

Abstract
Mutations in the LIM homeobox transcription factor 1-beta (LMX1B) are a cause of nail patellar syndrome, a condition characterized by skeletal changes, glaucoma and focal segmental glomerulosclerosis. Recently, a missense mutation (R246Q) in LMX1B was reported as a cause of glomerular pathologies without extra-renal manifestations, otherwise known as nail patella-like renal disease (NPLRD). We have identified two additional NPLRD families with the R246Q mutation, though the mechanisms by which LMX1BR246Q causes a renal-specific phenotype is unknown. In this study, using human podocyte cell lines overexpressing either myc-LMX1BWT or myc-LMX1BR246Q, we observed dominant negative and haploinsufficiency effects of the mutation on the expression of podocyte genes such as NPHS1, GLEPP1, and WT1. Specifically, we observed a novel LMX1BR246Q-mediated downregulation of WT1(−KTS) isoforms in podocytes. In conclusion, we have shown that the renal-specific phenotype associated with the LMX1BR246Q mutation may be due to a dominant negative effect on WT1(−KTS) isoforms that may cause a disruption of the WT1 (−KTS):(+KTS) isoform ratio and a decrease in the expression of podocyte genes. Full delineation of the LMX1B gene regulon is needed to define its role in maintenance of glomerular filtration barrier integrity.
Steroid resistant nephrotic syndrome (SRNS) is a condition characterized by defects in the glomerular filtration barrier (GFB) which leads to proteinuria, hypoalbuminemia, edema and an inability to achieve clinical remission following standard corticosteroid therapy. SRNS is associated with a number of renal pathologic changes, the most common being focal segmental glomerulosclerosis (FSGS). Globally, SRNS is a major cause of end stage kidney disease (ESKD) requiring dialysis and kidney transplantation. The pathogenesis of SRNS is still being unraveled, however evidence from studies of the familial form of the disease points to defects in glomerular podocytes as central to disease pathogenesis. Familial forms of SRNS are genetically heterogeneous and are associated with mutations in over 25 genes1. One of the genes previously associated with familial SRNS is LIM homeobox transcription factor 1-beta (LMX1B)2,3,4,5.
LMX1B is a transcription factor in the LIM-homeodomain family of proteins. The LIM-homeodomain proteins are vital for the normal development of dorsal limb structures, components of the GFB, and the anterior segment of the eyes. Mutations in LMX1B are a cause of nail patellar syndrome (NPS) (OMIM 161200), a condition that is characterized by skeletal dysplastic changes, glaucoma and FSGS with type III collagen fibrils in the glomerular basement membrane2. It has previously been observed that patients with NPS caused by a LMX1B mutation located in the homeodomain showed significantly more frequent and more severe renal involvement compared with patients with mutations outside of this domain3. More recently, a renal specific missense mutation (R246Q) in the homeobox domain of LMX1B (LMX1BR246Q) has been reported by different groups in France, United Kingdom and Japan6,7,8,9. In each of these families, all of the affected individuals presented with glomerular pathologies without skeletal or other extra-renal manifestations. However, the full spectrum of renal phenotypes associated with the LMX1BR246Q mutation is unknown and the reason why the mutation causes kidney-limited disease has not been fully elucidated. As part of our effort to fully characterize genes that are important for maintaining the functional integrity of the GFB, we carried out genome-wide linkage studies (GWLS) and whole-exome sequencing (WES) in families with familial SRNS. We identified two families with the renal-limited LMX1BR246Q mutations with widely variable renal phenotypes. We uncovered patterns of expression that suggest differential effects of the mutation on a panel of podocyte-expressed genes by comparing immortalized human podocyte control cells to immortalized lines stably overexpressing either wild-type LMX1B (LMX1BWT) or LMX1BR246Q. We observed reduced transcription of WT1, TRPC6, NPHS1, and GLEPP1 in LMX1BR246Q-expressing cells in a pattern suggesting a dominant negative effect by the R246Q mutation on WT1 gene expression and a haploinsufficiency effect on TRPC6, NPHS1, and GLEPP1 gene expression. Additionally, we discovered a novel LMX1BR246Q-mediated downregulation of WT1(−KTS) isoforms in podocytes which may result in an imbalance of the WT1(−KTS) isoforms and subsequently this renal-limited phenotype. This study expands our knowledge of the phenotypes associated with the LMX1BR246Q mutation and provides further insight into the effects of the mutation on the transcriptional regulation of key podocyte genes.
Results
Families and linkage analysis
We identified six families with familial SRNS with segregation patterns consistent with autosomal dominant (AD) inheritance. The majority of the affected individuals were diagnosed with FSGS on renal biopsy, and others with findings such as membranous nephropathy, mesangial proliferative glomerulonephritis and minimal change disease. We performed genome wide linkage studies (GWLS) on the six families, the analysis did not yield conclusive evidence of linkage (logarithm of the odds (LOD) score >3) for any individual family. Two families (DUK35705 and DUK34319) however had suggestive logarithm of the odds (LOD) scores of 1.7 and 1.4 on chromosome 9q respectively (Supplementary Figure 1a,b). The pedigrees of these two families are shown in Fig. 1a,b. To identify the smallest candidate region, we performed combined GWLS analysis on the two families and identify a minimal candidate region on chromosome 9q region that spans a physical distance of 58.4 Mb between rs966097 and rs3811133 (Supplementary Figure 2a).
Figure 1. Pedigree of two families with FSGS.

(a and b) There are at least 18 affected individuals in the two families. There is clear male-to-male transmission in both families consistent with an autosomal dominant mode of transmission. Arrowheads identify the proband in both families. DNA samples from individuals 1, 805 and 1000 in Fig. 2A were subjected to whole exome sequencing.
Phenotypes of the two families with linkage to Chromosome 9q locus
A summary of clinical findings in affected individuals is shown in Table 1. Most of the affected individuals presented with early onset kidney disease with a median age of 12 (range: 3–22) years. Three of the eight affected individuals in Family DUK35705 presented with early onset FSGS while kidney biopsy in the affected individuals in Family DUK34319 showed a wide variety of morphological changes including C1q nephropathy, immune complex glomerulonephritis, and focal global glomerulosclerosis among others (Table 1). In addition to renal diseases, two individuals in DUK35705 have a history of glaucoma and autism spectrum disorder (Table 1). Five out of the thirteen affected individuals in the two families developed ESKD before the age of 25 years. None of the affected individuals who received kidney transplantation had recurrence of the primary kidney disease after transplantation (Table 1).
Table 1. Phenotype associated with LMX1B mutation in two families with FSGS.
| Study ID | Gender | Age onset (yrs) | Proteinuria Y/N | Renal biopsy | Extra renal manifestations | Age at ESKD (yrs) | Transplant Y/N Recurrence Y/N |
|---|---|---|---|---|---|---|---|
| 35705: 1 | F | <18 | Yes | FSGS | No | 18 | Y/N |
| 35705: 101 | M | <22 | Yes | UNK | No | 22 | Y/N |
| 35705: 102 | F | <16 | Yes | UNK | Glaucoma | 16 | Y/N |
| 35705: 801 | M | 5 | Yes | FSGS | Autism | NA | N/NA |
| 35705: 802 | M | 3 | Yes | FSGS | Autism | NA | N/NA |
| 35705: 803 | F | 6 | Yes | NA | No | NA | N/NA |
| 35705: 805 | M | <14 | Yes | NA | No | NA | N/NA |
| 35705: 1000 | M | 18 | Yes | Memb | Glaucoma | 58 | Y/N |
| 34319: 1 | F | 12 | UNK | Familial nephropathy | No | 22 | N/NA |
| 34319: 100 | M | 4 | Yes | FGGS | No | NA | N/NA |
| 34319: 101 | F | 5 | Yes | UNK | No | N/NA | |
| 34319: 102 | M | 2 | Yes | Immune complex GN | No | 5 | Y/N |
| 34319: 8002 | F | 10 | Yes | C1q nephropathy | No | NA | N/NA |
F: Female, M: Male, UNK: Unknown, NA: Not applicable, Memb: Membranous nephropathy, FSGS: Focal segmental glomerulosclerosis, FGGS: Focal global glomerulosclerosis, GN: Glomerulonephritis, Y: Yes, N: No, YRS: Years.
Whole exome sequencing (WES) and targeted sequencing
The DNA from three affected individuals in Family DUK35705 were subjected to whole-exome sequencing (WES) using the Illumina TruSeq platform. After excluding variants with minor allele frequencies >1% in the dbGaP database, 68 shared variants remained between the three individuals, of which one variant is the missense variant c.737G > A p.R246Q in LIM homeobox transcription factor 1-β (LMX1B) gene (Supplementary Figure 2b). The complete list of the 68 shared variants is provided in Supplementary Table 1 and the filtering algorithm in Supplementary Table 2. LMX1B gene mutations are the cause of nail patellar syndrome (OMIM 161200). Direct sequencing of all affected members of Families DUK35705 and DUK34319 showed that the variant segregated with the disease in both families. We sequenced exons 4 to 6 of LMX1B that encode for the DNA binding domain of LMX1B protein in 130 additional families with non-syndromic FSGS, we did not find additional families with mutations in LMX1B, thus mutations in exons 4 to 6 of LMX1B are responsible for 1–2% of AD FSGS in our cohort. We did not find any other segregating variants in the chromosome 9q region (Supplementary Figure 2a).
Modifier genes
Since it has been suggested that variants in other genes may be responsible for the variability in renal phenotype associated with LMX1B mutations5, we searched for presumed pathogenic rare variants (MAF < 1%) in all known FSGS genes, COL4A3, COL4A4 and COL4A5 in the exomes of three individuals in 35705. We found a rare variant G545A in the COL4A4 gene in one individual in Family DUK35705. Direct sequencing of all other affected individuals in this family showed that only one individual carried this variant. This individual was reported as having membranous nephropathy on light microscopy. There was no obvious difference in the phenotype of this individual and the other members of the family, therefore it appears that modifier genes play a limited (if any) role in these families.
Phenotypes associated with kidney specific LMX1B mutations
We conducted an extensive literature search to identify studies describing the renal limited phenotype associated with LMX1B mutations6,7,8,9. We identified four studies reporting three mutations in the LMX1B DNA binding homeodomains, these studies reported on six families with LMX1B mutations. Four out of the six families had the R246Q mutation while the other two families carried R246P and R249Q mutations. These four studies reported a diversity of renal biopsy findings including minimal change disease (MCD), FSGS, mesangial proliferative glomerulonephritis, C1q nephropathy among others (Table 2).
Table 2. Renal biopsy findings in 45 individuals from eight families with renal limited LMX1B mutations.
| Renal biopsy findings | N (%) | Mutations | References |
|---|---|---|---|
| FSGS/FGGS | 14 (31.1) | R246Q | 6, 9, and present study |
| None specific (CKD) | 4 (8.9) | R249Q | 6 and 7 |
| MCD | 2 (4.4) | R246P | 6 |
| Nail patellar like renal disease | 1 (2.2) | R246Q | 8 |
| Mesangial proliferative GN/C1q nephropathy | 2 (4.4) | R246Q, R249Q | 7, and present study |
| Membranous GN | 1(2.2) | R246Q | Present study |
| Immune complex GN | 1 (2.2) | R246Q | Present study |
| Familial nephropathy | 1 (2.2) | R246Q | Present study |
| Unknown | 19 (42.2) | R246Q, R249Q | 6, 7, and, present study |
GN: Glomerulonephritis, MCD: Minimal change disease.
R246Q mutation and the expression of key glomerular filtration barrier (GFB) genes
To characterize the functional effects of the LMX1B R246Q mutation, we examined the changes in LMX1B transcriptional regulation induced by the R246Q mutation. To accomplish this, we generated conditionally immortalized human podocyte cell lines stably overexpressing either myc-LMX1BWT or myc-LMX1BR246Q via lentiviral infection. Total RNA was collected from both LMX1B-overexpressing lines and from differentiated controls (i.e. uninfected podocytes and tGFP-expressing podocytes). The infection was verified by immunoblot analysis for myc-tag expression and quantification of LMX1B mRNA levels in LMX1B-overexpressing podocytes. The levels of LMX1B overexpression were similar between the myc-LMX1BWT and myc-LMX1BR246Q infected cells (Fig. 2a and Supplementary Figure 3). We did not detect any obvious differences in the morphology of the F-actin cytoskeleton in the myc-LMX1BWT and myc-LMX1BR246Qpodocytes by immunofluorescence examination. In addition, subcellular localization of Nephrin, CD2AP, GLEPP1 and WT1 were similar between the two cell lines (Supplementary Figure 4).
Figure 2. Effect of R246Q mutation on podocyte related gene transcription.

(a) Western blot confirming expression of the viral transgene with the presence of the MYC tag in myc-LMX1BWT or myc-LMX1BR246Q infected immortalized human podocyte cells compared to uninfected and lentiviral control podocytes (top). β-actin was used as a loading control for all samples (bottom). Blots were cropped to exclude lanes that are not relevant to this paper, the uncropped blot is displayed in Supplementary Figure 6. (b) Graph depicting average Relative Quantity (RQ) values for NPHS1, GLEPP 1, INF2, and SYNPO in myc-LMX1BWT or myc-LMX1BR246Q cells (N ≥ 3). Error bars = SEM *p ≤ 0.05, **p ≤ 0.001 using t-test. (c) Immunoblotting showing reduced translation of nephrin (n = 7, p = 0.027) but not GLEPP1 (n = 4, p > 0.05), CD2AP (n = 3, p > 0.05), synaptopodin (n = 3, p > 0.05) and INF2 (n = 3, p > 0.05) in myc-LMX1BR246Q podocytes compared to myc-LMX1BWTexpressing cells. β-actin was used as a loading control for all samples and unedited blots are shown in Supplementary Figure 6.
We evaluated for alterations in the transcription of several key podocyte genes in the myc-LMX1BWT and myc-LMX1BR246Q-overexpressing podocytes by quantitative Real Time PCR (qPCR). NPHS1, TRPC6, and GLEPP1 gene expression was upregulated in both myc-LMX1BWT and myc-LMX1BR246Q-expressing podocytes. However, the expression of NPHS1, TRPC6 and GLEPP1 was significantly reduced in myc-LMX1BR246Q-expressing podocytes compared to myc-LMX1BWT-expressing podocytes, suggesting that the mutation may exert a haploinsufficiency effect on the transcriptional regulation of these genes. (Fig. 2b and Supplementary Figure 5). Conversely, no significant changes in gene expression were observed in INF2, CD2AP, SYNPO or COL4A4 between myc-LMX1BWT and myc-LMX1BR246Q-overeexpressing podocytes. (Fig. 2b and Supplementary Figure 5). The reduction in NPHS1 expression and lack of change in CD2AP, SYNPO and CD2AP was confirmed at the protein level by immunoblot analysis (Fig. 2c). There was no difference in expression of GLEPP1 between the wild type and the mutant cell line (Fig. 2c).
R246Q mutation and the expression of WT1 gene
A different pattern of gene expression was uncovered for the regulation of the Wilms’ Tumor 1 gene (WT1). In myc-LMX1BWT-expressing podocytes, WT1 was modestly increased compared to the control cell line (RQ of 1.26 vs 1.00, p = 0.0032) (WT1 probe 1 Fig. 3). Conversely, in myc-LMX1BR246Q-expressing podocytes, WT1 was significantly decreased relative to wild-type (RQ of 0.53 vs 1.26, p = 0.0002) and controls (RQ of 1.00 vs 0.53, p < 0.00001) (WT1 probe 1 Fig. 3) suggesting a dominant negative effect of the mutation on the regulation of WT1 gene expression. This reduction of WT1 expression in myc-LMX1BR246Q-expressing podocytes compared to myc-LMX1BWT-expressing podocytes was confirmed at the protein level by immunoblot analysis (Fig. 3).
Figure 3. Dominant negative WT1 isoform specific reduction due to R246Q mutation in LMX1B.

(a) The specific reduction of WT1(−KTS) isoform can be seen in this graph depicting average (RQ) values for WT1 (+KTS and −KTS) (WT1 Probe 1) and WT1 (+KTS only) (WT1 Probe 2) expression in myc-LMX1BWT or myc-LMX1BR246Q podocyte cells compared to control podocyte cells (uninfected and lentiviral control) (N ≥ 3). Error bars = SEM, *p ≤ 0.05, **p ≤ 0.001 using t-test. (b) The reduction in expression of WT1 protein in myc-LMX1BR246Qcells compared to myc-LMX1BWT cells is shown by immunoblotting (n = 6, p = 0.038). β-actin was used as a loading control for all samples and unedited blots are shown in Supplementary Figure 6.
In a previous study, we identified synaptopodin (SYNPO) as a target gene of WT1D, a +KTS isoform of WT110. Despite the apparent dominant negative effect of LMX1BR246Q on WT1 gene expression, there was no effect on SYNPO mRNA expression. This finding suggests that LMX1B may selectively modulate WT1-mediated gene transcription via the isoform-specific regulation of WT1 expression in podocytes. To test this hypothesis, we performed qPCR using an assay designed to detect only the +KTS isoforms (WT1 probe 2 Fig. 3). The expression of +KTS isoforms was increased in myc-LMX1BWTand myc-LMX1BR246Q expressing podocytes relative to controls and did not show the dramatic LMX1BR246Q induced reduction that was observed in the initial WT1 screen. (WT1 probe 1 Fig. 3). Taken together, these findings suggest that LMX1B regulates WT1 expression in an isoform-specific manner to selectively influence the expression profile of key podocyte genes (Fig. 4).
Figure 4. Schematic representation of the proposed mechanism for development of NPLRD due to the LMX1BR246Qmutation.
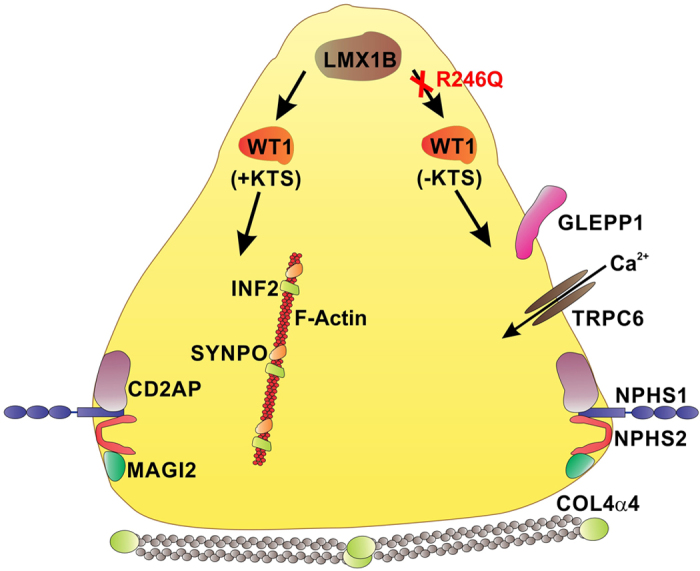
The R246Q mutation causes a specific reduction of the WT1(−KTS) isoform in podocytes leading to a down regulation of key podocyte genes such as NPHS1, GLEPP1, and TRPC6. This reduction of podocyte gene expression may also be amplified by a reduction in other transcription factors and reduced transcriptional activity of the LMX1B transcription factor itself. Through distinct or redundant mechanisms the presence of WT1(+ KTS) isoforms along with potential contributions from LMX1B and other transcription factors is able to maintain expression of some podocyte genes such as CD2AP, INF2, SYNPO and MAGI2.
Discussion
Advances in genome science and the availability of powerful next-generation sequencing tools for genomic research have accelerated the pace of gene discovery in hereditary nephrotic syndromes. To date more than 25 single genes have been linked to the development of SRNS1. Their discovery not only improves our understanding of disease mechanisms, but also provides data allowing better classification and treatment of diseases11. Mutations in LMX1B was previously identified as a cause of NPS. More recently, missense mutations in the homeodomain of LMX1B have been shown to cause a kidney-limited phenotype without extra renal manifestations. So far, twenty-seven cases from six families have been reported with kidney-limited phenotypes due to mutations in exons of LMX1B that encode for the homeodomain of the protein. In the present study, we report two additional families with 18 affected individuals. The predominant LMX1B mutation in the present report and the six families in previous reports is the R246Q mutation (6 of 8 families). Families with this mutation are predominantly of European descent, suggesting that this may be a founder mutation in this population. In our cohort of patients with non-syndromic AD SRNS, mutations in the LMX1B homeodomain are responsible for up to 2% of cases, similar to the report of Boyer et al.6.
The phenotypes associated with mutations of the homeodomain have been described using varied terminology including the term nail patella-like renal disease (NPLRD)8. NPLRD is associated with classic transmission electron microscopy findings of diffuse irregular thickening of the glomerular basement membrane with patchy electron lucent areas and irregular deposition of type III collagen fibrils. However, because light microscopy is a more widely available modality for examining renal biopsy especially in resource poor countries, it is important to report the spectrum of light microscopy findings associated with these mutations. In this study different light microscopic findings, including FSGS, MCD, membranous nephropathy and immune complex glomerulonephritis, were described in our patients. Reasons for this observation are not known but it may be due to factors such as the timing of kidney biopsy and/or the presence of modifier genes affecting phenotypes. In line with this speculation, we searched for rare variants in key podocyte and GBM genes that have been associated with SRNS. We identified a single rare variant in COL4A4 (G545A) in only one individual with no significant phenotypic variability from other affected family members without the variant. Further studies in a larger cohort of patients are needed to further elucidate the role of modifier genes.
The mechanisms by which mutations in the homeodomain region of LMX1B induce renal specific phenotypes have not been fully investigated. Using luciferase assay, Isojima et al. suggested that the mutation resulted in LMX1B haploinsufficiency8. However, a comprehensive survey of the LMX1B gene regulon in podocytes has not been reported. In this study we generated human podocyte cell lines stably expressing myc-LMX1BWT and myc-LMX1BR246Q to evaluate the functional effects of the R246Q mutation on LMX1B-mediated transcription on collagen alpha 4 and a panel of key podocyte genes. We showed a reduced expression of NPHS1, TRPC6, and GLEPP1 in immortalized podocyte cell line expressing the myc-LMX1BR246Q compared to the myc-LMX1BWT, suggesting that the mutation may exert a haploinsufficiency effect on the expression of these genes. We were able to confirm this LMX1BR246Q induced reduction in NPHS1 and WT1 expression at the protein level by immunoblot however, there was no change observed in GLEPP1 protein expression. The reason for the disparity in GLEPP1 expression is unclear, but may be due to a prolonged half-life of the GLEPP1 protein in podocytes. Conversely, we did not identify any significant differences in RNA or protein expression between the myc-LMX1BWT and myc-LMX1BR246Q overexpressing cell lines in other podocyte genes such as CD2AP, SYNPO, and INF2. Our expression findings are consistent with the observations by Isojima et al.8. Notably, the pattern of WT1 gene regulation is different from all the other genes examined. Specifically, the LMX1BR246Q mutation exerts a haploinsufficiency effect on TRPC6, NPHS1, and GLEPP1 expression while for WT1(−KTS) isoforms, the LMX1BR246Q mutation exerts a clear dominant negative effect.
The alternate splicing of leucine, threonine and serine (KTS) amino acids in a critical area between the third and fourth zinc finger domains of WT1 is a highly evolutionarily conserved splicing event12,13,14. However, this archetypal program of translational processing may be insufficient to explain the transcriptional effects of the LMX1BR246Q mutation on WT1 expression. A possible explanation for LMX1B-mediated isoform-specific WT1 gene regulation is the use of alternative promoters. These cis-regulatory elements facilitate diversification of gene expression through influences on the efficiency of transcription initiation, pre-mRNA processing, transcript composition, and mRNA turnover15. Additionally, these promoter elements have been shown to influence tissue-specific gene expression and developmental gene activity16,17. It is estimated that 53% of human genes contain alternative promoters and Pal et al. have demonstrated that alternative transcription exceeds alternative splicing in generating transcriptome diversity in the developing and adult cerebellum18,19. Although intragenic alternative promoters have been identified within the WT1 gene, the role of LMX1B in the activation of these regulatory elements is unknown12.
Previous studies have shown distinct functional roles and transcriptional targets for WT1(+KTS) and WT1(−KTS) isoforms20,21,22,23,24. The WT1(−KTS) isoforms have been shown to act as traditional regulators of transcription for genes such as NPHS1, NPHS2, and SYNPO23,25,26,27. Alternatively, the WT1(+KTS) isoforms display reduced DNA binding potential and have been shown to be required for transcription of other podocyte genes such as SYNPO, MAGI2 and SCRIB and may also participate in RNA processing10,21,22,23,24,25,26,27. It is clear that the ratio of WT1(−KTS) and WT1(+KTS) isoforms is crucial for proper kidney development and function, as a decrease in the +KTS isoform is believed to be responsible for the development of FSGS in Frasier syndrome28,29. Frasier syndrome is characterized by male pseudohermaphroditism and progressive glomerulopathy due to a mutation in intron 9 of the WT1 gene that results in reduced WT1 (+KTS) splicing events. In mouse models of KTS isoform specific knockdown, elimination of either isoform leads to a severe disruption of glomerular development and podocyte differentiation23,24. These findings suggest that an altered WT1 isoform ratio may be one of the mechanisms by which the LMX1BR246Q mutation produces a kidney-specific effect in NPLRD patients (Fig. 4). Additional studies will be required to further examine the potential alterations in KTS isoform ratio and to unravel which of these podocyte gene expression changes are due to direct alterations in LMX1B transactivation potential versus alterations in other transcriptional regulators such as WT1.
Contrary to previous reports, we found some individuals with extra renal pathology such as autism and glaucoma in one of the two families. Glaucoma is one of the manifestation of classical NPS, but we are not aware of the report of an association of autism with LMX1B mutation in NPS. An association between autism and common variants in LMX1B has been reported in a small cohort of patients30. It is therefore likely that the finding of autism in our patients may represent effects of other variants in LMX1B and other modifier genes. Alternatively, it is possible that the renal-specific mutations may dysregulate the transcription of genes that are important during the development of the podocytes and basement membranes in the kidney, eyes and brain.
In conclusion, we reported two additional families with renal specific mutations in LMX1B and showed wide variability in renal pathologic findings within family and between families. Based on these series of experiments, we proposed that the constellation of different glomerular changes in a family with hereditary kidney disease may be a pointer to a diagnosis of kidney specific LMX1B mutation. We provide evidence that the renal specific phenotype associated with this mutation may be due to dominant negative effect on WT1(−KTS) specific isoforms causing a disruption of the WT1 isoform ratio and subsequent decrease in key podocyte related genes. These findings suggest that a comprehensive delineation of the LMX1B regulon in podocytes may uncover novel transcriptional regulatory patterns and improve our understanding of podocyte LMX1B transcriptional activity in health and disease.
Materials and Methods
Institutional review board approval and participants enrollment
Institutional Review Board approval was obtained from the Duke University Medical Center (Durham, NC, USA) and informed consent was obtained from all subjects. All methods were performed in accordance with the relevant guidelines and regulations. Inclusion criteria and determination of affection status are as previously reported10,31,32.
Linkage analysis
A genome-wide linkage scan was performed using the informative markers in the Illumina HumanOmniExpress-24 genotyping beadchip assay (Illumina Inc., San Diego, California). The list of the informative markers is available on request. Genotyping was performed on 60 individuals from six families comprising of 36 affected individuals and 24 unaffected individuals (unaffected and unknown status). Two-point and multipoint LOD scores were calculated for all the informative SNP markers. A rare dominant model was assumed and a conservative “affecteds-only” analysis was performed.
Whole-exome sequencing
Whole exome sequencing was performed on three affected individuals in family Duke 35705 using standard protocol. We used the Agilent All Exon 50MB kit and sequenced to at least 75x coverage using one lane of a HiSeq 2000 sequencer. Reads were aligned to the Human Reference genome (HG 18) using the BWA software33. Single nucleotide variants were called usingSAMtools34. The variants were annotated to Ensembl 50_36 l using SequenceVariantAnalyzer (SVA) and were analyzed using the ATAV software33.
Sanger sequencing
Potential disease causing variants identified by whole-exome sequencing were confirmed by Sanger sequencing. All sequences were analyzed with the Sequencher software (Gene Codes Corp Ann Arbor, MI).
Conditionally Immortalized Human Podocyte Culture
Conditionally immortalized human podocytes were cultured as described previously10.
mRNA Extraction and Quantitative Real Time PCR
Total RNA was manually extracted from differentiated podocyte cells using an RNeasy Mini kit (Qiagen, Valencia, CA). Subsequently, 0.5 μg of total RNA was reverse transcribed into cDNA utilizing the RT system (Promega Corporation, Madison, WI) with oligo(dT) primers, according to the manufacturer’s protocol. Quantification of mRNA by real-time PCR was performed using the ABI VIAA7 system (Applied Biosystems, Foster City, CA). PCR reactions were performed in a final volume of 10 μl, consisting of 2 μl cDNA, 2.5 μl RNAse and DNAse free water, 0.5 μl of 20× TaqMan Gene Expression Assays, and 5 μl of TaqMan 2× PCR Master Mix (both Applied Biosystems). Probe sequences are listed in Supplementary Table 3. Each individual experiment was performed at least three times, in triplicate or quadruplicate. Relative expression of the target genes was analyzed by normalizing to the housekeeping gene β-Actin.
Lentiviral Constructs and Infection
Standard molecular cloning methods were used to replace the ubiquitin-EGFP of FUGW 19 with CMV-turboGFP, CMF-myc-hLMX1BWT and CMV-LMX1BR246Q. Lentivirus was made and conditionally immortalized human podocytes were transduced as described previously32. Infected and uninfected controls were treated with 2 ug/ml puromycin to select for cells expressing the transgene construct.
Immunoblotting
Conditionally immortalized podocyte were differentiated per established protocols (PMID: 19955187), harvested, and treated with lysis buffer (Pierce Biotechnology, Rockford, IL) supplemented with phosphatase/protease inhibitor cocktail, 1:100 dilution (Cell Signaling Technology) and 1:100 PMSF (Sigma-Aldrich) for 15 min on ice. Cells were then spun at 14,000 RPM for 10 mins at 4 °C (Eppendorf). Protein Immunoblotting was performed as described previously using a MYC tag antibody 1:000 (Cell Signaling Technology), rabbit polyclonal nephrin antibody1:1000 (Abcam), mouse monoclonal Glepp1 antibody 1:1000 (Santa Cruz), mouse monoclonal WT1 antibody 1:300 (Santa Cruz), rabbit polyclonal CD2AP 1:1000 (Abcam), rabbit polyclonal Synaptopodin antibody 1:800 (Santa Cruz), rabbit polyclonal INF2 antibody 1:1000 (Bethyl Laboratories), and mouse monoclonal β-actin antibody 1:3000 (Sigma-Aldrich)10.
Immunofluorescence
Conditionally immortalized human podocytes were differentiated per established protocols on collagen I–coated coverslips (BD Biosciences) and processed as described previously10. Cells were incubated with mouse monoclonal WT1 antibody (Santa Cruz), mouse monoclonal nephrin antibody (Santa Cruz), or polyclonal rabbit CD2AP (Abcam) overnight at 4 °C. Cells were then washed with ice-cold PBS and secondary Alexa Flora 488 antibody was applied (Invitrogen) at a concentration of 1:1000 for 1 hour at room temperature. Cells were then washed four times with PBS at room temperature before addition of phalloidin alexa-568 (Invitrogen) for one hour at RT followed by two PBS washes and then 4′,6-diamidino-2-phenylindole stain at a concentration of 1:20,000 diluted in PBS. Immunofluorescence imaging was performed using a Carl Zeiss AxioImager and the ZenBlue Bioimaging Software.
Statistical Analyses
All data are represented as the mean ± SEM. Group differences were assessed by the t test with unequal variances. Statistical significance was established at P < 0.05.
Additional Information
How to cite this article: Hall, G. et al. Dysregulation of WTI (−KTS) is Associated with the Kidney-Specific Effects of the LMX1B R246Q Mutation. Sci. Rep. 7, 39933; doi: 10.1038/srep39933 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
NIH NIDDK 5R01DK098135 and 5R01DK094987 to RG. GH receives funding through the American Society of Nephrology/Amos Medical Faculty Development Program and the P&F grant of the Duke O’Brien Center for Kidney Research (DOCK). We acknowledge support from the Renal Genomics Core (Core B) of the DOCK supported by NIH NIDDK P30DK096493 award. We would like to thank Dr. David Corcoran and Dr. Olivier Fedrigo and the personnel of the Duke Center for Genomic and Computational Biology for assistance with sequencing. We would also like to thank Ms. Marguerita Klein of the Duke Viral Vector Core Facility for the lentivirus preparation services, the personnel of the genomic core facilities of the Duke Molecular Physiology Institute and most importantly the family members of the Duke FSGS project.
Footnotes
Author Contributions R.G. and G.H. designed the overall study and identified families with LMX1B mutations. B.L. and G.W. performed the experiments. R.G., G.H., M.C.-L., J.-J.L., X.Q., and E.R.H. phenotyped the families and generated the linkage analysis data. R.G., G.H., B.L., and M.C.-L. generated the figures. R.G., G.H., B.L., J.-J.L., and E.R.H. contributed to writing the manuscript and all authors reviewed the manuscript.
References
- Sadowski C. E. et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 26, 1279–1289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer S. D. et al. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat. Genet. 19, 47–50 (1998). [DOI] [PubMed] [Google Scholar]
- Rohr C. et al. The LIM-homeodomain transcription factor Lmx1b plays a crucial role in podocytes. J. Clin. Invest. 109, 1073–1082 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongers E. M. et al. Genotype-phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur J Hum. Genet. 13, 935–946 (2005). [DOI] [PubMed] [Google Scholar]
- Lemley K. V. Kidney disease in nail-patella syndrome. Pediatr. Nephrol. 24, 45–54 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer O. et al. LMX1B mutations cause hereditary FSGS without extra renal involvement. J. Am. Soc. Nephrol. 24, 1216–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards N. et al. A novel LMX1B mutation in a family with end-stage renal disease of ‘unknown cause’. Clin. Kidney. J. 8, 113–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isojima T. et al. LMX1B mutation with residual transcriptional activity as a cause of isolated glomerulopathy. Nephrol. Dial. Transplant. 29, 81–8 (2014). [DOI] [PubMed] [Google Scholar]
- Konomoto T. et al. Clinical and histological findings of autosomal dominant renal-limited disease with LMX1B mutation. Nephrology 21, 765–773 (2016). [DOI] [PubMed] [Google Scholar]
- Hall G. et al. A novel missense mutation of Wilms’ Tumor 1 causes autosomal dominant FSGS. J. Am. Soc. Nephrol. 26, 831–43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone A. F. et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 86, 1253–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenstein P. & Hastie N. The many facets of the Wilms’ tumour gene, WT1. Hum. Mol. Genet. 15, R196–R201 (2006). [DOI] [PubMed] [Google Scholar]
- Kent J., Coriat A. M., Sharpe P. T. & Hastie N. D. The evolution of WT1 sequence and expression pattern in the vertebrates. Oncogene 11, 1781–92 (1995). [PubMed] [Google Scholar]
- Miles C. et al. Complete sequencing of the Fugu WAGR region from WT1 to PAX6: dramatic compaction and conservation of synteny with human chromosome 11p13. Proc. Natl. Acad. Sci. 95, 13068–13072 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoubi T. A. & Van De Ven W. J. Regulation of gene expression by alternative promoters. FASEB J. 10, 453–60 (1996). [PubMed] [Google Scholar]
- Kamat A., Hinshelwood M. M., Murry B. A. & Mendelson C. R. Mechanisms in tissue-specific regulation of estrogen biosynthesis in humans. Trends Endocrino.l Metab. 13, 122–128 (2002). [DOI] [PubMed] [Google Scholar]
- Saleh A., Makrigiannis A. P., Hodge D. L. & Anderson S. K. Identification of a novel Ly49 promoter that is active in bone marrow and fetal thymus. J. Immunol. 168, 5163–9 (2002). [DOI] [PubMed] [Google Scholar]
- Kimura K. et al. Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 16, 55–65 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S. et al. Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res. 21, 1260–72 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes A. et al. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell 106, 319–329 (2001). [DOI] [PubMed] [Google Scholar]
- Davies R. C. et al. WT1 interacts with the splicing factor U2AF65 in an isoform-dependent manner and can be incorporated into spliceosomes. Genes Dev. 12, 3217–3225 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson S. H. et al. Subnuclear localization of WT1 in splicing or transcription factor domains is regulated by alternative splicing. Cell 81, 391–401 (1995). [DOI] [PubMed] [Google Scholar]
- Lefebvre J. et al. Alternatively spliced isoforms of WT1 control podocyte specific gene expression. Kidney Int. 88, 321–31 (2015). [DOI] [PubMed] [Google Scholar]
- Wells J., Rivera M. N., Kim W. J., Starbuck K. & Haber D. A. The predominant WT1 isoform (+KTS) encodes a DNA binding protein targeting the planar cell polarity gene Scribble in renal podocytes. Molecular cancer research 8, 975–985 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann M. et al. Genome-wide analysis of Wilms’ Tumor 1-controlled gene expression in podocytes reveals key regulatory mechanisms. JASN. 26, 2097–2104, doi: 10.1681/asn.2014090940 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L. et al. Integration of Cistromic and Transcriptomic Analyses Identifies Nphs2, Mafb, and Magi2 as Wilms’ Tumor 1 Target Genes in Podocyte Differentiation and Maintenance. J. Am. Soc. Nephrol. 26, 2118–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L., Pietsch S. & Englert C. Towards an understanding of kidney diseases associated with WT1 mutations. Kidney Int. 88, 684–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaux S. et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat. Genet. 17, 467–470 (1997). [DOI] [PubMed] [Google Scholar]
- Klamt B. et al. Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/−KTS splice isoforms. Hum. Mol. Genet. 7, 709–714 (1998). [DOI] [PubMed] [Google Scholar]
- Thanseem I. et al. Association of transcription factor gene LMX1B with autism. PLoS One 6, e23738 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gbadegesin R. et al. A new locus for familial FSGS on chromosome 2p. J. Am. Soc. Nephrol. 21, 1390–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gbadegesin R. A. et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J. Am. Soc. Nephrol. 25, 1991–2002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. & Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. 1000 Genome Project Data Processing Subgroup: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.