

Abstract
The extracellular matrix (ECM) is a guiding force that regulates various developmental stages of the breast. In addition to providing structural support for the cells, it mediates epithelial-stromal communication and provides cues for cell survival, proliferation, and differentiation. Perturbations in ECM architecture profoundly influence breast tumor progression and metastasis. Understanding how a dysregulated ECM can facilitate malignant transformation is crucial to designing treatments to effectively target the tumor microenvironment. Here, we address the contribution of ECM mechanics to breast cancer progression, metastasis, and treatment resistance and discuss potential therapeutic strategies targeting the ECM.
Keywords: Extracellular matrix (ECM), Breast cancer, Desmoplasia, Mechanosignaling, Metastasis, Treatment resistance
1 Introduction
The breast tissue is unique in its ability to evolve structurally throughout embryonic development, puberty, and pregnancy [1, 2]. The extracellular matrix (ECM) of the breast is a guiding force that regulates the various stages of breast development and differentiation [3]. For example, collagens and glycosaminoglycans are important during mammary gland development [3, 4]. During branching morphogenesis, ECM composition is delicately balanced between deposition of ECM components and their concomitant degradation by matrix metalloproteinases (MMPs) [5]. The ECM during lactation is highly compliant, a structural requirement for differentiation of mammary epithelial cells [6, 7]. Involution is characterized by enhanced deposition of collagen, together with proteolysis of laminin and fibronectin and expression of MMPs [8, 9]. Thus, the role of the extracellular matrix (ECM) goes significantly beyond providing architectural support to the cells.
The ECM regulates diverse cellular behavior such as proliferation, differentiation, and migration [10, 11], in addition to regulating the survival and differentiation of the neighboring endothelial cells [12–14], as well as immune responses within the tissue [15]. Moreover, the ECM mediates communication between the epithelial and stromal cells, and perturbations in this process can lead to breast cancer. In this review, we will discuss the influence of ECM mechanics on breast cancer progression, formation of the pre-metastatic niche and metastasis, and discuss its implications on mechano-signaling, immune infiltration, and the tumor vasculature. We will additionally address mechanisms by which ECM contributes to treatment resistance in breast cancer and potential therapeutic avenues that can circumvent ECM-regulated tumorigenesis and metastasis.
2 ECM architecture of the breast tissue
The basic mammary structure comprises of luminal epithelial cells around a central lumen, surrounded by a layer of contractile myoepithelial cells, encased within the basement membrane (BM), which separates the epithelium from the stroma (Fig. 1). The stromal compartment or the interstitial matrix surrounds the cells and the BM and contributes to the tensile strength of the tissue. The BM and the interstitial matrix together make up the ECM. The BM is produced jointly by the epithelial, endothelial, and stromal cells and is largely composed of type IV collagens, laminin, fibronectin, and linker proteins such as nidogen and entactin [16, 17]. The interstitial matrix is rich in fibrillar collagen, proteoglycans, glycoproteins (such as tenascin C), and fibronectin [16]. Majority of the stroma surrounding the normal mammary cells is composed of fibrillar collagen [18]. However, during pathogenesis, the composition of the ECM is significantly altered [19–21].
Fig. 1.
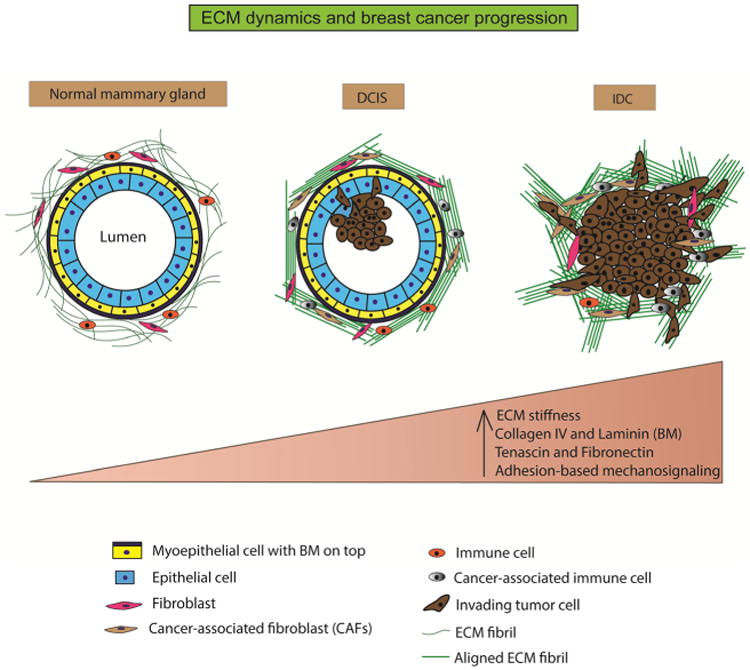
ECM dynamics and breast cancer progression. The normal mammary gland is characterized by a well-defined layer of epithelial cells around a central lumen. It is surrounded by a layer of contractile myoepithelial cells, which, in turn, is encased by the basement membrane. The interstitial matrix or the stroma surrounds this structure and comprises of randomly organized fibrillar collagen. The stroma also hosts fibroblasts and immune cells that function in ECM maintenance, immune surveillance, and mammary homeostasis. During ductal carcinoma in situ (DCIS), the epithelial cells undergo unregulated proliferation and start infiltrating into the central lumen. The ECM fibrils (collagen) are cross-linked and organized in bundles parallel to the tumor boundary. Stromal composition is altered with the appearance of cancer-associated fibroblasts and immune cells. During invasive ductal carcinoma (IDC), the lumen is almost completely filled with epithelial cancer cells. The ECM fibrils undergo further cross-linking and organized themselves perpendicular to the tumor boundary to provide migration tracks for the tumor cells to invade into neighboring tissue and blood vessels. Overall, the progression from normal mammary tissue to IDC is accompanied by increasing ECM stiffness, altered ECM composition, and aberrant mechanosignaling
3 Altered ECM during breast cancer progression
The ECM of the normal mammary gland is compliant and becomes progressively stiffer and more collagen-rich during tumor progression in a process known as “desmoplasia” [22]. The aberrant ECM remodeling is carried out predominantly by cancer-associated fibroblasts (CAFs), with pro-tumor immune cells and epithelial cells contributing during later stages of tumor progression [23]. Imaging modalities like sonoelastography, MRI elastography, TDI analysis, and physical palpatation routinely use tissue stiffness as a measure of breast tumorigenesis [24]. While these techniques are limited by their resolution in deciphering the cellular from the acellular components within the tissue, they provide a conclusive link between rigidity and malignancy. Alterations in mechanics of the breast tumor tissue occur as a result of increased interstitial pressure within the tumor, compression stress as a result of enhanced tumor cell proliferation, ECM stiffening due to increased deposition and cross-linking of ECM components, and increased cellular contractility [6]. While each of these factors contributes to altered tumor mechanics, we will focus on factors that affect ECM rigidity, thereby facilitating breast cancer progression. Breast tumors can be classified based on morphological (tumor size, lymph node status, and metastases) as well as molecular phenotypes (based on presence of hormone receptors).
3.1 ECM dynamics in morphological breast tumor subtypes
In terms of morphological features, mammary tumor progression can be classified into two major stages (1) ductal in situ carcinoma (DCIS), which is characterized as the pre-malignant stage and (2) invasive ductal carcinoma (IDC), characterized by an invasive phenotype, which includes breaching of the BM and migration of the tumor cells into neighboring tissues. The main criterion distinguishing IDC from DCIS is the loss of myoepithelial layer and BM [25, 26]. Each stage is accompanied by changes in the ECM, which facilitate infiltration and colonization of pro-tumorigenic cells, allowing for tumors to progress to malignancy (Fig. 1). DCIS originates in regions of the breast that are mammographically dense [27], and its progression involves incremental increases in collagen deposition, linearization, and thickening of the collagen fibers [28], all of which result in increased ECM stiffness. Various collagens including collagen I, II, III, V, and IX show increased deposition as breast cancer progresses [19–21].
Studies on MMTV-PyMT mice using AFM show that the elastic modulus (measure of tissue stiffness) of the mammary gland in normal, non-invasive breast tissue was approximately 400 Pa. In contrast, it was 1200 Pa in pre-malignant tumors and 3000 Pa in malignant tumors [24], indicating a significant increase in tumor tissue stiffness at different stages of breast cancer. Studies done on human samples reveal that as breast tumors progress from DCIS to IDC, the ECM undergoes increased collagen fiber linearization and thickening due to deposition and cross-linking of the collagen [28]. Additionally, the orientation of the collagen fibers is profoundly altered [29]. In DCIS, the linearized and thickened collagen I fibers are oriented adjacent to the tumor boundary [18, 28, 30]. In IDC, additional thickening and linearization of the collagen fibers are observed [28]. Moreover, in IDC, the collagen fibers are aligned perpendicular to the tumor boundary, forming migration tracks for invasive tumor cells to exit the tumor tissue and enter the blood stream [18, 28].
In terms of mechanical heterogeneity within the tumor tissue, a fourfold increase in stiffness was observed, specifically at the invasive front of the IDC phenotype. While the elastic modulus in normal tissue was approximately 400 Pa, it increased by more than tenfold in the invasive regions to approximately 5000 Pa. Moreover, the tumor core was found to be less stiff compared to the invasive front [28], implying that invading cells need a stiffer platform compared to proliferating cells. Interestingly, the difference in the elastic moduli between normal and invasive breast cancer phenotypes decreased substantially (200 and 300 Pa, respectively), when cells were freshly isolated from mice, as compared to when the measurements were performed in situ [24]. Whether these isolated cells demonstrated differential tumorigenic potential in vitro still needs to be investigated. These data show that the ECM is important in providing the optimal niche for tumor cells to feel and respond to mechanical stresses, distinct from those within the normal breast tissue, in order to acquire oncogenic and malignant properties.
3.2 ECM dynamics in molecular breast tumor subtypes
An additional classification of breast cancer subtypes is based on expression of hormone receptors, in addition to their histopathological features. These include the less aggressive luminal A and luminal B and the more aggressive HER2+ and basal (also referred to as triple-negative breast cancer (TNBC)). Previous studies on breast density and tumor subtypes have shown inconclusive and often confounding results. For example, in separate studies, estrogen receptor (ER) expression is found to be both enriched and lost in dense breast tissue [31–33]. Closer analysis of the stroma in breast cancer subtypes has revealed that HER2+ and TNBC have a significant enrichment for collagen deposition and matrix stiffness compared with the luminal subtypes [28], lending credence to the idea that increased tissue stiffness is associated with tumor aggression. Additionally, the tumor epithelium displayed increased integrin expression and mechanosignaling [28]. Given that these two subtypes represent the greatest threat to mortality among breast cancer patients as well as a significantly reduced time to relapse, the knowledge of the contribution of the stroma and tissue mechanics towards this phenotype is essential [34, 35].
While a stromally dense microenvironment can promote disease progression, different subtypes likely create these environments through different means. TNBCs show a significant influx of myeloid cells and an increase in the number of fibroblasts, which drive ECM remodeling by increasing matrix stiffness [28, 36, 37]. HER2+ breast tumors have a lower inflammatory response and higher number of fibroblasts suggesting a more direct fibroblast-tumor cell interaction driving stromal desmoplasia [28, 36]. This is supported by gene expression analysis of CAFs, showing that HER2+ CAFs are enriched for a gene expression signature closely related to the interaction and remodeling of the ECM, including integrin signaling and actin cytoskeleton regulation [38]. Thus, enhanced ECM stiffness appears to be a common denominator in aggressive breast cancers, stratified both morphologically and molecularly. The mechanisms by which a stiff ECM may contribute to tumor progression are multifold and often interrelated.
4 Effects of ECM rigidity on tumor progression
While it is being increasingly recognized that altered ECM stiffness contributes, in part, to tumor progression, we will now highlight some of the mechanisms underlying this process. Increased ECM rigidity results in altered mechanosignaling, a modified vascular landscape and pro-tumorigenic immune infiltration, all of which may facilitate transition to an invasive phenotype.
4.1 Altered mechanosignaling
Tumor-stroma communication entails tumor cells responding physically and biochemically to mechanical changes in the ECM in a process known as “mechanosignaling” [7]. Tumor cells sense the biophysical alterations in the ECM via mechanosensors such as the integrin family of receptors [39]. Integrins are heterodimeric transmembrane receptors, which serve as mediums for ECM-tumor cell dialogue. ECM stiffening leads to upregulation and clustering of integrins, such as β1 integrin, which, in turn, can initiate tumorigenesis as well as maintain proliferation of late-stage tumor cells [40]. The downstream effector of β1 integrin is the focal adhesion kinase (FAK), the activated form (pY397 FAK) of which is elevated in stiffer matrices [22, 41]. Increased levels of pY397 FAK lead to increased intracellular contractility via the Rho-ROCK pathway [42], allowing tumor cells to pull on the matrix during invasion. Additionally, elevated FAK activates the MAP kinase pathway, promoting tumor cell proliferation [42]. In high-grade DCIS, increased levels of β1 integrin and FAK are observed [28]. Expression of another mechanically activated kinase, p130Cas, is also increased in high-grade DCIS [28]; however, the functional implications of this are still not clearly understood. At the stiffest invasive tumor front, the mechanosensing proteins β1 integrin, vinculin, activated Akt, and FAK colocalize and the degree of expression and colocalization increases progressively from normal to DCIS to IDC phenotypes [43]. Increased breast tissue stiffness also upregulates oncogenic microRNAs that inhibit tumor-suppressive pathways and promote invasion [44] and numerous other proliferative/invasive pathways implicated in breast carcinogenesis [7, 45, 46].
4.2 Altered immune landscape
ECM rigidity regulates the number and nature of immune infiltrates in the tumor tissue by facilitating increased adhesion and migration of immune cells. For example, stiff, linear collagen provides tracks for the immune cells to move into the tumor [16, 17], much like it facilitates the outward movement of tumor cells. Additionally, ECM components express β1 integrins, non-integrin receptor DDR1, and leukocyte-associated LAIR-binding sites that anchor the immune cells, allowing their recruitment into the tumor microenvironment [47, 48]. ECM rigidity regulates the activation, maturation, and differentiation of immune T cells [17] and ECM degradation generates fragments, such as elastin, that are chemo-attractive specifically to monocytes [49] and implicated in breast cancer progression [50]. Recently, it was shown that a collagen-dense breast tumor environment recruits tumor-associated neutrophils (TANs) due to altered cytokine signaling, and depletion of TANs reduces metastasis in a stiffness-dependent manner [51]. However, the mechanism by which neutrophils promote metastasis in a collagen-rich tumor environment is still unclear. Interestingly, in this study, none of the other immune cell populations such as T lymphocytes and myeloid cells were found to be different between collagen-dense and normal mammary tissue [51]. Macrophages are often recruited during the later stages of breast cancer [52]. Indeed, during late stages of breast tumor progression, increased stiffness, inflammation, and infiltration of CD45+ immune cells are observed [28]. CD163+ activated macrophages, a subset of CD45+ cells, are most abundant in the IDC phenotype and express TGF-β, which further induces migration and ECM deposition [28], likely as part of a tumor feedback circuit.
4.3 Altered vasculature
Tumor proliferation and invasion require formation of new blood vessels. Endothelial cells lining the blood vessels are highly mechanosensitive, suggesting an intimate connection between ECM rigidity and vascular remodeling [53]. Consistently, microvascular density is significantly higher in invasive DCIS as compared to low-grade DCIS [54]. In addition to vascular density, the tumor vasculature is stiffer than the normal vasculature [24]. Moreover, the blood vessels within the tumor core are stiffer and thinner as compared to those at the invasive front and stain negative for lectin, indicating no vascular function in the tumor core [24]. Endothelial cells cultured on stiff collagen matrices show extensive branching compared to those on softer matrices [55]. Additionally, increasing stiffness leads to increased endothelial permeability and leukocyte transmigration [56], which potentially mediates tumor progression and invasion. The mechanisms by which the ECM regulates the vascular network in breast cancer are still poorly understood. Further studies are needed to establish the mechanistic underpinnings of breast tumor stiffness and angiogenesis/vasculogenesis to predict patient response to anti-angiogenic treatments.
5 ECM and metastasis
5.1 ECM facilitates formation of the pre-metastatic niche
Thus far, we have addressed the role of the ECM in primary breast tumor progression. These primary tumors must now acquire additional properties to be able to metastasize to distant organs. The formation of the pre-metastatic niche is crucial to the process of metastasis. Pre-metastatic niche formation involves the preparation of distant sites for oncogenic colonization, prior to the arrival of the disseminated tumor cells [57]. Most often, there is a pattern by which tumor cells from a given primary site metastasize to a secondary site. In breast cancer, upon breaching the BM, the tumor cells enter systemic circulation and colonize specifically in the lung, liver, brain, or bone [58, 59]. The pertinent question is what makes these secondary organs specifically habitable to the primary tumor cells? Emerging evidence implicates the ECM in creating tumor-hospitable pre-metastatic niches. For example, the collagen cross-linking protein LOX is crucial to invasion and its inhibition leads to decreased metastasis [60, 61]. Additionally, increased fibronectin expression observed at the secondary sites regulates the development of the pre-metastatic niche [62].
In breast cancer, the recruitment of myeloid-derived suppressor cells (MDSCs) to the pre-metastatic site is necessary for the remodeling of the ECM to potentially render it “pro-metastatic.” The MDSCs mobilized to the pre-metastatic niche serve to create an immune-suppressive microenvironment in the lung, to facilitate proliferation of the arriving tumor cells [63]. In mice models of breast cancer, the CD11b+ Ly6Chigh MDSCs recruited to the pre-metastatic lung secrete the ECM proteoglycan versican, which facilitates mesenchymal-to-epithelial transition of breast tumor cells, enhances their proliferation, and accelerates metastases [64]. Importantly, knockdown of versican significantly decreases the metastatic burden in the lung and versican is found to be upregulated more so in metastatic lungs of patients compared to healthy controls [64], implicating the importance of ECM in creating a tumor-hospitable niche.
LOX, a potent modulator of the ECM, cross-links collagen fibers and increases matrix stiffness [30, 65, 66]. Hypoxic breast cancer cells secrete LOX, which accumulates with fibronectin in the pre-metastatic niche and cross-links collagen IV in the lung BM [67]. The CD11b+ Gb1+ MDSCs recruited to the pre-metastatic sites [68] adhere to the remodeled ECM and secrete matrix metalloproteinases (MMPs), which degrade the matrix collagen releasing collagen IV peptides. These chemoattractive peptides create a positive loop, attracting more MDSCs to the pre-metastatic lung, thereby creating a pre-metastatic niche. Enzymatic inhibition of LOX decreases MDSC recruitment and metastasis [67, 69]. In orthotopic breast cancer models in mice, knockdown of LOX and LOXL4 led to decreased collagen cross-linking and reduced recruitment of CD11b+ cells in the pre-metastatic lungs, ultimately inhibiting metastasis [70]. More recently, hypoxia-regulated LOX was seen to increase bone metastasis in ER-negative breast cancer patient samples [60]. Interestingly, fibronectin upregulates the enzymatic activity of LOX [71], potentially contributing to LOX-induced ECM stiffening of the pre-metastatic niche.
Fibronectin is often upregulated in the pre-metastatic niche [62]. Fibroblasts secrete fibronectin in distant target organs, which binds to the integrin α4β1 (also known as VLA-4) expressed on the infiltrating VEGFR1+ bone marrow-derived hematopoietic progenitor cells (HPCs) [68]. The interaction between fibronectin and α4β1 is essential for formation of VEGFR1+ HPC clusters, and inhibition of these clusters prevents metastasis [68]. Additionally, fibronectin-integrin interaction in these pre-metastatic clusters leads to upregulation of MMPs, which further remodel the ECM, making it conducive to tumor cell infiltration [68]. ECM-integrin interactions facilitate the homing in of the tumor cells to the pre-metastatic niche [72]. As mentioned earlier, breast tumor cells specifically home into the lung, liver, bone, and brain. The sorting of the breast tumor cells into these different organs is still not well understood. Emergent knowledge attributes this to the presence of specific integrins on the exosomes that dictate the precise location of the pre-metastatic niche [73]. Exosomal integrins (ITGs) guide organ-specific metastasis by fusing with the target cells and initiating the pre-metastatic niche formation [73]. Exosomes directed to the lung express integrin alpha 6 (ITGα6), while those directed to the liver express ITGβ5. The lung-tropic ITGs fused with resident S100A4+ fibroblasts within the laminin-rich lung microenvironments. These exosomes serve to “educate” the lung by allowing ITGs to adhere to specific ECM environments, priming them for the incoming breast cancer cells [73].
5.2 Stromal remodeling is essential for metastasis
The pre-metastatic niche creates a tumor-hospitable environment at the secondary metastatic site prior to metastatic colonization. After their arrival at a second site of growth, tumor cells further remodel the environment around them to create favorable growth conditions. The contribution of the ECM to the formation of pre-metastatic and metastatic niches is depicted in Fig. 2. Experimental evidence suggests that tumor cell dissemination occurs early in tumorigenesis and a large proportion of patients present with metastatic disease at the time of diagnosis [74, 75]. Interestingly, remodeling of the lymph node ECM occurs in models of experimental metastasis without a primary tumor mass, suggesting that these systemic changes occur directly through the tumor cell secretome [76]. A number of ECM proteins are involved in the formation of a metastatic niche in breast cancer, including collagen, fibronection, tenascin C, versican, and periostin, among others [72]. The deposition of collagens has been noted in the secondary sites of organs common to breast cancer metastasis such as the lymph node and the bone [76, 77].
Fig. 2.
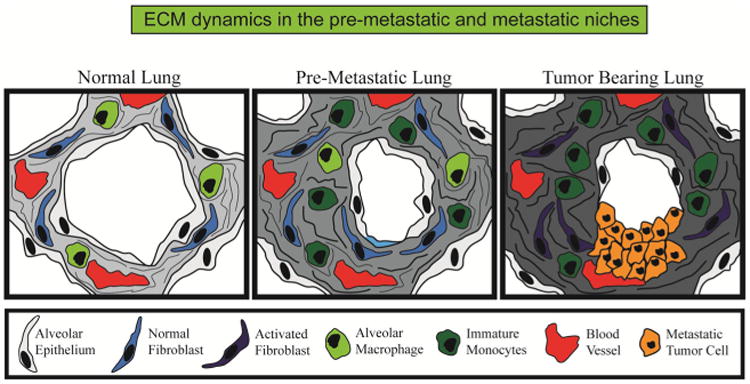
Stromal dynamics in the pre-metastatic and metastatic niches. Using the lung as an example, the normal lung contains mostly alveolar epithelium with high penetration of blood vessels and little stromal content. Systemic changes are elicited by the primary tumor prior to dissemination and formation of overt metastatic lesions including the influx of specific immune cells such as immature monocytes, activation of fibroblasts, and increased deposition of ECM components such as collagen and fibronectin. Such features are progressively exacerbated during the formation of tumors at the secondary site and essential to continued growth
It has been proposed that the acquisition of stem-like characteristics is necessary for metastasis to the lung, a phenotype promoted via optimal stromal cues [78]. Tenascin C, via the notch and wnt pathways, regulates this stem-like phenotype, and its expression by metastatic tumor cells is important to successful colonization [79]. S100A4+ fibroblasts mediate stromal tenascin C deposition in the lung and metastatic outgrowth of breast cancer cells [80]. Additionally, expression of periostin, via wnt signaling, is enhanced in the lungs following metastatic colonization and essential for the outgrowth of tumor cells [81]. The disruption of interactions between the tumor cell integrins and the newly created ECM of the secondary site prevents acquisition of stem-like phenotype and limits metastatic tumor growth [82, 83].
While the contribution of specific ECM components to the formation of the pre-metastatic and metastatic niches is better understood, much less is known about how tissue stiffness facilitates metastasis. For example, while it is known that LOX is crucial to the formation of the pre-metastatic niche [67] and that LOX leads to increased stiffness, the mechanical properties of the target site remain unclear. Does the pre-metastatic lung become stiffer with increased deposition of LOX, periostin, fibronectin, tenascin C, versican, and collagen? Does this stiffness further perpetrate, or even initiate, infiltration of both tumor cells as well as pro-metastatic immune and stromal cells? What is the individual or combined contribution of these components to the pre-metastatic and the metastatic ECM topology and architecture? What is the interplay between the matrix-building ECM constituents (such as LOX) and the matrix-degrading MMPs, and how are they spatially and temporally regulated to fine-tune the metastatic process? The knowledge of the biophysical nature of the ECM encompassing stiffness, porosity, linearization and cross-linking, in the pre-metastatic and metastatic niches, will allow detection of the degree of metastatic spread as well as shed light on the ECM-mediated therapeutic resistance.
6 ECM mediates treatment resistance
The biophysical characteristics of the ECM can affect the treatment outcome of patients with breast cancer. Using shear-wave elastography, it was found that patients with softer breast tumors were more responsive to treatment as compared to those with stiffer tumors [84]. Moreover, the ECM secreted protein acidic and rich in cysteine (SPARC) signature was associated with resistance to chemotherapy in Her2+ breast cancers [85]. Indeed, as described previously, a stiffer substrate can perpetuate cell proliferation, invasion, vasculogenesis, and pro-oncogenic immune infiltration. Thus, it is logical to infer that a stiff tumor microenvironment facilitates resistance to therapy, allowing the tumor niche to thrive despite exposure to cytotoxic insult. There are a number of ways in which the ECM can mediate treatment resistance (Fig. 3).
Fig. 3.
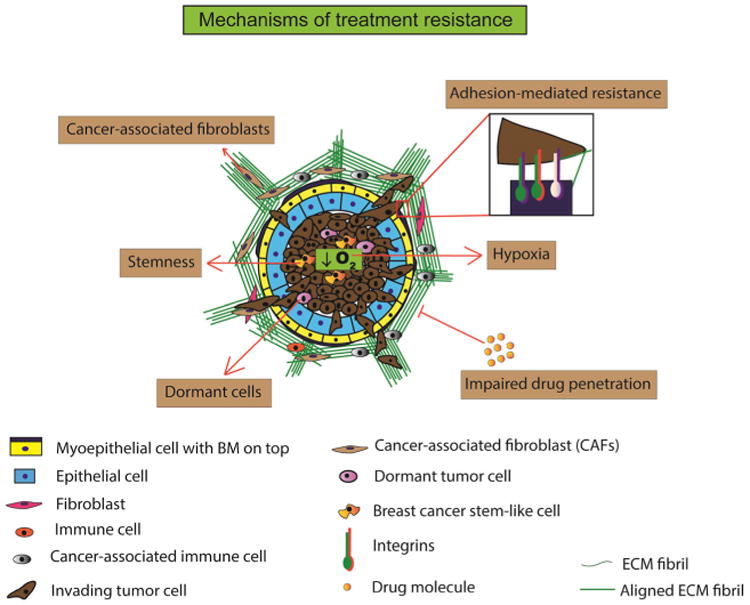
Mechanisms of treatment resistance. The breast cancer ECM mediates resistance to chemotherapy and radiation therapy in a number of ways. (i) Adhesion-mediated resistance entails aberrant mechanosignaling, upregulation of certain tumor-promoting integrins and non-integrins, and integrin clustering resulting in chemotherapeutic resistance. (ii) Hypoxia and stemness are known perpetrators of chemotherapeutic resistance. ECM is postulated to be involved in feedforward mechanisms wherein increased ECM stiffness increases hypoxia and stemness and subsequently resistance to anti-cancer agents. (iii) Impaired drug penetration occurs due to increasingly rigid ECM that physically inhibits cytotoxic molecules to reach the tumor cells. (iv) Dormant cells, induced by ECM mechanics, are inherently resistant to chemotherapeutic agents that target actively proliferating cells. Thus, dormancy within the tumor microenvironment ensures survival of these resistant tumor cells and their activation and proliferation at a suitable time. (v) Chemotherapeutic drugs mediate conversion of normal fibroblasts to cancer-associated fibroblasts (CAFs). These secrete fibronectin and collagen causing extensive ECM remodeling and facilitating chemotherapy-induced alterations in the ECM and resistance to treatment
6.1 Limiting drug penetration
Collagen, glycosaminoglycans, and decorin are a few of the ECM components that create a protective scaffold around and throughout the tumor, limiting the diffusion of chemotherapeutic agents [86, 87]. This may be due, in part, to increased ECM stiffness as a result of increased deposition and cross-linking of components, such as collagen [87]. Drug penetrance was enhanced when collagen-rich tumors were treated with collagenase [87]. Interestingly, it was found that the amount of collagen, but not its orientation, was essential to impede drug penetration [88]. Additionally, the high interstitial pressure and compression forces within the tumor micro-environment [6] can contribute to increased ECM stiffness and ineffective drug delivery to the target site. A stromal gene expression signature consisting of ECM proteins such as collagens, SPARC, periostin, thrombospondins, and decorin is associated with poor therapeutic outcome in breast cancer patients treated with anthracycline-based drugs [89]. ECM is also implicated in perpetrating resistance to other chemotherapeutic agents such as doxorubicin [90], tamoxifen [91], and antibody-based agents [92]. Thus, the ECM is likely to provide a protective encasement for the tumor against anti-cancer agents, thereby promoting carcinogenic progression.
6.2 Contact-mediated resistance
Integrin and non-integrin adhesions anchor the tumors cells to the ECM and provide proliferation, differentiation, and invasion signals. Additionally, they perpetuate chemoresistance in tumors [93]. In breast cancer, differential expression of certain integrins dictates the degree of tumor aggression [94]. For example, αVβ3 integrin mediates metastasis of breast cancer to bone [95]. Additionally, β3 integrins mediate resistance to EGFR inhibitors erlotinib and lapatinib, in addition to driving a stem-like phenotype [96], which further thwarts the cytotoxic effects of chemotherapeutic agents. β1 integrin is associated with dormant cells, which are particularly resistant to both chemotherapy and radiation [97]. Much less is known about the role of non-integrin discoidin receptors in mediating resistance to therapy in breast cancer. It is shown that the discoidin receptor, DDR1, facilitates chemoresistance via the NF-κβ pathway [98]. Given that discoidin receptors promote tumor progression independently, as well as through interactions with integrins, such as α2β1 integrin [99], it will be worthwhile to investigate their role, together with that of integrins, in promoting chemoresistance.
6.3 Increased hypoxia and stemness
As previously discussed, breast tumor cells secrete LOX, which stiffens the ECM and can promote the formation of a pre-metastatic niche in the lung [67]. Additionally, LOX is essential for driving hypoxia-induced metastasis as evidenced by using LOX inhibitors that inhibit metastasis even in the presence of hypoxia [69]. While the expression of LOX is regulated by hypoxia-inducible factor-1 (HIF-1) [69], the possibility of a feedback mechanism wherein hypoxia drives LOX expression and vice versa cannot be discounted. Tumor stiffness can lead to increased tumor cell proliferation and vascular remodeling, which could maintain, if not initiate, a hypoxic environment. Presence of hypoxia is correlated with poor prognosis and treatment resistance in breast cancer patients [100]. Recently, it was shown that hypoxic and stiff breast tumors promote proliferation of breast cancer stem-like cells [101], which, in turn, can mediate resistance to therapy [102].
6.4 Induction of dormancy
Tumor dormancy is a state in which primary or disseminated tumor cells exit the proliferative cell cycle phase and enter a quiescent, non-dividing phase. Dormant breast tumor cells are highly resistant to chemotherapy drugs, which essentially target actively proliferating cells, allowing these quiescent cells to persist and form micrometastases at a later time [103]. Fibronectin production, phosphorylated myosin light chain kinase (pMLCK), and β1 integrin mediate the switch from dormant to proliferative state [104]. The resultant proliferating cells display actin stress fibers making them highly contractile [104], possibly in response to a stiffening ECM due to fibronectin deposition and increased β1 integrin-mediated adhesion. Interestingly, in hepatocellular carcinoma, softer substrates are shown to induce cellular dormancy while stiffer substrates can facilitate proliferation and therapeutic resistance [105]. This was also shown in vitro in breast and lung cancer cell lines [106]. It is postulated that at the primary site, the tumor cells encounter a stiff matrix which facilitates invasion, whereas at the secondary site, the cells encounter a softer matrix which triggers their switch to dormancy [105]. These studies provide interesting perspectives on ECM heterogeneity. Additionally, they provide rationale for the premise that not all therapies targeting a stiff ECM will lead to effective treatment outcomes, as softer matrices may play complementary roles in treatment resistance. Thus, when developing therapeutic strategies, it is imperative to consider the heterogeneity of the oncogenic ECM rather than isolated characteristics such as increased stiffness.
6.5 Treatment-induced ECM remodeling
A number of anti-cancer agents can induce ECM remodeling, which serves as a compensatory mechanism to promote treatment resistance. In breast cancer models, doxorubicin increases the expression of the ECM protein fibulin, which, in turn, can promote resistance of breast cancer cells to doxorubicin [107]. Chemotherapeutic agents can also potentially modulate ECM remodeling via other indirect mechanisms such as by promoting the conversion of normal stromal fibroblasts to CAFs [108], which secrete fibronectin and collagen leading to extensive ECM remodeling [109]. In addition to chemotherapy, radiation therapy is also documented to induce stiffness of tumor tissue leading to treatment resistance [110]. Ionizing radiation leads to increased LOX secretion in a hypoxia-dependent manner [110], which may provide an explanation as to the increased frequency of tumor recurrence after radiation treatment. Thus, chemotherapeutic drugs themselves, along with radiation, can lead to ECM remodeling, which can further promote tumor progression, resistance to treatment, and possible recurrence of the disease.
7 Therapeutic targeting of tumor ECM
Given the profound effect that the ECM and matrix mechanics have on tumor progression, targeting the ECM may prove highly tractable. Such a goal can be achieved in two different ways: (1) by targeting the stroma and (2) by targeting the cellular responses to the stroma. Targeting the stroma involves reversing the tumor-induced changes to the ECM to subdue increased tissue mechanics and likely tumor progression. In pre-clinical models, normalizing the tumor stroma through the inhibition of TGF-β signaling decreases collagen deposition and increases vascular permeability, significantly slowing down tumor growth and enhancing therapeutic response [111]. However, while pre-clinical systems show beneficial outcomes, the pleiotropic nature of TGF-β signaling makes clinical trials far more complicated. The use of agents specifically targeting ECM components to reduce tissue rigidity has also been shown to be efficacious in slowing tumor progression [69, 112]. The use of hyaluronidase to degrade extracellular hyaluronic acid is effective in not only slowing tumor growth but also relieving solid stresses to enhance penetrance and response to chemotherapeutic treatment [113, 114]. Given the positive potential of the pre-clinical studies in breast and other cancer types, numerous clinical trials have been initiated using neoadjuvant hyaluronidase, including for metastatic breast cancer (NCT02753595). Approaches targeting the maturation of collagen through the inhibition of cross-linking enzymes have also shown promise in pre-clinical models. The enzymatic LOX inhibitor Beta-aminoproprionitrile (BAPN) significantly reduces collagen deposition, tumor cell invasion, and metastasis in spontaneous and orthotopic models of breast cancer [69, 115]. The allosteric LOXL2 inhibitor diminishes desmoplastic response and prevents tumor cell metastasis in numerous cancer models [116]. Novel approaches of depleting copper with tetrathiomolybdate in phase I and phase II clinical trials decrease lung metastasis, LOX activity, and collagen cross-linking in high-risk breast cancer patients [117]. These approaches have yet to be investigated in clinical trials but offer novel approaches with significant potential for clinical application.
A second means by which to prevent the potential pro-tumorigenic effects of ECM deposition and tissue mechanics is to inhibit the cellular responses to these cues. The most prominent means of converting mechanical cues into biochemical responses is through integrin-mediated FAK phosphorylation. The inhibition of this phosphorylation event prevents the induction of downstream signaling events. Currently, FAK inhibitors have shown significant pre-clinical efficacy both through genetic and pharmacological manipulations [118–121]. The targeted attenuation of FAK activity through either direct kinase blocking or phosphorylation blocking has been tested and showed significant promise in phase I clinical trials (NCT00666926 and NCT00996671). In a neoadjuvant setting, loss of FAK activity in tumor cells holds significant promise, yet the systemic effects and compensatory mechanisms employed by the tumor have yet to be established.
8 Conclusion
The heterogeneity of breast cancer is largely due to the dynamic nature of its ECM. During normal mammary development, the ECM undergoes constant remodeling, albeit under tightly regulated conditions. Breast cancer results when there is an imbalance in this process and the ECM becomes dysregulated and disorganized. Fundamental differences exist in the ECM of the normal breast versus that of breast tumor, in terms of rigidity, composition, organization, and cross-linking. It is now being widely acknowledged that the ECM is not a passive bystander during cancer progression. Rather, it participates actively, both physically and biochemically, in the evolution from pre-malignant DCIS to malignant IDC. Additionally, it can provide the necessary ingredients for creation of a pre-metastatic niche in a distant organ such as the lung, brain, or bone, followed by metastatic colonization by the primary tumor cells.
Tumor growth and invasion can occur when there is a balance between ECM deposition and degradation. CAFs and epithelial cells contribute to deposition by secreting ECM components such as fibronectin and collagen. Interestingly, these cells also secrete ECM-degrading enzymes such as MMPs and collagenases, when it is suitable for the tumor cells to breakdown the BM and begin the process of invasion. Thus, ECM homeostasis while dysregulated in cancer is still governed by feedback mechanisms that balance deposition and degradation. The contributions of ECM dynamics to breast cancer and metastasis are now beginning to be unraveled, and understanding the feedback circuits that control the ECM architecture will facilitate optimal design of therapeutic interventions that can target these dynamics and potentially prevent breast tumors from advancing to malignancy.
Key unanswered questions.
Does the ECM contribute to cancer initiation and, if so, how early in the process of oncogenesis is the ECM involved? Can tumorigenesis be prevented by targeting early tumor-stroma interactions?
What is the interplay between ECM deposition and degradation? What are the mechanisms that control their spatial and temporal regulation during breast cancer?
While it is known that specific immune cells are recruited based on the ECM rigidity, how are these signals conveyed to the immune cells? Once recruited, how is their interaction with the matrix important in their regulation of tumorigenesis?
Given that the ECM is an important factor for tumor cell survival and migration, what happens to tumor cells in circulation while they are in transit between the primary and distant secondary sites?
What mechanisms control the long-range communication between the primary and the secondary sites, wherein cues procured from the primary breast tumor allow ECM remodeling in the lung? Additionally, what factors decide whether the distant site for breast cancer metastasis will be lung, bone, brain, or liver?
While stiff substrates can facilitate breast tumor proliferation, softer substrates can promote dormancy. How do we design optimal therapeutic strategies to combat both rigid and compliant matrices without affecting normal breast tissue?
Acknowledgments
The authors apologize to all colleagues whose work could not be cited due to space limitations. The authors would like to thank Dr. Janna Mouw for her insightful review of the manuscript. This work was supported by NIH grant RO1CA192914, USMRAA (DOD) grant BC122990, U54 grant CA210184, and UO1 grant CA202241-01.
References
- 1.Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer. Science (New York, NY) 2002;296(5570):1046–1049. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson GW, Karpf aB, Kratochwil K. Regulation of mammary gland development by tissue interaction. Journal of Mammary Gland Biology and Neoplasia. 1999;4(1):9–19. doi: 10.1023/A:1018748418447. [DOI] [PubMed] [Google Scholar]
- 3.Wicha MS, Liotta LA, Vonderhaar BK, Kidwell WR. Effects of inhibition of basement membrane collagen deposition on rat mammary gland development. Developmental Biology. 1980;80(2):253–266. doi: 10.1016/0012-1606(80)90402-9. [DOI] [PubMed] [Google Scholar]
- 4.Silberstein GB, Daniel CW. Glycosaminoglycans in the basal lamina and extracellular matrix of the developing mouse mammary duct. Developmental Biology. 1982;90(1):215–222. doi: 10.1016/0012-1606(82)90228-7. [DOI] [PubMed] [Google Scholar]
- 5.Fata JE, Werb Z, Bissell MJ. Regulation of mammary gland branching morphogenesis by the extracellular matrix and its remodeling enzymes. Breast cancer research: BCR. 2004;6(1):1–11. doi: 10.1186/bcr634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. Journal of Mammary Gland Biology and Neoplasia. 2004 doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- 7.Schedin P, Keely PJ. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harbor Perspectives in Biology. 2011;3(1):1–22. doi: 10.1101/cshperspect.a003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schedin P, Keely PJ. and Mechanosignaling in normal development and tumor progression. 2011:1–22. doi: 10.1101/cshperspect.a003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schedin P, Mitrenga T, McDaniel S, Kaeck M. Mammary ECM composition and function are altered by reproductive state. Molecular Carcinogenesis. 2004;41(4):207–220. doi: 10.1002/mc.20058. [DOI] [PubMed] [Google Scholar]
- 10.Hynes RO. The extracellular matrix: not just pretty fibrils. Science (New York, NY) 2009;326(5957):1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer TL - 15. EMBO Reports. 2014;15 VN-r(12) doi: 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sweet DT, Chen Z, Wiley DM, Bautch VL, Tzima E. The adaptor protein Shc integrates growth factor and ECM signaling during postnatal angiogenesis. Blood. 2012;119(8):1946–1955. doi: 10.1182/blood-2011-10-384560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman aC, Nakatsu MN, Chou W, Gershon PD, Hughes CCW. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Molecular Biology of the Cell. 2011;22(20):3791–3800. doi: 10.1091/mbc.E11-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers KA, Applegate KT, Danuser G, Fischer RS, Waterman CM. Distinct ECM mechanosensing pathways regulate microtubule dynamics to control endothelial cell branching morphogenesis. Journal of Cell Biology. 2011;192(2):321–334. doi: 10.1083/jcb.201006009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorokin L. The impact of the extracellular matrix on inflammation. Nature reviews Immunology. 2010;10(10):712–723. doi: 10.1038/nri2852. [DOI] [PubMed] [Google Scholar]
- 16.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. Journal of Cell Biology. 2012;196(4):395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Current Opinion in Cell Biology. 2010 doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Medicine. 2006;4(1):38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huijbers IJ, Iravani M, Popov S, Robertson D, Al-Sarraj S, Jones C, Isacke CM. A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PloS One. 2010;5(3) doi: 10.1371/journal.pone.0009808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu GG, Risteli L, Makinen M, Risteli J, Kauppila A, Stenback F. Immunohistochemical study of type I collagen and type I pN-collagen in benign and malignant ovarian neoplasms. Cancer. 1995;75(4):1010–1017. doi: 10.1002/1097-0142(19950215)75:4<1010∷AID-CNCR2820750417>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 21.Kauppila S, Stenbäck F, Risteli J, Jukkola A, Risteli L. Aberrant type I and type III collagen gene expression in human breast cancer in vivo. The Journal of Pathology. 1998;186(3):262–268. doi: 10.1002/(SICI)1096-9896(1998110)186:3<262∷AID-PATH191>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 22.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer cell. 2005;8(3):241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harbor Perspectives in Biology. 2011;3(12):1–24. doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez JI, Kang I, You WK, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integrative biology: quantitative biosciences from nano to macro. 2011;3(9):910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, et al. Polyak K. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13(5):394–406. doi: 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lerwill MF. Current practical applications of diagnostic immunohistochemistry in breast pathology. The American Journal of Surgical Pathology. 2004;28:1076–1091. doi: 10.1097/01.pas.0000126780.10029.f0. [DOI] [PubMed] [Google Scholar]
- 27.Ursin G, Hovanessian-Larsen L, Parisky YR, Pike MC, Wu AH. Greatly increased occurrence of breast cancers in areas of mammographically dense tissue. Breast Cancer Research. 2005;7(5):R605–R608. doi: 10.1186/bcr1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, et al. Weaver VM. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integrative biology: quantitative biosciences from nano to macro. 2015;7(10):1120–34. doi: 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Keely PJ. Aligned collagen is a prognostic signature for survival in human breast carcinoma. American Journal of Pathology. 2011;178(3):1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levental KR, Yu H, Kass L, Lakins JN, Erler JT, Fong SFT, et al. Weaver VM. NIH Public Access. 2010;139(5):891–906. doi: 10.1016/j.cell.2009.10.027.Matrix. [DOI] [Google Scholar]
- 31.Ding J, Warren R, Girling A, Thompson D, Easton D. Mammographic density, estrogen receptor status and other breast cancer tumor characteristics. The Breast Journal. 16(3):279–289. doi: 10.1111/j.1524-4741.2010.00907.x. (n.d) [DOI] [PubMed] [Google Scholar]
- 32.Conroy SM, Butler LM, Harvey D, Gold EB, Sternfeld B, Greendale GA, Habel LA. Metabolic syndrome and mammographic density: the Study of Women's Health Across the Nation. International Journal of Cancer. 2011;129(7):1699–1707. doi: 10.1002/ijc.25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yaghjyan L, Colditz GA, Collins LC, Schnitt SJ, Rosner B, Vachon C, Tamimi RM. Mammographic breast density and subsequent risk of breast cancer in postmenopausal women according to tumor characteristics. Journal of the National Cancer Institute. 2011;103(15):1179–1189. doi: 10.1093/jnci/djr225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hugh J, Hanson J, Cheang MCU, Nielsen TO, Perou CM, Dumontet C, et al. Vogel C. Breast cancer subtypes and response to docetaxel in node-positive breast cancer: use of an immunohistochemical definition in the BCIRG 001 trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27(8):1168–76. doi: 10.1200/JCO.2008.18.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang RF, Chen HH, Chang YC, Huang CS, Chen JH, Lo CM. Quantification of breast tumor heterogeneity for ER status, HER2 status, and TN molecular subtype evaluation on DCE-MRI. Magnetic Resonance Imaging. 2016;34(6):809–819. doi: 10.1016/j.mri.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Park SY, Kim HM, Koo JS. Differential expression of cancer-associated fibroblast-related proteins according to molecular subtype and stromal histology in breast cancer. Breast Cancer Research and Treatment. 2015;149(3):727–741. doi: 10.1007/s10549-015-3291-9. [DOI] [PubMed] [Google Scholar]
- 37.Afik R, Zigmond E, Vugman M, Klepfish M, Shimshoni E, Pasmanik-Chor M, et al. Varol C. Tumor macrophages are pivotal constructors of tumor collagenous matrix. Journal of Experimental Medicine. 2016 doi: 10.1084/jem.20151193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tchou J, Kossenkov AV, Chang L, Satija C, Herlyn M, Showe LC, et al. Gelmon K. Human breast cancer associated fibroblasts exhibit subtype specific gene expression profiles. BMC Medical Genomics. 2012;5(1):39. doi: 10.1186/1755-8794-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galbraith CG, Yamada KM, Sheetz MP. The relationship between force and focal complex development. Journal of Cell Biology. 2002;159(4):695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White DE, Kurpios NA, Zuo D, Hassell JA, Blaess S, Mueller U, Muller WJ. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004;6(2):159–170. doi: 10.1016/j.ccr.2004.06.025. doi:10.1016/j.ccr.2004.06.025\rS1535610804002077 [pii] [DOI] [PubMed] [Google Scholar]
- 41.Wozniak Ma, Desai R, Solski Pa, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. The Journal of Cell Biology. 2003;163(3):583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28(49):4326–4343. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rubashkin MG, Cassereau L, Bainer R, DuFort CC, Yui Y, Ou G, et al. Weaver VM. Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate. Cancer Research. 2014;74(17):4597–4611. doi: 10.1158/0008-5472.CAN-13-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mouw JK, Yui Y, Damiano L, Bainer RO, Lakins JN, Acerbi I, et al. Weaver VM. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nature medicine. 2014;20(4):360–7. doi: 10.1038/nm.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gehler S, Ponik SM, Riching KM, Keely PJ. Bidirectional signaling: extracellular matrix and integrin regulation of breast tumor progression. Critical Reviews in Eukaryotic Gene Expression. 2013;23(2):139–157. doi: 10.1615/CritRevEukarGeneExpr.2013006647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu J, Xiong G, Trinkle C, Xu R. Integrated extracellular matrix signaling in mammary gland development and breast cancer progression. Histology and Histopathology. 2014;29(9):1083–1092. doi: 10.1002/jcp.24872.The. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franco C, Hou G, Ahmad PJ, Fu EYK, Koh L, Vogel WF, Bendeck MP. Discoidin domain receptor 1 (Ddr1) deletion decreases atherosclerosis by accelerating matrix accumulation and reducing inflammation in low-density lipoprotein receptor-deficient mice. Circulation Research. 2008;102(10):1202–1211. doi: 10.1161/CIRCRESAHA.107.170662. [DOI] [PubMed] [Google Scholar]
- 48.Meyaard L. The inhibitory collagen receptor LAIR-1 (CD305) Journal of Leukocyte Biology. 2008;83(4):799–803. doi: 10.1189/jlb.0907609. [DOI] [PubMed] [Google Scholar]
- 49.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, et al. Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. Journal of Clinical Investigation. 2006;116(3):753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krishnan R, Cleary EG. Elastin gene expression in elastotic human breast cancers and epithelial cell lines. Cancer Research. 1990;50(7):2164–2171. [PubMed] [Google Scholar]
- 51.García-Mendoza MG, Inman DR, Ponik SM, Jeffery JJ, Sheerar DS, Van Doorn RR, Keely PJ. Neutrophils drive accelerated tumor progression in the collagendense mammary tumor microenvironment. Breast cancer research: BCR. 2016;18(1):49. doi: 10.1186/s13058-016-0703-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sieminski AL, Hebbel RP, Gooch KJ. The relative magnitudes of endothelial force generation and matrix stiffness modulate capillary morphogenesis in vitro. Experimental Cell Research. 2004;297(2):574–584. doi: 10.1016/j.yexcr.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 54.Teo NB, Shoker BS, Jarvis C, Martin L, Sloane JP, Holcombe C. Vascular density and phenotype around ductal carcinoma in situ (DCIS) of the breast. British Journal of Cancer. 2002;86(6):905–911. doi: 10.1038/sj.bjc.6600053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mason BN, Starchenko A, Williams RM, Bonassar LJ, Reinhart-King CA. Tuning three-dimensional collagen matrix stiffness independently of collagen concentration modulates endothelial cell behavior. Acta Biomaterialia. 2013;9(1):4635–4644. doi: 10.1016/j.actbio.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kohn JC, Zhou DW, Bordeleau F, Zhou AL, Mason BN, Mitchell MJ, et al. Reinhart-King CA. Cooperative effects of matrix stiffness and fluid shear stress on endothelial cell behavior. Biophysical Journal. 2015;108(3):471–478. doi: 10.1016/j.bpj.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaplan RN, Rafii S, Lyden D. Preparing the “soil”: the premetastatic niche. Cancer Research. 2006 doi: 10.1158/0008-5472.CAN-06-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nature reviews Cancer. 2002;2(8):563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 59.Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nature reviews. Cancer. 2009;9(4):274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 60.Cox TR, Rumney RM, Schoof EM, Perryman L, Hoye AM, Agrawal A, et al. Erler JT. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature. 2015;522(7554):106–110. doi: 10.1038/nature14492. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, et al. Sansom OJ. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med. 2015;7:1063–1076. doi: 10.15252/emmm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Erler JT, Weaver VM. Three-dimensional context regulation of metastasis. Clinical and Experimental Metastasis. 2009;26(1):35–49. doi: 10.1007/s10585-008-9209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giles AJ, Reid CM, De Wayne Evans J, Murgai M, Vicioso Y, Highfill SL, et al. Kaplan RN. Activation of hematopoietic stem/progenitor cells promotes immunosuppres-sion within the pre-metastatic niche. Cancer Research. 2016;76(6):1335–1347. doi: 10.1158/0008-5472.CAN-15-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Mittal V. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesen-chymal to epithelial transition. Cancer Research. 2012;72(6):1384–1394. doi: 10.1158/0008-5472.CAN-11-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kagan HM, Li W. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. Journal of Cellular Biochemistry. 2003;88(4):660–672. doi: 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- 66.Pfeiffer BJ, Franklin CL, Hsieh FH, Bank RA, Phillips CL. Alpha 2(I) collagen deficient oim mice have altered biomechanical integrity, collagen content, and collagen crosslinking of their thoracic aorta. Matrix Biology. 2005;24(7):451–458. doi: 10.1016/j.matbio.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 67.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Giaccia AJ. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15(1):35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaplan RN, Riba RD, Zacharoulis S, Anna H, Vincent L, Costa C, et al. Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–827. doi: 10.1038/nature04186.VEGFR1-positive. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Erler JT, Bennewith KL, Nicolau M, Dornhöfer N, Kong C, Le QT, et al. Giaccia AJ. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440(7088):1222–6. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 70.Wong CCL, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, et al. Semenza GL. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(39):16369–74. doi: 10.1073/pnas.1113483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fogelgren B, Polgár N, Szauter KM, Újfaludi Z, Laczkó R, Fong KSK, Csiszar K. Cellular fibronectin binds to lysyl oxidase with high affinity and is critical for its proteolytic activation. Journal of Biological Chemistry. 2005;280(26):24690–24697. doi: 10.1074/jbc.M412979200. [DOI] [PubMed] [Google Scholar]
- 72.Høye AM, Erler JT. Structural ECM components in the pre-metastatic and metastatic niche. American Journal of Physiology Cell Physiology. 2016;1(73) doi: 10.1152/ajpcell.00326.2015. ajpcell.00326.2015. [DOI] [PubMed] [Google Scholar]
- 73.Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Lyden D. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–35. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Klein CA. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 75.Kennecke H, Yerushalmi R, Woods R, Cheang MCU, Voduc D, Speers CH, et al. Gelmon K. Metastatic behavior of breast cancer subtypes. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28(20):3271–7. doi: 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- 76.Rizwan A, Bulte C, Kalaichelvan A, Cheng M, Krishnamachary B, Bhujwalla ZM, et al. Tyers M. Metastatic breast cancer cells in lymph nodes increase nodal collagen density. Scientific Reports. 2015;5:10002. doi: 10.1038/srep10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koyama T, Hasebe T, Tsuda H, Hirohashi S, Sasaki S, Fukutomi T, et al. Mukai K. Histological factors associated with initial bone metastasis of invasive ductal carcinoma of the breast. Japanese Journal of Cancer Research. 1999;90(3):294–300. doi: 10.1111/j.1349-7006.1999.tb00747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Werb Z. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526(7571):131–135. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oskarsson T, Acharyya S, Zhang XHF, Vanharanta S, Tavazoie SF, Morris PG, et al. Massagué J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nature medicine. 2011;17(7):867–74. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. Kalluri R. VEGF-A and tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(38):16002–7. doi: 10.1073/pnas.1109493108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481(7379):85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 82.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Molecular Biology of the Cell. 2001;12(4):863–879. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. European Journal of Cancer. 2010;46(7):1181–1188. doi: 10.1016/j.ejca.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hayashi M, Yamamoto Y, Ibusuki M, Fujiwara S, Yamamoto S, Tomita S, et al. Iwase H. Evaluation of tumor stiffness by elastography is predictive for pathologic complete response to neoadjuvant chemotherapy in patients with breast cancer. Annals of Surgical Oncology. 2012;19(9):3042–3049. doi: 10.1245/s10434-012-2343-1. [DOI] [PubMed] [Google Scholar]
- 85.Giussani M, Merlino G, Cappelletti V, Tagliabue E, Daidone MG. Tumor-extracellular matrix interactions: identification of tools associated with breast cancer progression. Seminars in Cancer Biology. 2015 doi: 10.1016/j.semcancer.2015.09.012. doi:10.1016/j. semcancer.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 86.Magzoub M, Jin S, Verkman aS. Enhanced macro-molecule diffusion deep in tumors after enzymatic digestion of extracellular matrix collagen and its associated proteoglycan decorin. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2008;22(1):276–284. doi: 10.1096/fj.07-9150com. [DOI] [PubMed] [Google Scholar]
- 87.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Research. 2000;60(9):2497–2503. doi: 10.1126/science.271.5252.1079. [DOI] [PubMed] [Google Scholar]
- 88.Erikson A, Andersen HN, Naess SN, Sikorski P, Davies CDL. Physical and chemical modifications of collagen gels: impact on diffusion. Biopolymers. 2008;89(2):135–143. doi: 10.1002/bip.20874. [DOI] [PubMed] [Google Scholar]
- 89.Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, et al. Delorenzi M. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nature medicine. 2009;15(1):68–74. doi: 10.1038/nm0209-220a. [DOI] [PubMed] [Google Scholar]
- 90.Misra S, Obeid LM, Hannun YA, Minamisawa S, Berger FG, Markwald RR, et al. Ghatak S. Hyaluronan constitutively regulates activation of COX-2-mediated cell survival activity in intestinal epithelial and colon carcinoma cells. Journal of Biological Chemistry. 2008;283(21):14335–14344. doi: 10.1074/jbc.M703811200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jansen MPHM, Foekens Ja, van Staveren IL, Dirkzwager-Kiel MM, Ritstier K, Look MP, et al. Berns EMJJ. Molecular classification of tamoxifen-resistant breast carcinomas by gene expression profiling. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23(4):732–40. doi: 10.1200/JCO.2005.05.145. [DOI] [PubMed] [Google Scholar]
- 92.Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Advanced Drug Delivery Reviews. 2008 doi: 10.1016/j.addr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Holle AW, Young JL, Spatz JP. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Advanced Drug Delivery Reviews. 2016 doi: 10.1016/j.addr.2015.10.007. doi::10.1016/j.addr.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 94.Chrenek Ma, Wong P, Weaver VM. Tumour-stromal interactions. Integrins and cell adhesions as modulators of mammary cell survival and transformation. Breast cancer research: BCR. 2001;3(4):224–229. doi: 10.1186/bcr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sloan EK, Pouliot N, Stanley KL, Chia J, Moseley JM, Hards DK, Anderson RL. Tumor-specific expression of αvβ3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Research. 2006;8(2):R20. doi: 10.1186/bcr1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, et al. Cheresh DA. An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nature cell biology. 2014;16(5):457–68. doi: 10.1038/ncb2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Barkan D, Chambers AF. B1-integrin: a potential therapeutic target in the battle against cancer recurrence. Clinical Cancer Research. 2011 doi: 10.1158/1078-0432.CCR-11-0642. [DOI] [PubMed] [Google Scholar]
- 98.Das S, Ongusaha PP, Yang YS, Park JM, Aaronson Sa, Lee SW. Discoidin domain receptor 1 receptor tyrosine kinase induces cyclooxygenase-2 and promotes chemoresistance through nuclear factor-kappaB pathway activation. Cancer Research. 2006;66:8123–8130. doi: 10.1158/0008-5472.CAN-06-1215. [DOI] [PubMed] [Google Scholar]
- 99.Rammal H, Saby C, Magnien K, Van-Gulick L, Garnotel R, Buache E, Morjani H. Discoidin domain receptors: potential actors and targets in cancer. Frontiers in Pharmacology. 2016 doi: 10.3389/fphar.2016.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nature reviews Cancer. 2008;8(12):967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pang MF, Siedlik MJ, Han S, Stallings-Mann M, Radisky DC, Nelson CM. Tissue stiffness and hypoxia modulate the integrin-linked kinase ILK to control breast cancer stem-like cells. Cancer research. 2016 doi: 10.1158/0008-5472.CAN-16-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Al-Ejeh F, Smart CE, Morrison BJ, Chenevix-Trench G, López JA, Lakhani SR, Khanna KK. Breast cancer stem cells: treatment resistance and therapeutic opportunities. Carcinogenesis. 2011 doi: 10.1093/carcin/bgr028. [DOI] [PubMed] [Google Scholar]
- 103.Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VHC, Groom AC, Chambers AF. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metas-tases. Breast Cancer Research and Treatment. 2003;82(3):199–206. doi: 10.1023/B:BREA.0000004377.12288.3c. [DOI] [PubMed] [Google Scholar]
- 104.Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, et al. Green JE. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Research. 2008;68(15):6241–6250. doi: 10.1158/0008-5472.CAN-07-6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S, et al. Iredale JP. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53(4):1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tilghman RW, Blais EM, Cowan CR, Sherman NE, Grigera PR, Jeffery ED, Parsons JT. Matrix rigidity regulates cancer cell growth by modulating cellular metabolism and protein synthesis. PLoS ONE. 2012;7(5) doi: 10.1371./journal.pone.0037231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pupa SM, Giuffré S, Castiglioni F, Bertola L, Cantú M, Bongarzone I, et al. Tagliabue E. Regulation of breast cancer response to chemotherapy by fibulin-1. Cancer Research. 2007;67(9):4271–4277. doi: 10.1158/0008-5472.CAN-06-4162. [DOI] [PubMed] [Google Scholar]
- 108.Peiris-Pagès M, Sotgia F, Lisanti MP. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog-GLI signalling in breast cancer cells. Oncotarget. 2015;6(13):10728–10745. doi: 10.18632/oncotarget.3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature reviews Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 110.Shen CJ, Sharma A, Vuong DV, Erler JT, Pruschy M, Broggini-Tenzer A. Ionizing radiation induces tumor cell lysyl oxidase secretion. BMC Cancer. 2014;14(1):532. doi: 10.1186/1471-2407-14-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K, et al. Xu L. TGF-β blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(41):16618–23. doi: 10.1073/pnas.1117610109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bondareva A, Downey CM, Ayres F, Liu W, Boyd SK, Hallgrimsson B, Jirik FR. The lysyl oxidase inhibitor, beta-aminopropionitrile, diminishes the metastatic colonization potential of circulating breast cancer cells. PloS One. 2009;4(5):e5620. doi: 10.1371/journal.pone.0005620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Beckenlehner K, Bannke S, Spruss T, Bernhardt G, Schönenberg H, Schiess W. Hyaluronidase enhances the activity of adriamycin in breast cancer models in vitro and in vivo. Journal of Cancer Research and Clinical Oncology. 1992;118(8):591–596. doi: 10.1007/BF01211802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shuster S, Frost GI, Csoka AB, Formby B, Stern R. Hyaluronidase reduces human breast cancer xenografts in SCID mice. International Journal of Cancer. 2002;102(2):192–197. doi: 10.1002/ijc.10668. [DOI] [PubMed] [Google Scholar]
- 115.Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, et al. Moses HL. Stromally derived lysyl oxi-dase promotes metastasis of transforming growth factor-β-deficient mouse mammary carcinomas. Cancer Research. 2013;73(17):5336–5346. doi: 10.1158/0008-5472.CAN-13-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, et al. Smith V. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nature medicine. 2010;16(9):1009–17. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]
- 117.Chan N, Willis A, Kornhauser N, Ward MM, Lee SB, Nackos E, et al. Vahdat L. Influencing the tumor microen-vironment: phase 2 study of copper depletion with tetrathiomolybdate in high risk breast cancer and preclinical models of lung metastases. Clinical Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-16-1326. [DOI] [PubMed] [Google Scholar]
- 118.Golubovskaya VM, Cance WG. Focal adhesion kinase and p53 signaling in cancer cells. International Review of Cytology. 2007;263(7):103–153. doi: 10.1016/S0074-7696(07)63003-4. [DOI] [PubMed] [Google Scholar]
- 119.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Current Opinion in Cell Biology. 2006;18(5):516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 120.Tanjoni I, Walsh C, Uryu S, Tomar A, Nam JO, Mielgo A, et al. Schlaepfer DD. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer biology & therapy. 2010;9(10):764–77. doi: 10.4161/cbt.9.10.11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Walsh C, Tanjoni I, Uryu S, Tomar A, Nam JO, Luo H, et al. Schlaepfer DD. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer biology & therapy. 2010;9(10):778–90. doi: 10.4161/cbt.9.10.11433. [DOI] [PMC free article] [PubMed] [Google Scholar]