

The maturation of tomato fruits is accompanied by widespread reprogramming of the epigenome.
Eating a brilliantly colored, aromatic piece of fruit that has been ripened to perfection remains one of life’s delights. Given how important the ripening process is in determining the quality, taste and aroma of our produce, it is surprising that understanding of this process has not matured further in recent years. In this issue, Zhong et al.1 report that the developmental trigger of fruit ripening in tomato is an epigenetic switch. Using whole-genome bisulfite sequencing, they delineate how the methylome changes during wild-type fruit development, providing a resource for researchers and breeders alike.
The chemical trigger that controls ripening of climacteric fruits, such as banana and tomato, is the ‘fruit ripening’ hormone ethylene. Plants produce ethylene gas (together with reaction co-products CO2 and hydrogen cyanide) from methionine by way of a cyclic amino acid intermediate, 1-aminocyclopropane-1-carboxylic acid. Through the expression of specific ethylene receptors and a conserved signaling pathway, plants can ‘smell’ minute quantities of ethylene and respond by activating transcription of many thousands of genes. Some of the proteins produced in response to ethylene detection promote fruit degreening, tissue softening and the release of volatile compounds, resulting in an aroma and sweet taste that attract both humans and various seed-dispersing herbivores. However, plants with fleshy fruits, such as banana and tomato, have a developmental brake that prevents premature ripening until the seeds have matured, regardless of how much ethylene the plant produces or detects. In tomato, this elusive developmental switch, which is a crucial point of no return in the ripening process, occurs just before the ‘breaker stage’ (the time at which the fruit begins to ripen, visible as a change of color), and it ensures that ethylene-induced ripening does not occur before seed maturation2.
A possible link between ripening and DNA methylation was suggested in a previous study3 that identified a natural epigenetic mutation (or epiallele) in the tomato colorless nonripening gene (Cnr). Except for rare reversion events, Cnr epimutants do not ripen and their fruits remain forever green (Fig. 1a). This nonripening phenotype could not be attributed to any detectable genetic alteration in the Cnr gene, which codes for a SQUAMOSA promoter binding protein–box transcription factor. Rather, the phenotype was due to heritable cytosine hypermethylation of the Cnr gene promoter and inhibition of its expression.
Figure 1.
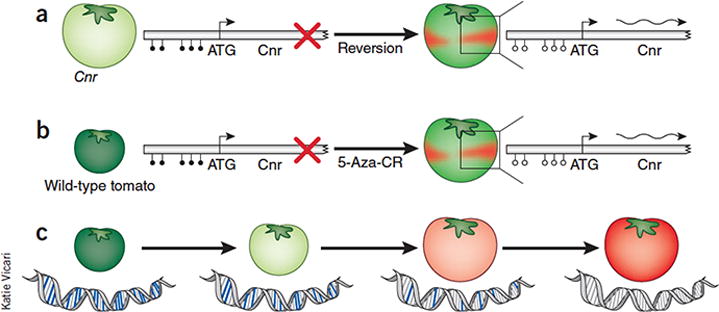
Evidence for epigenetic control of ripening. (a) A natural epiallele in Cnr prevents ripening, resulting in colorless fruit. The Cnr mutant is caused by an epimutation that blocks fruit ripening. Bisulfite sequencing revealed hypermethylation (filled circles) of the Cnr promoter, which resulted in inhibition of RIN transcription factor binding, preventing Cnr gene expression and fruit ripening. Very rare reversion events result in partial ripening and wild-type sectors (red) in the green fruit. Bisulfite sequencing of the Cnr promoter revealed a demethylated state (open circles), allowing binding of RIN to the promoter, Cnr expression (wavy arrow) and activation of ripening3. (b). Unripe tomato fruit is injected with 5-azacytidine, an inhibitor of DNA methylation. Before injection, the Cnr gene promoter is hypermethylated, the RIN protein is inhibited from promoter binding and the gene is not transcribed. Thirteen days after drug injection, a time still too early for normal ripening, the previously green tomato is partially ripe (red stripes), indicating drug-induced premature ripening. In ripe tissue (red), Cnr is transcribed and the promoter is unmethylated, whereas in adjacent, unripe tissue (green), the Cnr promoter is heavily methylated and the gene is not expressed1. 5-Aza-CR, 5-azacytidine. (c) Progressive states of tomato fruit ripening are accompanied by a developmental program of promoter demethylation in which the promoters of hundreds of fruit ripening genes show a gradual decrease in promoter methylation (indicated in blue in the DNA of the figure), which is accompanied by increased binding of RIN (and other transcription factors) to their promoters and a concomitant increase in RNA expression as fruit ripening progresses1.
Zhong et al.1 use several experimental approaches to show that cytosine methylation likely has a regulatory role in normal fruit development that functions by restricting the timing of the ripening process. First, they demonstrate that injection of immature (green) fruit with 5-azacytidine, a well-known inhibitor of cytosine DNA methylation in mammalian cells and a first-line treatment for myelodysplastic syndromes4, prematurely induces fruit ripening (Fig. 1b). Using bisulfite sequencing to detect cytosine DNA methylation5 in selected target gene promoters, they find concomitant demethylation of the Cnr gene promoter, Cnr gene expression and expression of various fruit-ripening genes in red sectors of the 5-azacytidine–ripened tomatoes. Altered cytosine methylation and increased expression of ripening genes are not observed in adjacent, unripened, green fruit tissue. These findings may indicate that inhibition of promoter methylation is sufficient to remove the developmental restriction on ripening.
To examine whether more widespread changes in genome methylation occur during the progression from green to red fruit, the authors use a whole-genome bisulfite sequencing method6 to produce the first base pair–resolution methylome maps of the tomato epigenome. Profiling of both methylomes and transcriptomes at four stages of tomato fruit development from immature to fully ripe (and of two fruit-ripening mutants and leaf tissues as controls) reveals that DNA methylation is substantially altered in ~1% of the 900-Mb tomato genome during fruit development. Interestingly, the average level of methylation in the 5′ ends of genes (that is, predicted promoters) gradually declines during fruit ripening, whereas promoter methylation remains elevated in the two ripening-deficient mutants carrying Cnr and rin (ripening inhibitor). As the latter gene encodes a MADS-box transcription factor7, this provides further evidence of a link between DNA methylation and developmental control of fruit ripening.
Through a detailed analysis of the Cnr promoter in wild-type fruits, the authors identify two differentially methylated regions that are demethylated during ripening; these two promoter elements remain hypermethylated in the Cnr epiallele and in rin loss-of-function mutants. Similar observations of progressive demethylation during ripening are made for the putative promoters of hundreds of known ripening-associated genes, further strengthening the connection between promoter hypomethylation and ripening. A previous study showed that the binding of RIN to a limited set of promoters was inhibited in the Cnr epimutant7, indicating that promoter hypermethylation may prevent RIN binding. To further explore this possibility, the authors use genome-wide location analysis (chromatin immunoprecipitation sequencing; ChIP-seq) to map RIN transcription factor binding sites in wild-type and rin mutant fruits. Combining the CHIP-seq data with gene expression data yields a high-confidence set of 262 RIN target genes; strikingly, these genes include the vast majority of all known fruit-ripening genes.
The average methylation level of these genes at RIN binding sites is lower (hypomethlyated) than that of neighboring genomic regions, and methylation further decreases during fruit maturation. Moreover, RIN target gene transcription negatively correlates with the methylation status of RIN binding sites (Fig. 1c). These findings are consistent with studies of mammalian genes, where hypomethylation of gene-regulatory regions is commonly observed at sites of DNA-protein interaction8. Interestingly, the authors observe very little change in DNA methylation state on transposable elements during fruit maturation, in stark contrast to the novel developmental demethylation events recently reported in the endosperm and pollen of Arabidopsis, which occur mainly on transposable elements9,10.
As with other studies of widespread changes in DNA methylation and gene regulation, the results of Zhong et al.1 are largely correlative, and one should be cautious in drawing conclusions about a cause-and-effect relationship11. Nevertheless, three key observations support the hypothesis that genome methylation contributes to repression of fruit ripening before seed maturation: first, promoters of ripening genes become demethylated during development but are hypermethylated in ripening-deficient mutants; second, pharmacological studies reveal that 5-azacytidine induces early ripening; and third, RIN does not bind hypermethylated Cnr promoters.
Fortunately, direct testing of the role of DNA methylation during fruit development may soon be made possible by new technologies for epigenome editing. For example, the importance of cytosine methylation in the Cnr promoter (or any other promoter) could be tested by fusing proteins that write (methyltransferases) or erase (demethylases) cytosine base modifications to custom-designed DNA binding transcription activator-like effector proteins. Regulated expression of such transgenes in plants might provide a means of targeting cytosine methylation or demethylation events to specific cis-elements (e.g., RIN binding sites) in order to assess the functions of epigenetic marks in specific developmental contexts such as ripening. ‘Epigenetic engineering’ might prove especially useful for trait improvement in crops that have little genetic diversity owing to breeding bottlenecks, such as the domesticated soybean. For breeders, the main outcome of this study is the realization that the identification of epigenetic variation in genes that encode economically important plant traits might provide an important new resource for creating improved crop varieties.
Footnotes
Editor’s note: Dr. Ecker reviewed a preprint of this paper before acceptance but had no role in revision of the manuscript.
COMPETING FINANCIAL INTERESTS
The author declares no competing financial interests.
References
- 1.Zhong S, et al. Nat Biotechnol. 2013;31:154–159. doi: 10.1038/nbt.2462. [DOI] [PubMed] [Google Scholar]
- 2.Klee HJ, Giovannoni JJ. Annu Rev Genet. 2011;45:41–59. doi: 10.1146/annurev-genet-110410-132507. [DOI] [PubMed] [Google Scholar]
- 3.Manning K, et al. Nat Genet. 2006;38:948–952. doi: 10.1038/ng1841. [DOI] [PubMed] [Google Scholar]
- 4.Yang X, Lay F, Han H, Jones PA. Trends Pharmacol Sci. 2010;31:536–546. doi: 10.1016/j.tips.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciark SJ. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lister R, et al. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martel C, Vrebalov J, Tafelmeyer P, Giovannoni JJ. Plant Physiol. 2011;157:1568–1579. doi: 10.1104/pp.111.181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lister R, et al. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ibarra CA, et al. Science. 2012;337:1360–1364. doi: 10.1126/science.1224839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calarco JP, et al. Cell. 2012;151:194–205. doi: 10.1016/j.cell.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schubeler D. Science. 2012;338:756–757. doi: 10.1126/science.1227243. [DOI] [PubMed] [Google Scholar]