
None.
References
- 1.Brook RD, Rajagopalan S, Pope CA, III, et al. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–78. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 2.Franklin BA, Brook R, Pope CA., III Air pollution and cardiovascular disease. Curr Probl Cardiol. 2015;40:207–238. doi: 10.1016/j.cpcardiol.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Dockery DW, Pope CA, III, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG, Speizer FE. An association between air pollution and mortality in six U.S. cities. N Engl J Med. 1993;329:1753–9. doi: 10.1056/NEJM199312093292401. [DOI] [PubMed] [Google Scholar]
- 4.Pope CA, III, Burnett RT, Thun MJ, Calle EE, Krewski D, Ito K, Thurston GD. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. J Am Med Assoc. 2002;287:1132–41. doi: 10.1001/jama.287.9.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med. 2007;356:447–58. doi: 10.1056/NEJMoa054409. [DOI] [PubMed] [Google Scholar]
- 6.Kaufman JD, Adar SD, Barr RG, et al. Association between air pollution and coronary artery calcification within six metropolitan areas in the USA (the Multi-Ethnic Study of Atherosclerosis and Air Pollution): a longitudinal cohort study. Lancet. 2016 May 24; doi: 10.1016/S0140-6736(16)00378-0. E-pub ahead of print http://dx.doi.org/10.1016/S0140-6736(16)00378-0. [DOI] [PMC free article] [PubMed]
- 7.Brook RD, Cakmak S, Turner MC, et al. Long-term fine particulate matter exposure and mortality from diabetes in Canada. Diabetes Care. 2013;36:3313–20. doi: 10.2337/dc12-2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pope CA, III, Turner MC, Burnett RT, Jerrett M, Gapstur SM, Diver WR, Krewski D, Brook RD. Relatioships between fine particulate air pollution, cardiometabolic disorders, and cardiovascular mortality. Circ Res. 2015;116:108–15. doi: 10.1161/CIRCRESAHA.116.305060. [DOI] [PubMed] [Google Scholar]
- 9.Pope CA, III, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, Godleski JJ. Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation. 2004;109:71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. [DOI] [PubMed] [Google Scholar]
- 10.Pope CA, III, Ezzati M, Dockery DW. Fine-particulate air pollution and life expectancy in the United States. N Engl J Med. 2009;360:376–86. doi: 10.1056/NEJMsa0805646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samet JM, Dominici F, Curriero FC, Coursac I, Zeger SL. Fine particulate air pollution and mortality in 20 U.S. cities, 1987–1994. N Engl J Med. 2000;343:1742–9. doi: 10.1056/NEJM200012143432401. [DOI] [PubMed] [Google Scholar]
- 12.Mustafić H, Jabre P, Caussin C, Murad MH, Escolano S, Tafflet M, Périer MC, Marijon E, Vernerey D, Empana JP, Jouven X. Main air pollutants and myocardial infarction: a systematic review and meta-analysis. J Am Med Assoc. 2012;307:713–21. doi: 10.1001/jama.2012.126. [DOI] [PubMed] [Google Scholar]
- 13.Shah ASV, Lee KK, McAllister DA, Hunter A, Nair H, Whiteley W, Langrish JP, Newby DE, Mills NL. Short term exposure to air pollution and stroke: systematic review and meta-analysis. BMJ. 2015;350:h1295. doi: 10.1136/bmj.h1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pope CA, III, Muhlestein JB, May HT, Renlund DG, Anderson JL, Horne BD. Ischemic heart disease events triggered by short-term exposure to fine particulate air pollution. Circulation. 2006;114:2443–8. doi: 10.1161/CIRCULATIONAHA.106.636977. [DOI] [PubMed] [Google Scholar]
- 15.Rich DQ, Kipen HM, Zhang J, Kamat L, Wilson AC, Kostis JB. Triggering of transmural infarctions, but not nontransmural infarctions, by ambient fine particles. Environ Health Perspect. 2010;118:1229–34. doi: 10.1289/ehp.0901624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pope CA, III, Muhlestein JB, Anderson JL, Cannon JB, Hales NM, Meredith KG, Le V, Horne BD. Short-term exposure to fine particulate matter air pollution is preferentially associated with the risk of ST-segment elevation acute coronary events. J Am Heart Assoc. 2015;4:e002506. doi: 10.1161/JAHA.115.002506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–60. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Neill MS, Veves A, Zanobetti A, Sarnat JA, Gold Dr, Economides PA, Horton ES, Schwartz J. Diabetes enhances vulnerability to particulate air pollution-associated impairment in vascular reactivity and endothelial function. Circulation. 2005;111:2913–20. doi: 10.1161/CIRCULATIONAHA.104.517110. [DOI] [PubMed] [Google Scholar]
- 19.Krishnan RM, Adar SD, Szpiro AA, Jorgensen NW, Van Hee VC, Barr RG, O’Neill MS, Herrington DM, Polak JF, Kaufman JD. Vascular responses to long- and short-term exposure to fine particulate matter: MESA Air (Multi-Ethnic Study of Atherosclerosis and Air Pollution) J Am Coll Cardiol. 2012;60:2158–66. doi: 10.1016/j.jacc.2012.08.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider A, Neas LM, Graff DW, Herbst MC, Cascio WE, Schmitt MT, Buse JB, Peters A, Devlin RB. Association of cardiac and vascular changes with ambient PM2.5 in diabetic individuals. Part Fibre Toxicol. 2010;7:14. doi: 10.1186/1743-8977-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siponen T, Yli-Tuomi T, Aurela M, Dufva H, Hillamo R, Hirvonen MR, Huttunen K, Pekkanen J, Pennanen A, Salonen I, Tiitanen P, Salonen RO, Lanki T. Source-specific fine particulate air pollution and systemic inflammation in ischaemic heart disease patients. Occup Environ Med. 2015;72:277–83. doi: 10.1136/oemed-2014-102240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hajat A, Allison M, Diez-Roux AV, Jenny NS, Jorgensen NW, Szpiro AA, Vedal S, Kaufman JD. Long-term exposure to air pollution and markers of inflammation, coagulation, and endothelial activation: a repeat-measures analysis in the Multi-Ethnic Study of Atherosclerosis (MESA) Epidemiology. 2015;26:310–20. doi: 10.1097/EDE.0000000000000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patti G, Pasceri V, Melfi R, Goffredo C, Chello M, D’Ambrosio A, Montesanti R, Di Sciascio G. Impaired flow-mediated dilation and risk of restenosis in patients undergoing coronary stent implantation. Circulation. 2005;111:70–5. doi: 10.1161/01.CIR.0000151308.06673.D2. [DOI] [PubMed] [Google Scholar]
- 24.Weiner SD, Ahmed HN, Jin Z, Cushman M, Herrington DM, Nelson JC, Di Tullio MR, Homma S. Systemic inflammation and brachial artery endothelial function in the Multi-Ethnic Study of Atherosclerosis (MESA) Heart. 2014;100:862–6. doi: 10.1136/heartjnl-2013-304893. [DOI] [PubMed] [Google Scholar]
- 25.Tsuchiya K, Nakayama C, Iwashima F, Sakai H, Izumiyama H, Doi M, Hirata Y. Advanced endothelial dysfunction in diabetic patients with multiple risk factors; importance of insulin resistance. J Atheroscler Thromb. 2007;14:303–9. doi: 10.5551/jat.e525. [DOI] [PubMed] [Google Scholar]
- 26.Henry RM, Ferreira I, Kostense PJ, Dekker JM, Nijpels G, Heine RJ, Kamp O, Bouter LM, Stehouwer CD. Type 2 diabetes is associated with impaired endothelium-dependent, flow-mediated dilation, but impaired glucose metabolism is not; The Hoorn Study. Atherosclerosis. 2004;174:49–56. doi: 10.1016/j.atherosclerosis.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Amabile N, Rautou PE, Tedgui A, Boulanger CM. Microparticles: key protagonists in cardiovascular disorders. Semin Throm Hemost. 2010;36:907–16. doi: 10.1055/s-0030-1267044. [DOI] [PubMed] [Google Scholar]
- 28.Rautou PE, Vion AC, Amabile N, Chironi G, Simon A, Tedgui A, Boulandger CM. Microparticles, vascular function, and atherothrombosis. Circ Res. 2011;109:593–606. doi: 10.1161/CIRCRESAHA.110.233163. [DOI] [PubMed] [Google Scholar]
- 29.Suades R, Padro T, Badimon L. The role of blood-borne microparticles in inflammation and hemostasis. Semin Thromb Hemost. 2015;41:590–606. doi: 10.1055/s-0035-1556591. [DOI] [PubMed] [Google Scholar]
- 30.Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Throm Basc Biol. 2011;31:27–33. doi: 10.1161/ATVBAHA.110.218123. [DOI] [PubMed] [Google Scholar]
- 31.Schiro A, Wilkinson FL, Weston R, Smyth JV, Serracino-Inglott F, Alexander MY. Endothelial microparticles as conveyors of information in atherosclerotic disease. Atherosclerosis. 2014;234:295–302. doi: 10.1016/j.atherosclerosis.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 32.van Eeden SF, Tan WC, Suwa T, Mukae H, Terashima T, Fujii T, Qui D, Vincent R, Hogg JC. Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM10) Am J Respir Crit Care Med. 2001;164:826–30. doi: 10.1164/ajrccm.164.5.2010160. [DOI] [PubMed] [Google Scholar]
- 33.Shoenfelt J, Mitkus RJ, Zeisler R, Spatz RO, Powell J, Fenton MJ, Squibb KA, Medvedev AE. Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J Leukoc Biol. 2009;86:303–12. doi: 10.1189/jlb.1008587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alfaro-Moreno E, Torres V, Miranda J, Martínez L, García-Cuellar C, Nawrot TS, Vanaudenaerde B, Hoet P, Ramírez-López P, Rosas I, Nemery B, Osornio-Vargas AR. Induction of IL-6 and inhibition of IL-8 secretion in the human airway cell line Calu-3 by urban particulate matter collected with a modified method of PM sampling. Environ Res. 2009;109:528–35. doi: 10.1016/j.envres.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 35.Huang SL, Hsu MK, Chan CC. Effects of submicrometer particle compositions on cytokine production and lipid peroxidation of human bronchial epithelial cells. Environ Health Perspect. 2003;111:478–82. doi: 10.1289/ehp.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jimenez JJ, Jy W, Mauro LM, Soderland C, Horstman LL, Ahn YS. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res. 2003;109:175–80. doi: 10.1016/s0049-3848(03)00064-1. [DOI] [PubMed] [Google Scholar]
- 37.Lee ST, Chu K, Jung KH, Kim JM, Moon HJ, Bahn JJ, Im WS, Sunwoo J, Moon J, Kim M, Lee SK, Roh JK. Circulating CD62E+ microparticles and cardiovascular outcomes. PLoS One. 2012;7:e35713. doi: 10.1371/journal.pone.0035713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gordon C, Gudi K, Krause A, Sackrowitz R, Harvey BG, Strulovici-Barel Y, Mezey JG, Crystal RG. Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers. Am J Respir Crit Care Med. 2011;184:224–32. doi: 10.1164/rccm.201012-2061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116:1231–44. doi: 10.1161/CIRCRESAHA.116.302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gospodarowicz D, Brown KD, Birdwell CR, Zetter BR. Control of proliferation of human vascular endothelial cells. Characterization of the response of human umbilical vein endothelial cells to fibroblast growth factor, epidermal growth factor, and thrombin. J Cell Biol. 1978;77:774–88. doi: 10.1083/jcb.77.3.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson RA, Teneyck CJ, Hart MN. Growth control in cerebral microvessel-derived endothelial cells. Brain Res. 1986;384:114–20. doi: 10.1016/0006-8993(86)91226-6. [DOI] [PubMed] [Google Scholar]
- 42.Smith S, Francis R, Guilbert L, Baker PN. Growth factor rescue of cytokine mediated trophoblast apoptosis. Placenta. 2002;23:322–30. doi: 10.1053/plac.2001.0783. [DOI] [PubMed] [Google Scholar]
- 43.Bruns CJ, Solorzano CC, Harbison MT, Ozawa S, Tsan R, Fan D, Abbruzzese J, Traxler P, Buchdunger E, Radinsky R, Fidler IJ. Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res. 2000;60:2926–35. [PubMed] [Google Scholar]
- 44.Suffee N, Hlawaty H, Meddahi-Pelle A, Maillard L, Louedec L, Haddad O, Martin L, Laguillier C, Richard B, Oudar O, Letourneur D, Charnaux N, Sutton A. RANTES/CCL5-induced pro-angiogenic effects depend on CCR1, CCR5 and glycosaminoglycans. Angiogenesis. 2012;15(4):727–44. doi: 10.1007/s10456-012-9285-x. [DOI] [PubMed] [Google Scholar]
- 45.Suffee N, Richard B, Hlawaty H, Oudar O, Charnaux N, Sutton A. Angiogenic properties of the chemokine RANTES/CCL5. Biochem Soc Trans. 2011;39:1649–53. doi: 10.1042/BST20110651. [DOI] [PubMed] [Google Scholar]
- 46.Caunt M, Hu L, Tang T, Brooks PC, Ibrahim S, Karpatkin S. Growth-regulated oncogene is pivotal in thrombin-induced angiogenesis. Cancer Res. 2006;66:4125–32. doi: 10.1158/0008-5472.CAN-05-2570. [DOI] [PubMed] [Google Scholar]
- 47.Fimmel S, Devermann L, Herrmann A, Zouboulis C. GRO-alpha: a potential marker for cancer and aging silenced by RNA interference. Ann N Y Acad Sci. 2007;1119:176–89. doi: 10.1196/annals.1404.016. [DOI] [PubMed] [Google Scholar]
- 48.Reinders ME, Sho M, Robertson SW, Geehan CS, Briscoe DM. Proangiogenic function of CD40 ligand-CD40 interactions. J Immunol. 2003;171:1534–41. doi: 10.4049/jimmunol.171.3.1534. [DOI] [PubMed] [Google Scholar]
- 49.Dormond O, Contreras AG, Meijer E, Datta D, Flynn E, Pal S, Briscoe DM. CD40-induced signaling in human endothelial cells results in mTORC2- and Akt-dependent expression of vascular endothelial growth factor in vitro and in vivo. J Immunol. 2008;181:8088–95. doi: 10.4049/jimmunol.181.11.8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gangur V, Birmingham NP, Thanesvorakul S, Joseph S. CCR3 and CXCR3 as drug targets for allergy: principles and potential. Curr Drug Targets Inflamm Allergy. 2003;2:53–62. doi: 10.2174/1568010033344453. [DOI] [PubMed] [Google Scholar]
- 51.Bodnar RJ, Yates CC, Wells A. IP-10 blocks vascular endothelial growth factor-induced endothelial cell motility and tube formation via inhibition of calpain. Circ Res. 2006;98:617–25. doi: 10.1161/01.RES.0000209968.66606.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qi XF, Kim DH, Yoon YS, Jin D, Huang XZ, Li JH, Deung YK, Lee KJ. Essential involvement of cross-talk between IFN-gamma and TNF-alpha in CXCL10 production in human THP-1 monocytes. J Cell Physiol. 2009;220:690–7. doi: 10.1002/jcp.21815. [DOI] [PubMed] [Google Scholar]
- 53.Hohensinner PJ, Rychli K, Zorn G, Hülsmann M, Berger R, Mörtl D, Richter B, Huber K, Wojta J, Pacher R, Riessner A. Macrophage-modulating cytokines predict adverse outcome in heart failure. Thromb Haemost. 2010;103:435–41. doi: 10.1160/TH09-06-0399. [DOI] [PubMed] [Google Scholar]
- 54.Cavusoglu E, Marmur JD, Yanamadala S, Chopra V, Hegde S, Nazli A, Singh KP, Zhang M, Eng C. Elevated baseline plasma IL-8 levels are an independent predictor of long-term all-cause mortality in patients with acute coronary syndrome. Atherosclerosis. 2015;242:589–94. doi: 10.1016/j.atherosclerosis.2015.08.022. [DOI] [PubMed] [Google Scholar]
- 55.Edsfeldt A, Grufman H, Asciutto G, Nitulescu M, Persson A, Nilsson M, Nilsson J, Gonçalves I. Circulating cytokines reflect the expression of pro-inflammatory cytokines in atherosclerotic plaques. Atherosclerosis. 2015;241:443–9. doi: 10.1016/j.atherosclerosis.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 56.Carr MW, Roth SJ, Luther E, Rose SS, Springer TA. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci U S A. 1994;91:3652–6. doi: 10.1073/pnas.91.9.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168:3195–204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- 58.Taraboletti G, D’Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol. 2002;160:673–80. doi: 10.1016/S0002-9440(10)64887-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levitzky YS, Guo CY, Rong J, Larson MG, Walter RE, Keaney JF, Jr, Sutherland Pa, Vasan A, Lipinska I, Evans JC, Benjamin EJ. Relation of smoking status to a panel of inflammatory markers: the framingham offspring. Atherosclerosis. 2008;201:217–24. doi: 10.1016/j.atherosclerosis.2007.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de-Torres JP, Blanco D, Alcaide AB, Levänen B, Christenson K, Jirholt P, Åhrén C, Qvarfordt I, Ekberg-Jansson A, Lindén A. Smokers with CT detected emphysema and no airway obstruction have decreased plasma levels of EGF, IL-15, IL-8 and IL-1ra. PloS One. 2013;8:e60260. doi: 10.1371/journal.pone.0060260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andelid K, Tengvall S, Andersson A, et al. Systemic cytokine signaling via IL-17 in smokers with obstructive pulmonary disease: a link to bacterial colonization? Int J Chron Obstruct Pulmon Dis. 2015;10:689. doi: 10.2147/COPD.S76273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Symvoulakis EK, Vardavas CI, Fountouli P, Stavroulaki A, Antoniou KM, Duijker G, Tzatzarakis MN, Sfiridaki K, Bolonaki E, Alegakis T, Tsatsakis AM. Time interval from cigarette smoke exposure to blood donation and markers of inflammation: should a smoking cut-off be designated? Xenobiotica. 2010;40:613–20. doi: 10.3109/00498254.2010.500745. [DOI] [PubMed] [Google Scholar]
- 63.Conklin BS, Zhao W, Zhong DS, Chen C. Nicotine and cotinine up-regulate vascular endothelial growth factor expression in endothelial cells. Am J Pathol. 2002;160:413–8. doi: 10.1016/S0002-9440(10)64859-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garlichs CD, Cicha I, Raaz D, Meyer L, Stumpf C, Klinghammer L, Yilmaz A, Daniel WG. CD40/CD154 system and pro-inflammatory cytokines in young healthy male smokers without additional risk factors for atherosclerosis. Inflamm Res. 2009;58:306–11. doi: 10.1007/s00011-008-8084-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.