

Abstract
Autophagy is a cellular self-eating process essential for stress response and maintaining tissue homeostasis by lysosomal degradation of unwanted or damaged proteins and organelles. Here, we show that cells with defective mitochondria induce autophagy to promote cell survival through activating the AMPK pathway. Loss of mitochondrial complex III protein cytochrome b activates the AMPK signaling and induced autophagy. Inhibiting mitochondria energetics by mitochondria-targeted agents activates the AMPK signaling and induced autophagy. Genetic inhibition of AMPK inhibits autophagy induction in cells with defective mitochondria, while genetic inhibition of autophagy has no effect on AMPK activation. Mitochondria dysfunction has no effect of DNA repair of UV-induced DNA damage. However, mitochondria dysfunction sensitizes cells to apoptosis induced by UV radiation. Genetic inhibition of autophagy or AMPK sensitized cells to apoptosis in cells with defective mitochondria. Our results demonstrate that AMPK and autophagy senses mitochondria dysfunction and serves as a mechanism for survival. Our findings may provide new insights into the interplay between mitochondria function and autophagy process in maintaining tissue homeostasis, and suggest that this interaction may play important roles in diseases such as cancer and neurodegeneration.
Keywords: AMPK, Apoptosis, Autophagy, DNA repair, Mitochondria, Ultraviolet radiation, UV
Introduction
Macroautophagy (hereafter autophagy) is a cellular self-eating process, in which cellular proteins, cytoplasm, and organelles are captured through a double-membrane structure known as the autophagosome and then targeted for proteolytic degradation in lysosomes.1, 2 Dysfunction of autophagy can affect susceptibility to multiple diseases, including neurodegeneration, microbial infection, metabolic diseases, cardiovascular diseases, aging, and cancer.2, 3, 4 Occurring at a low basal level, autophagy can be induced to maintain tissue homeostasis in response to a variety of physiological and pathological stresses,5 including energy starvation, DNA damage, and infections.
One of the energy-generating organelle is the mitochondrion via oxidative phosphorylation. Emerging evidence support a critical role of mitochondria dysfunction in the pathogenesis of several diseases, including neurodegeneration,6, 7 diabetes,8 and cancer.9 In response to diverse stresses including starvation, photodamage, and persistent mitochondrial DNA damage, damaged or dysfunctional mitochondria can be removed by a selective autophagy pathway called mitophagy.10, 11 However, it is poorly understood how cells respond to mitochondrial dysfunction.
Here we show that inhibiting mitochondrial function genetically or by mitochondria-targeted antioxidant activates the AMPK signaling and induces autophagy. Induction of autophagy is critical for cell survival, since inhibition of autophagy sensitizes cells with defective mitochondria to apoptosis.
Materials & methods
Cell culture
Human HaCaT keratinocytes (kindly provided by Dr. Fusenig), AMPK wild-type (WT) and knockout (KO) mouse embryonic fibroblast (MEF) cells,12 ATG5 WT and KO MEF cells (kindly provided by Dr. Mizushima), and 143B WT and cytochrome b null (CYTB KO) cells (kindly provided by Dr. Chandel)13, 14 were maintained in monolayer culture in 95% air/5% CO2 at 37 °C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units per mL penicillin, and 100 mg per mL streptomycin (Invitrogen, Carlsbad, California). Normal human epidermal keratinocytes (NHEK) cells were obtained from Clonetics (Lonza, Walkersville, MD) and cultured in KGM Gold BulletKit medium (Lonza, Walkersville, MD) according to the manufacturer's instructions. MEF and HaCaT cells were cultured for less than 20 passages. NHEK cells were cultured for less than 4 passages. No authentication was done.
UV radiation
UV radiation was performed as described previously.15 Our UV radiation was monitored every other week to measure the exposure output and dose. Our UV system emits UVB radiation (51%) and UVA (49%) but does not emit UVC radiation.
Western blotting
Western blotting was performed as described previously.16, 17 Antibodies used were as follows: Ampk, p-Ampk (Thr172), LC3-I/II, ACC, p-ACC (Ser79), p4EBP1 (Ser65), 4EBP1, LC3-I/II (Cell Signaling), and PARP and GAPDH (Santa Cruz). Cytosolic and nuclear proteins were isolated as described previously.16
Determination of two major forms of UV-induced DNA damage in genomic DNA by slot blot assay
Slot blot assays of CPD and 6-4 PP were performed as described previously.18 Briefly, cells were collected at different time points post-UV and DNA was isolated using a QIAamp DNA Mini Kit (Qiagen, 51304). The DNA concentration was calculated from the absorbance at 260 nm using NanoDrop 1000 (NanoDrop products, Wilmington, DE). The CPD and 6-4 PP in DNA were quantified by slot blot (Bio-Rad) with antibodies (COSMO BIO Co., TDM-2 for CPD and 64 M-2 for 6-4 PP). For examining repair kinetics, the percentage (%) of repair was calculated by comparing the optical density at the indicated time to that of the corresponding absorbance at time zero when there was no opportunity for repair and 100% of CPDs (or 6-4PPs) were present post-UV.
Statistical analyses
Statistical analyses were performed using Prism 6 (GraphPad software, San Diego, CA, USA). Data were expressed as the mean of at least three independent experiments, and analyzed by Student's t-test. Error bars indicate the standard deviations of the means (S.D.). A p-value of <0.05 was considered statistically significant.
Results
Loss of mitochondria function activates the AMPK signaling
To determine the role of mitochondria dysfunction in cellular response, we compared the difference in the molecular signaling between wild-type (WT) cells and cells containing a loss-of-function mutation in electron transport chain complex III (cytochrome b-c1 complex) (CYTB KO). Despite the loss of mitochondrial respiration function, both WT and CYTB KO cells proliferate at comparable rates.13, 14 As compared with WT cells, however, CYTB KO cells showed increased phosphorylation of AMPK (Thr 172) and ACC (Ser 79) (Fig. 1), a known AMPK target. These data indicate that loss of mitochondria respiration activates the AMPK signaling.
Fig. 1.
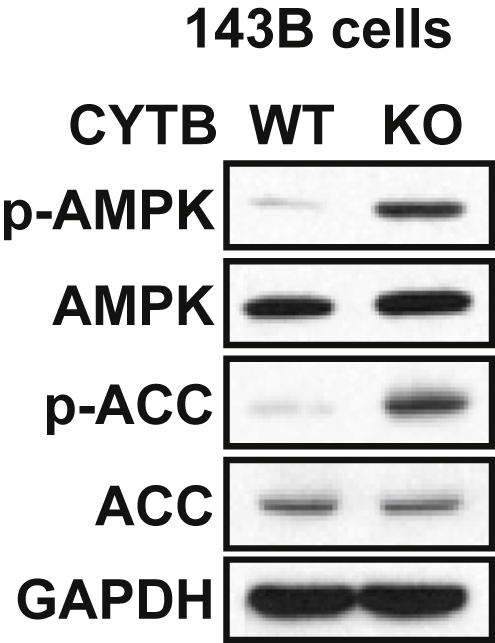
Loss of CYTB activates the AMPK signaling. Immunoblot analysis of p-AMPK (Thr 172), AMPK, p-ACC (Ser 79) and GAPDH in 143B cells with wild-type (WT) or loss of CYTB (KO, knockout).
Inhibition of mitochondria function by mitochondria-targeted antioxidants activates the AMPK signaling
Mitochondria-targeted antioxidants including Mito-carboxy proxyl (Mito-CP) have been shown to inhibit Cell proliferation and synergize with 2-deoxyglucose to trigger cell death in breast cancer cells.19, 20, 21 Mito-CP together with 2-deoxy-glucose (2-DG) inhibits cell proliferation and induces apoptosis of pancreatic cancer cells via modification of mitochondrial bioenergetics rather than its antioxidant properties.21 Therefore Mito-CP can have effect similar to loss of CYTB. Indeed, in both normal human epidermal keratinocytes (NHEK) and HaCaT cells, Mito-CP treatment increased the phosphorylation of AMPK (Thr 172) and ACC (Ser 79) (Fig. 2A,B), indicating that Mito-CP activates the AMPK signaling.
Fig. 2.

Inhibition of mitochondria function by Mito-CP activates the AMPK signaling. (A, B) Immunoblot analysis of p-AMPK (Thr 172), AMPK, p-ACC (Ser 79) and GAPDH in NHEK cells (A) and HaCaT cells (B) treated with vehicle or Mito-CP (1 μM) for 24 h.
Mitochondria dysfunction induces autophagy
Autophagy is a cellular catabolic mechanism to promote cell survival in response to diverse cellular stresses5 including energy stress and UV radiation.22, 23 To determine whether mitochondrial defects affect autophagy, we assessed the difference in LC3-II formation in cells with or without defective mitochondria treated with vehicle or the lysosome inhibitor bafilomycin A1 (BfA1). The conversion of LC3-I – LC3-II is one of the distinctive hallmarks of autophagy.24, 25 Following lysosome inhibition, loss of CYTB increased the formation of LC3-II, while WT cells showed no change in LC3-II level (Fig. 3A). Similarly, Mito-CP increased the LC3-II formation in HaCaT cells treated with bafilomycin A1 (Fig. 3B). These results indicate that defective mitochondria induce autophagy.
Fig. 3.

Mitochondria dysfunction activates autophagy. (A) Immunoblot analysis of LC3-I/II and GAPDH in WT and CYTB KO 143B cells treated with vehicle or bafilomycin A1 (BfA1, 20 nM) for 6 h. (B) Immunoblot analysis of LC3-I/II and GAPDH in HaCaT cells pretreated with vehicle or bafilomycin A1 (BfA1, 20 nM) for 2 h and in combination with vehicle or Mito-CP (1 μM) for 6 h.
AMPK activation mediates autophagy induction in cells with defect mitochondria
To determine the crosstalk between AMPK and autophagy in response to mitochondria dysfunction, we compared the effect of Mito-CP on the AMPK signaling in cells with or without loss of the essential autophagy gene ATG5 (ATG5 KO), and on autophagy induction in cells with wild-type (WT) or AMPK deficient cells (AMPK KO). Mito-CP increased AMPK phosphorylation in both WT and ATG5 KO cells (Fig. 4A), indicating autophagy does not affect Mito-CP-induced AMPK activation. Mito-CP induced AMPK activation and autophagy in WT cells, while it had little effect on autophagy induction in AMPK KO cells (Fig. 4B,C), indicating that Mito-CP induced autophagy is mediated through AMPK activation.
Fig. 4.

AMPK activation is required for autophagy induction in cells with defective mitochondria. (A) Immunoblot analysis of p-AMPK (Thr 172), AMPK, LC3-I/II and GAPDH in mouse embryonic fibroblasts (MEF) with wild-type (WT) or ATG5 knockout (KO) treated with vehicle or Mito-CP (1 μM) for 6 h. (B) Immunoblot analysis of p-AMPK (Thr 172), AMPK, LC3-I/II and GAPDH in MEF cells with wild-type (WT) or AMPK knockout (KO) treated with vehicle or Mito-CP (1 μM) for 6 h. (C) Immunoblot analysis of LC3-I/II and GAPDH in cells same as in (B) except treatment with bafilomycin A1 (BfA1, 20 nM) for 6 h.
Mitochondria defects do not affect repair of UV-induced DNA damage
Recently we have shown that AMPK activation and autophagy promotes repair of UV-induced DNA damage.26, 27 To determine whether Mito-CP affect repair of UV-induced DNA damage, we compared DNA repair kinetics between cells with or without defective mitochondria. Neither Mito-CP nor loss of CYTB had effect on repair of either cyclobutane pyrimidine dimer (CPD) or 6-4 photoproducts (6-4 PP) (Fig. 5A–D), two major DNA damage products induced by UV-irradiation. These data demonstrate that although defective mitochondria induce AMPK activation and autophagy, they do not affect UV-induced DNA damage repair.
Fig. 5.

Mitochondria dysfunction does not affect DNA repair following UV-induced damage. (A, B) Slot blot analysis of CPD levels (A) and 6-4 PP (B) levels in HaCaT cells at 0, 6, and 24 h post UV-irradiation (20 mJ/cm2) after pretreatment with vehicle or Mito-CP (1 μM) for 1 h. (C–D) Slot blot analysis of CPD levels (C) and 6-4 PP (D) levels in WT and CYTB KO 143B cells at 0, 6, and 24 h post UV-irradiation (20 mJ/cm2). The quantification of DNA repair in percentage is shown below the corresponding slot blot assay.
Mitochondria dysfunction interacts with autophagy and AMPK to promote cell survival
To determine the function of AMPK or autophagy in cellular response to mitochondrial dysfunction, we assessed the effect of mitochondria inhibition on apoptosis between cells with or without AMPK inhibition or autophagy inhibition. Loss of CYTB sensitizes cells to UV-induced apoptosis as indicated by PARP cleavage (Fig. 6A). Inhibiting autophagy by its inhibitor 3-MA increased UV-induced apoptosis (Fig. 6A). Mito-CP induced apoptosis in WT cells, while CYTB-deficient cells were resistant to Mito-CP-induced apoptosis (Fig. 6B). Autophagy deficiency sensitized cells to Mito-CP-induced apoptosis (Fig. 6C). Loss of AMPK sensitized cells to Mito-CP-induced apoptosis (Fig. 6D). These results demonstrate that AMPK activation and autophagy induction promote cell survival.
Fig. 6.

AMPK-mediated autophagy promotes cell survival in cells with defective mitochondria. (A) Immunoblot analysis of PARP and GAPDH in WT and CYTB KO 143B cells at 24 h post-UV (20 mJ/cm2) after pretreatment with the autophagy inhibitor 3-MA (5 mM) for 3 h. (B) Immunoblot analysis of PARP and GAPDH in WT and CYTB KO 143B cells treated with vehicle or Mito-CP (1 μM) for 24 h. (C) Immunoblot analysis of PARP and GAPDH in WT and ATG5 KO MEF cells treated with vehicle or Mito-CP (1 μM) for 24 h. (D) Immunoblot analysis of PARP and GAPDH in WT and AMPK KO MEF cells treated with vehicle or Mito-CP (1 μM) for 24 h. (E) Schematic for the regulation of AMPK and autophagy by mitochondrial dysfunction in cell survival.
Discussion
Autophagy is a cellular self-eating process. In response to stresses, the cells can activate both autophagy and apoptosis pathways. On one hand, autophagy induction can suppress cell death such as apoptosis; on the other hand, it can also lead to the alternative form of cell death, i.e. autophagic cell death.28 In this study, we show that cells with defective mitochondria induce autophagy to promote cell survival through activating the AMPK pathway (Fig. 6E). Mitochondria dysfunction has no effect of DNA repair of UV-induced DNA damage. However, mitochondria dysfunction sensitizes cells to apoptosis induced by UV radiation, possibly due to reduced energy production. Genetic inhibition of autophagy or AMPK sensitized cells to apoptosis in cells with defective mitochondria. Our results demonstrate that AMPK and autophagy senses mitochondria dysfunction and serves as a mechanism for survival. Our findings may provide new insights into the connection between mitochondria function and autophagy process in maintaining tissue homeostasis, and suggest that this interplay may play important roles in diseases such as cancer and neurodegeneration.
Mitochondria carry out diverse functions, producing ATP and many biosynthetic intermediates. Mitochondrial dysfunction is implicated in multiple common diseases, including neurodegeneration, cardiomyopathies, metabolic syndromes, cancer, and obesity.29 Mitochondria can be degraded through autophagy pathway called mitophagy. Inhibiting mitochondria respiration or bioenergetics induces autophagy through AMPK. It is likely that depletion of ATP triggered the activation of the energy-sensing AMPK signaling, which subsequently induced autophagy.30
Autophagy has been demonstrated to mediate cell survival under stress conditions,5 including energy stress and UV radiation.22, 23 On one hand, autophagy defects in the central nervous system lead to neurodegeneration in mice.31, 32 On the other hand, autophagy has been utilized by some cancer cells for metabolic and survival advantages.4, 33, 34, 35 We found that Mito-CP and genetic loss of mitochondrial respiration induces autophagy to promote cell survival. Indeed, a recent report demonstrated that another mitochondria-targeted agent mitoquinone induces autophagy in breast cancer cells; such autophagy induction by mitoquinone is positively correlated with growth arrest and cell death.36
Mitochondria-targeted agents including Mito-carboxy proxyl (Mito-CP) has been synthesized and found as a mitochondria-targeted antioxidant.37 Mito-CP also decreases ATP production. The effect of Mito-CP on cell proliferation and apoptosis is found to be mediated through inhibiting mitochondria bioenergetics rather than its antioxidant function.21 Here, we found that Mito-CP activated AMPK signaling and induced autophagy similar to genetic loss of mitochondria respiration function. Cells with genetic loss of CYTB were refractory to Mito-CP-induced apoptosis, indicating that Mito-CP targeted mitochondria respiration.
Opposite to what we predicted, mitochondrial dysfunction did not affect DNA repair of UV-induced genomic damage. We have recently shown that both AMPK and autophagy positively regulates UV-induced DNA damage repair.26, 27 It is possible that induction of AMPK and autophagy counteracted the potential negative regulation of DNA repair in cells with defective mitochondria.
In conclusion, we have demonstrated that cells with defective mitochondria induce autophagy to promote cell survival through activating the AMPK pathway. AMPK and autophagy senses mitochondria dysfunction and serves as a mechanism for survival. Our findings may provide new insights into the connection between mitochondria function and autophagy process in maintaining tissue homeostasis, and suggest important roles of mitochondria-autophagy interplay in diseases such as cancer and neurodegeneration.
Conflict of interest
The authors have no conflict of interest.
Financial disclosures
The authors have received no financial support from and have no interest in any commercial source that is related directly or indirectly to the scientific work presented in this article.
Acknowledgments
This work was supported by the NIH/NIEHS grant ES024373 and ES016936 (YYH), the American Cancer Society (ACS) grant RSG-13-078-01 (YYH), the University of Chicago Cancer Research Center (P30 CA014599), the CTSA (UL1 TR000430), and the University of Chicago Friends of Dermatology Endowment Fund. We thank Dr. Norbert Fusenig for providing the HaCaT cells (human keratinocytes and epithelial cells), Dr. Navdeep S. Chandel for providing the143B WT and cytochrome b null (CYTB KO) cells, and Dr. Noboru Mizushima for kindly providing WT and Atg5 KO MEFs.
Footnotes
Peer review under responsibility of Chongqing Medical University.
References
- 1.Klionsky D.J. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 2.Mizushima N., Levine B., Cuervo A.M. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi A.M., Ryter S.W., Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 4.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kroemer G., Marino G., Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bossy-Wetzel E., Barsoum M.J., Godzik A. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 7.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 8.Lowell B.B., Shulman G.I. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 9.Brandon M., Baldi P., Wallace D.C. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–4662. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 10.Youle R.J., Narendra D.P. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bess A.S., Crocker T.L., Ryde I.T. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Res. 2012;40:7916–7931. doi: 10.1093/nar/gks532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laderoute K.R., Amin K., Calaoagan J.M. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinberg F., Hamanaka R., Wheaton W.W. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U. S. A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullen A.R., Wheaton W.W., Jin E.S. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ming M., Han W., Maddox J. UVB-induced ERK/AKT-dependent PTEN suppression promotes survival of epidermal keratinocytes. Oncogene. 2010;29:492–502. doi: 10.1038/onc.2009.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ming M., Shea C.R., Guo X. Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc Natl Acad Sci U. S. A. 2010;107:22623–22628. doi: 10.1073/pnas.1010377108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiang L., Zhao B.Z., Ming M. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U. S. A. 2014;111:9241–9246. doi: 10.1073/pnas.1322913111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Ming M., Soltani K., Shea C.R. Dual role of SIRT1 in UVB-induced skin tumorigenesis. Oncogene. 2015;34:357–363. doi: 10.1038/onc.2013.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng G., Lopez M., Zielonka J. Mitochondria-targeted nitroxides exacerbate fluvastatin-mediated cytostatic and cytotoxic effects in breast cancer cells. Cancer Biol Ther. 2011;12:707–717. doi: 10.4161/cbt.12.8.16441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng G., Zielonka J., Dranka B.P. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012;72:2634–2644. doi: 10.1158/0008-5472.CAN-11-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng G., Zielonka J., McAllister D. Antiproliferative effects of mitochondria-targeted cationic antioxidants and analogs: role of mitochondrial bioenergetics and energy-sensing mechanism. Cancer Lett. 2015;365:96–106. doi: 10.1016/j.canlet.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiang L., Wu C., Ming M. Autophagy controls p38 activation to promote cell survival under genotoxic stress. J Biol Chem. 2013;288:1603–1611. doi: 10.1074/jbc.M112.415224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L.H., Chu P.M., Lee Y.J. Targeting protective autophagy exacerbates UV-triggered apoptotic cell death. Int J Mol Sci. 2012;13:1209–1224. doi: 10.3390/ijms13011209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabeya Y., Mizushima N., Ueno T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klionsky D.J., Abeliovich H., Agostinis P. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu C.L., Qiang L., Han W. Role of AMPK in UVB-induced DNA damage repair and growth control. Oncogene. 2013;32:2682–2689. doi: 10.1038/onc.2012.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiang L., Zhao B., Shah P. Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy. 2015 Nov 13;0 doi: 10.1080/15548627.2015.1110667. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiuri M.C., Zalckvar E., Kimchi A. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 29.Nunnari J., Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mihaylova M.M., Shaw R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komatsu M., Waguri S., Chiba T. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 32.Nixon R.A. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 33.Kimmelman A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galluzzi L., Pietrocola F., Bravo-San Pedro J.M. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhi X., Zhong Q. Autophagy in cancer. F1000Prime Rep. 2015;7:18. doi: 10.12703/P7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao V.A., Klein S.R., Bonar S.J. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J Biol Chem. 2010;285:34447–34459. doi: 10.1074/jbc.M110.133579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhanasekaran A., Kotamraju S., Karunakaran C. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: role of mitochondrial superoxide. Free Radic Biol Med. 2005;39:567–583. doi: 10.1016/j.freeradbiomed.2005.04.016. [DOI] [PubMed] [Google Scholar]