

Abstract
Background
Long-acting muscarinic antagonist/long-acting β2-agonist combinations are recommended for patients whose chronic obstructive pulmonary disease (COPD) is not managed with monotherapy. We assessed the efficacy and safety of glycopyrrolate (GP)/formoterol fumarate (FF) fixed-dose combination delivered via a Co-Suspension™ Delivery Technology-based metered dose inhaler (MDI) (GFF MDI).
Methods
This was a Phase IIb randomized, multicenter, placebo-controlled, double-blind, chronic-dosing (7 days), crossover study in patients with moderate-to-very severe COPD (NCT01085045). Treatments included GFF MDI twice daily (BID) (GP/FF 72/9.6 μg or 36/9.6 μg), GP MDI 36 μg BID, FF MDI 7.2 and 9.6 μg BID, placebo MDI, and open-label formoterol dry powder inhaler (FF DPI) 12 μg BID or tiotropium DPI 18 μg once daily. The primary endpoint was forced expiratory volume in 1 s area under the curve from 0 to 12 h (FEV1 AUC0–12) on Day 7 relative to baseline FEV1. Secondary endpoints included pharmacokinetics and safety.
Results
GFF MDI 72/9.6 μg or 36/9.6 μg led to statistically significant improvements in FEV1 AUC0–12 after 7 days’ treatment versus monocomponent MDIs, placebo MDI, tiotropium, or FF DPI (p ≤ 0.0002). GFF MDI 36/9.6 μg was non-inferior to GFF MDI 72/9.6 μg and monocomponent MDIs were non-inferior to open-label comparators. Pharmacokinetic results showed glycopyrrolate and formoterol exposure were decreased following administration via fixed-dose combination versus monocomponent MDIs; however, this was not clinically meaningful. GFF MDI was well tolerated.
Conclusions
GFF MDI 72/9.6 μg and 36/9.6 μg BID improve lung function and are well tolerated in patients with moderate-to-very severe COPD.
Trial registration
ClinicalTrials.gov NCT01085045. Registered 9 March 2010.
Electronic supplementary material
The online version of this article (doi:10.1186/s12931-016-0491-8) contains supplementary material, which is available to authorized users.
Keywords: COPD, LAMA, LABA, Lung function, Bronchodilators, COPD maintenance, Co-Suspension™ Delivery Technology, Metered dose inhaler
Background
Concurrent use of long-acting muscarinic antagonist (LAMA) and long-acting β2-agonist (LABA) therapy has been shown to maximize the bronchodilator response [1], and dual LAMA/LABA combination therapy is now recommended as an alternative option for patients whose chronic obstructive pulmonary disease (COPD) is not well managed with bronchodilator monotherapy [2, 3]. In addition to providing improvements in airflow limitation and symptom control, dual bronchodilator therapy reduces the risk of adverse effects that may be associated with increased dosages of a single bronchodilator [1]. The use of a fixed-dose combination (FDC), in which the two agents are combined in a single device, may improve adherence, leading to improved outcomes and reduced costs [4, 5].
There are currently options for both once-daily (QD) and twice-daily (BID) dosing of LAMA/LABA FDCs [6], with some evidence suggesting that BID dosing may be preferable for patients who experience night-time symptoms [7]. However, choice of delivery device has been limited to LAMA/LABA FDCs delivered by dry powder inhalers (DPIs) or Soft Mist™ devices. Thus there is an opportunity to expand patient choice by developing a metered dose inhaler (MDI) formulation-based LAMA/LABA FDC.
A Co-Suspension™ Delivery Technology has been developed to overcome the variability and instability associated with drug delivery via traditional hydrofluoroalkane propellant-based MDI devices. This technology uses a novel formulation technique in which active-agent particles (typically in the form of micronized drug crystals) form strong and non-specific associations within the propellant with specially engineered porous microparticles, made from distearoylphosphatidylcholine and calcium chloride. These formulations possess excellent stability and dose uniformity, and allow simultaneous delivery of multiple drugs from one MDI without one drug affecting the delivery of the others, and maintain consistency of dose and aerosol properties between monotherapies and their combinations [8].
This Phase IIb study investigated the efficacy and safety of a LAMA/LABA FDC MDI, GFF MDI, containing glycopyrrolate (GP; equivalent to the bromide salt, glycopyrronium bromide) and formoterol fumarate (FF) formulated using the Co-Suspension delivery technology, using the dose of glycopyrrolate identified in a dose-response study in patients with moderate-to-severe COPD [9]. Earlier Phase I/IIa studies evaluated the efficacy and safety of the individual components, using a previous formulation of phospholipid porous particles [10, 11].
The primary objective of this two-part study was to assess lung function, specifically the improvement in forced expiratory volume in 1 s (FEV1) area under curve (AUC) from 0 to 12 h post-dose (FEV1 AUC0–12). Firstly to assess improvements in FEV1 AUC0–12 with GFF MDI 72/9.6 μg or 36/9.6 μg BID compared with individual component MDIs, placebo MDI (suspension of porous microparticles only), or the open-label comparators FF DPI and tiotropium bromide DPI in patients with COPD. Secondly, to assess improvements in FEV1 AUC0–12 reported with FF 9.6 μg MDI versus placebo MDI in patients with COPD. The safety profiles of GFF MDI and FF MDI, as well as the pharmacokinetic (PK) profiles of both glycopyrrolate and formoterol after chronic administration of GFF MDI, were also investigated.
Methods
Patients
Patients were 40–80 years of age with a diagnosis of COPD and were current or former smokers, with a smoking history of at least 10 pack-years. Key lung function criteria were: pre- and post-bronchodilator FEV1/forced vital capacity (FVC) ratio <0.7; post-bronchodilator FEV1 ≥750 mL, and ≥30% and <80% of the predicted value at screening; and pre-bronchodilator FEV1 ≤ 80% at baseline. Key exclusion criteria were: pregnancy or lactation; respiratory disease other than COPD; poorly managed COPD that had required hospitalization within 3 months of screening, or treatment with corticosteroids or antibiotics within 6 weeks of screening. In addition, patients who did not meet American Thoracic Society criteria for acceptable spirometry were excluded. Patients provided informed consent before undergoing any screening assessments.
Study design
This was a Phase IIb randomized, multicenter, placebo-controlled, double-blind, chronic dosing (7 days), four-period, eight-treatment, incomplete-block, crossover study, conducted in two parts in the USA, Australia, and New Zealand (NCT01085045). Patients recruited to Part A were not eligible for Part B. The design of the study is depicted in Additional file 1: Figure S1.
Part A was a four-period, eight-treatment, incomplete-block crossover study, designed to evaluate eight treatments: (i) GFF MDI 72/9.6 μg BID; (ii) GFF MDI 36/9.6 μg BID; (iii) GP MDI 36 μg BID; (iv) FF MDI 9.6 μg BID; (v) FF MDI 7.2 μg BID; (vi) placebo MDI BID; (vii) FF DPI 12 μg BID; and (viii) tiotropium DPI 18 μg QD. In this report, GP was expressed as glycopyrrolate (also known as glycopyrronium bromide) for which ex-actuator doses of 36 μg and 72 μg are equivalent to glycopyrronium (active moiety) 28.8 μg and 57.6 μg, respectively. Similarly, FF was expressed as formoterol fumarate, for which the dose of 9.6 μg (ex-actuator) is equivalent to formoterol fumarate dihydrate 10 μg. Each patient received four of eight possible treatments. A given treatment sequence included a GP MDI or an FF MDI component in no more than two treatment periods, whether administered as an FDC or as a single agent. Six combinations of four treatments were chosen for the study and then 48 treatment sequences created. Patients were randomized to one of the 48 treatment sequences which were generated centrally using an Interactive Web-based Response System based on Williams Square layouts.
Part B was a four-period, four-treatment, full-crossover study designed to evaluate: (i) FF MDI 9.6 μg BID; (ii) FF MDI 7.2 μg BID; (iii) placebo MDI BID; and (iv) FF DPI 12 μg BID. Patients were randomized to one of 24 possible treatment sequences, which were generated in the same way as Part A.
Patients administered each of their four assigned treatments for 1 week, followed by a 7- to 21-day washout period between treatments. All inhalers were dispensed in a blinded manner, with the exception of FF DPI and tiotropium DPI, which were provided open-label. The first dose of study drug was administered at the clinic under the supervision of a study coordinator (patients had been assessed previously for correct use of the MDI by study staff when using an albuterol MDI for the bronchodilator reversibility assessment). Self-administration continued at home. Each dose comprised two MDI actuations. Patients used their study drug BID for 1 week. FF DPI and open-label tiotropium DPI were administered for 7 days, according to the manufacturer’s instructions.
This study was conducted in accordance with International Conference on Harmonization guidelines, the Declaration of Helsinki and the US Code of Federal Regulations. The protocol, its amendments and patient informed consent form were approved by an Independent Ethics Committee or Institutional Review Board.
Efficacy endpoints
In both parts of the study, the primary endpoint was FEV1 AUC0–12 on Day 7 relative to baseline FEV1. In Part A, there were two primary comparisons: (i) GFF MDI 72/9.6 μg BID versus GP MDI 36 μg BID and (ii) GFF MDI 72/9.6 μg BID versus FF MDI 9.6 μg BID. To demonstrate efficacy for GFF MDI, superiority to both monocomponent MDIs was required. In Part B, the primary endpoint was based on the comparison of FF MDI 9.6 μg BID with placebo MDI BID.
The secondary efficacy endpoints of the study included measurements on both Day 1 and Day 7: peak change from baseline in FEV1, time to onset of action (≥10% improvement in FEV1 relative to baseline), proportion of patients achieving ≥12% improvement in FEV1 relative to baseline, peak change from baseline in inspiratory capacity (IC) and change in morning pre-dose trough FEV1 and IC. An exploratory endpoint included change from baseline in FVC. These endpoints provided additional information on the dose-response of bronchodilator effects on lung function for GFF MDI and FF MDI by exploring two doses for each versus active comparators and placebo MDI: GFF MDI 72/9.6 μg and 36/9.6 μg, and FF MDI 7.2 μg and 9.6 μg.
Pharmacokinetics
PK parameters were derived from the plasma concentrations of glycopyrrolate and formoterol fumarate obtained on approximately Day 7 (Day 7 ± 2) of each treatment regimen during Study Parts A and B. In Part A, the PK profiles of glycopyrrolate and formoterol after chronic administration of GFF MDI were compared with those after chronic administration of the monocomponent MDIs. In Part B, the PK profile of formoterol after chronic administration of two dose levels of FF MDI was compared with those after chronic administration of FF DPI. PK samples were collected at pre-dose, at 2, 6 and 20 min, and at 1, 2, 4, 8, 10 and 12 h post-dose. PK analyses were performed by Pharsight Inc using a validated version of WinNonlin® Enterprise (Version 5.2).
Safety evaluations
In addition to monitoring adverse events (AEs) and serious AEs (SAEs), the following safety evaluations were performed: 12-lead electrocardiogram (ECG), vital signs, physical examination, clinical laboratory tests, and evaluation for symptoms of AEs of interest including dry mouth, tremor and paradoxical bronchospasm.
Statistical analysis
Data processing, data screening, descriptive reporting and analysis of the efficacy and safety data were performed using SAS Version 9.2 (SAS Institute, Inc., Cary, NC). The PK data were analyzed using WinNonLin Version 5.2 (Pharsight Corp., USA). PK graphs were prepared using SigmaPlot for Windows Version 9.01 (Systatsoftware, Inc., San Jose, CA). Power calculations were performed using R software. Further details of statistical methods and analysis are detailed in the Additional file 1.
The primary efficacy analysis was based on a modified intent-to treat (mITT) population, defined as patients who completed at least two treatment periods up to at least 2 h post-dose on Day 7 (with no more than one missing data point from the 15-min to the 2-h post-dose timepoint, inclusive); patients whose baseline FEV1 at Visits 4, 6 and 8 was not within 15% of baseline FEV1 at Visit 2 (reproducibility criteria) were also excluded from the mITT population. A separate population, PK-mITT, was defined for use in the PK analyses.
Data from Part A and Part B were combined for analysis using a linear mixed-effects model of the primary endpoint FEV1 AUC0–12 (baseline FEV1 was included as a covariate). The AUC was calculated using trapezoidal integration on the available timepoints. Superiority testing was performed using a two-sided 0.05 level of significance; non-inferiority testing was performed using a 0.025 level of significance based on a one-sided confidence interval (CI). The pre-defined non-inferiority margin for continuous spirometry variables was 100 mL, selected on the basis that it is the minimally clinically significant difference, defined as the change in FEV1 that can be perceived by the patient [12]. As such, non-inferiority was only confirmed for a treatment group if the relevant bound of the two-sided 95% CI for the difference was above −100 mL or below 100 mL. Mean changes from baseline in FEV1 were provided with 95% CI to support any conclusions of non-inferiority.
Sample size calculations
Calculations to determine adequate sample size were based on the primary endpoint, FEV1 AUC0–12. For superiority testing of spirometry parameters, a difference of 100 mL was the pre-defined minimally clinically significant difference to be observed in FEV1 AUC0–12. Combined data from Part A and Part B gave the study the power of approximately 82–95% to detect the minimally clinically significant difference in FEV1 AUC0–12 between the treatment comparisons of interest.
Role of the funding source
The funder of the study was involved in study design, data collection, data analysis, data interpretation and writing of the report. All authors had full access to all the data in the study and the corresponding author had the final responsibility for the decision to submit for publication. No restrictions were placed on authors regarding the statements made in the manuscript.
Results
Patient disposition
A total of 169 patients were screened and 122 were randomized between 24 March 2010 and 28 October 2010 to receive treatment at sites in Australia, New Zealand and the USA. Following review of data from four sentinel patients, 118 patients were randomized: 68 patients into Part A and 50 patients into Part B (Fig. 1). All 118 patients received at least one dose of study drug and were included in the ITT population and 104 patients (88.1%) were included in the mITT population. Major reasons for exclusion from the mITT population were: (i) patient did not complete at least two treatment periods up to at least 2 h post-dose on Day 7 (Part A, 10.3% of patients and Part B, 14.0% of patients), and (ii) patient failed reproducibility criteria or had missing data on Day 7 (maximum of 3.4% patients in any one period of the study). There were 82 patients (69.5%) included in the per-protocol population (patients who completed all four treatment periods). The majority of patients (80.5 to 96.2% across treatment groups) received 80 to 100% of their assigned treatment regimen.
Fig. 1.
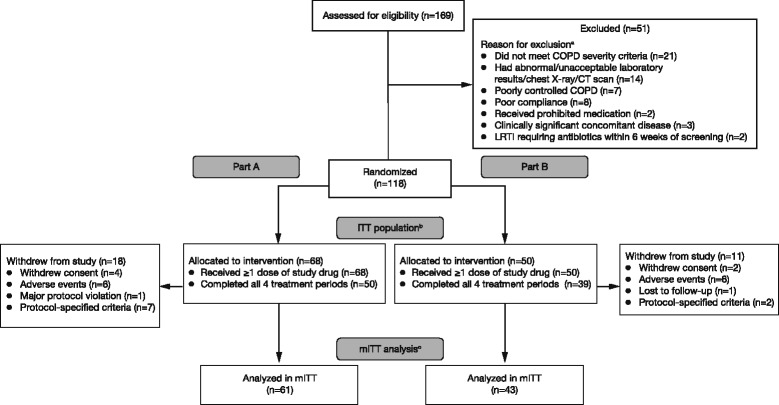
Patient disposition. In Part A, patients were randomized to receive any of the eight treatments in each of the four periods of the study in an incomplete block crossover design. In Part B, patients were randomized to receive all three formoterol doses and placebo in each of the four periods of the study in a full crossover design. aFive patients met multiple criteria for exclusion from randomization (not meeting inclusion criteria and/or meeting exclusion criteria). bPatients randomized to treatment, who received at least one dose of study drug. cPatients who completed at least two treatment periods with at least 2 h of post-dose data on Day 7, with no more than one missing data-point from 15 min to 2 h post-dose, inclusive. COPD, chronic obstructive pulmonary disease; CT, computed tomography; ITT, intent-to-treat; LRTI, lower respiratory tract infection; mITT, modified ITT
Baseline characteristics
Patients’ baseline and demographic characteristics are shown in Table 1 (mITT population). Briefly, the mean (± standard deviation [SD]) duration of patients’ history of COPD was 7.7 (±5.9) years; the mean post-bronchodilator FEV1 was 50.8 (±12.7) % of predicted; and the mean FEV1 bronchodilator reversibility was 16.9 (±14.9) %. Overall, 52.9% (55/104) of patients had moderate COPD, 44.2% (46/104) had severe COPD and 2.9% (3/104) had very severe COPD.
Table 1.
Baseline demographics (mITT population)
| Parameter | GFF MDI | GP MDI 36 μg (N = 41) | Open-label tiotropium 18 μg (N = 58) | FF MDI | Placebo MDI (N = 52) | Open-label FFa DPI 12 μg (N = 55) | ||
|---|---|---|---|---|---|---|---|---|
| 72/9.6 μg (N = 41) | 36/9.6 μg (N = 43) | 9.6 μg (N = 64) | 7.2 μg (N = 64) | |||||
| Age, years | ||||||||
| Mean (SD) | 62.4 (9.4) | 63.3 (8.3) | 66.3 (6.1) | 64.1 (7.9) | 63.4 (8.9) | 63.6 (8.9) | 62.8 (9.6) | 60.6 (9.0) |
| Gender, n (%) | ||||||||
| Male | 25 (61.0) | 24 (55.8) | 23 (56.1) | 34 (58.6) | 34 (53.1) | 36 (56.3) | 29 (55.8) | 34 (61.8) |
| Race, n (%) | ||||||||
| Black/African | 0 | 0 | 0 | 0 | 3 (4.7) | 3 (4.7) | 3 (5.8) | 3 (5.5) |
| White | 39 (95.1) | 42 (97.7) | 41 (100) | 57 (98.3) | 61 (95.3) | 61 (95.3) | 48 (92.3) | 52 (94.5) |
| Australia/New Zealand (indigenous) | 2 (4.9) | 1 (2.3) | 0 | 1 (1.7) | 0 | 0 | 1 (1.9) | 0 |
| Smoking status, n (%) | ||||||||
| Current | 16 (39.0) | 18 (41.9) | 15 (36.6) | 24 (41.4) | 29 (45.3) | 28 (43.8) | 24 (46.2) | 25 (45.5) |
| Former | 25 (61.0) | 25 (58.1) | 26 (63.4) | 34 (58.6) | 35 (54.7) | 36 (56.3) | 28 (53.8) | 30 (54.5) |
| Duration of COPD, years | ||||||||
| Mean (SD) | 7.6 (7.3)b | 6.2 (5.4)c | 7.8 (6.2)b | 7.4 (6.7)d | 8.6 (6.1)e | 7.7 (4.4)f | 8.3 (5.2)g | 7.3 (4.3)h |
| Mean % predicted FEV1 (SD) | ||||||||
| Pre-bronchodilator | 44.1 (13.9)b | 46.8 (14.1)c | 45.8 (13.5)b | 44.9 (13.9)d | 44.7 (12.6)e | 43.9 (12.0)f | 43.7 (11.6)g | 44.0 (13.3)h |
| Post-bronchodilator | 50.6 (13.0)b | 53.0 (13.1)c | 51.5 (13.3)b | 51.3 (13.4)d | 51.4 (12.5)e | 50.2 (12.6)f | 51.1 (12.4)g | 50.9 (12.9)h |
| Mean FEV1, L (SD) | ||||||||
| Pre-bronchodilator | 1.33 (0.48)b | 1.38 (0.47)c | 1.30 (0.41)b | 1.33 (0.47)d | 1.29 (0.43)e | 1.28 (0.40)f | 1.30 (0.41)g | 1.35 (0.46)h |
| Post-bronchodilator | 1.52 (0.47)b | 1.56 (0.47)c | 1.46 (0.40)b | 1.51 (0.46)d | 1.49 (0.46)e | 1.47 (0.43)f | 1.52 (0.47)g | 1.56 (0.48)h |
| FEV1 bronchodilator reversibility, L (SD)i | ||||||||
| Mean (SD) | 17.8 (16.3)b | 16.3 (17.2)c | 14.2 (14.5)b | 17.1 (16.2)d | 17.5 (14.7)e | 15.9 (12.7)f | 18.6 (12.9)g | 18.5 (15.5)h |
aForadil® Aerolizer®; b n = 38; c n = 39; d n = 56; e n = 58; f n = 63; g n = 45; h n = 54; ipercentage change from pre-albuterol at 30 min post-albuterol for FEV1
% = 100 × n/N, where n = number of patients in category and N = number of patients in the group
Duration of COPD = (date of first dose of study treatment in the study – COPD onset date)/365.25
Data from four sentinel patients were included in the mITT population in the analyses of demographic and baseline characteristics only
COPD chronic obstructive pulmonary disease, DPI dry powder inhaler, FEV 1 forced expiratory volume in 1 s, FF formoterol fumarate, GFF glycopyrrolate/formoterol fumarate, GP glycopyrrolate, MDI metered dose inhaler, mITT modified intent-to-treat, SD standard deviation
FEV1 AUC0–12 on Day 7
Figure 2a shows the least squares mean (LSM) change from baseline in FEV1 over 12 h on Day 7. All active treatments were superior to placebo MDI for FEV1 AUC0–12 on Day 7 (p < 0.0001) (Figure 2b). GFF MDI 36/9.6 μg was non-inferior to GFF MDI 72/9.6 μg in FEV1 AUC0–12 on Day 7 since the upper bound of the CI was <100 mL (LSM difference between treatments = 0.008 L; 95% CI = −0.039, 0.054 L). GFF MDI 72/9.6 μg and GFF MDI 36/9.6 μg each demonstrated superior bronchodilation of 101 to 124 mL compared with their individual component MDIs, GP MDI 36 μg, FF MDI 9.6 μg and FF MDI 7.2 μg, as well as superior bronchodilation compared with the open-label comparators FF DPI and tiotropium DPI (p ≤ 0.0002) for FEV1 AUC0–12 on Day 7 (Table 2). GP MDI 36 μg, demonstrated non-inferiority to the LAMA comparator open-label tiotropium DPI for FEV1 AUC0–12 on Day 7 (LSM difference between treatments = −0.006 L; 95% CI = −0.049, 0.038 L). Both doses of FF MDI (7.2 and 9.6 μg) demonstrated non-inferiority to the open-label comparator FF DPI (Table 2).
Fig. 2.

FEV1 AUC0–12 on Day 7 efficacy endpoint. a LSM change (95% CI) in FEV1 over 0–12 h on Day 7 by treatment; b LSM (95% CI) FEV1 AUC0–12 difference from placebo on Day 7 by treatment (mITT population). aForadil® Aerolizer® . bLSM allows for any imbalances in baseline covariates that relate to responses to be adjusted for in order to avoid bias in treatment effect estimates. AUC0–12, area under the curve from 0 to 12 h post-dose; DPI, dry powder inhaler; FEV1, forced expiratory volume in 1 s; FF, formoterol fumarate; GFF, glycopyrrolate/formoterol fumarate; GP, glycopyrrolate; LSM, least squares mean; MDI, metered dose inhaler; mITT, modified intent-to-treat
Table 2.
FEV1 AUC0–12 at Day 7: GFF MDI 72/9.6 μg and 36/9.6 μg comparisons (mITT population)
| LSM treatment differences for GFF MDI in FEV1 AUC0–12 at Day 7 | ||||||||
|---|---|---|---|---|---|---|---|---|
| GFF MDI | GP MDI 36 μg | Open-label tiotropium 18 μg | FF MDI | Placebo MDI | Open-label FFa DPI 12 μg | |||
| Comparator | 72/9.6 μg | 36/9.6 μg | 9.6 μg | 7.2 μg | ||||
| GFF MDI 72/9.6 μg | ||||||||
| LSMb difference (SE), L | NA | 0.008 (0.0236) | 0.109 (0.0250)† | 0.103 (0.0216)† | 0.116 (0.0245)† | 0.124 (0.0237)† | 0.298 (0.0261)† | 0.101 (0.0241)† |
| 95% CI | −0.039, 0.054 | 0.059, 0.158c | 0.060, 0.145 | 0.068, 0.165 | 0.078, 0.171 | 0.247, 0.349 | 0.053, 0.148 | |
| GFF MDI 36/9.6 μg | ||||||||
| LSMb difference (SE), L | See above | NA | 0.101 (0.0245)† | 0.095 (0.0213)† | 0.109 (0.0242)† | 0.116 (0.0236)† | 0.290 (0.0261)† | 0.093 (0.0241)*** |
| 95% CI | 0.053, 0.149 | 0.053, 0.137 | 0.061, 0.156 | 0.070, 0.163 | 0.239, 0.342 | 0.045, 0.140 | ||
*** p < 0.001; † p < 0.0001
aForadil® Aerolizer®; bLSM allows for any imbalances in baseline covariates that relate to responses to be adjusted for in order to avoid bias in treatment effect estimates; cnon-inferiority comparison
CI, confidence interval; DPI, dry powder inhaler; FEV1 AUC0–12, forced expiratory volume in 1 s area under the curve from 0 to 12 h post-dose; FF, formoterol fumarate; GFF, glycopyrrolate/formoterol fumarate; GP, glycopyrrolate; LSM, least squares mean; MDI, metered dose inhaler; mITT, modified intent-to-treat; NA, not available; SE, standard error
Secondary endpoints
All active treatments were superior to placebo MDI for the lung function secondary endpoints (peak change from baseline in FEV1; time to onset of action on Day 1 [≥10% improvement in FEV1 relative to baseline]; peak change from baseline FEV1 and change from baseline in morning pre-dose trough FEV1 and 12-h post-dose trough FEV1; peak change from baseline in IC, change from baseline in morning pre-dose trough IC and 12-h post-dose trough IC; mean daily peak flow readings) on Days 1 and 7 (p ≤ 0.0056). The percentage of patients achieving ≥12% improvement in FEV1 was 86.8% (GFF MDI 72/9.6 μg), 87.2% (GFF MDI 36/9.6 μg), 73.7% (GP MDI 36 μg) 66.1% (open-label tiotropium DPI 18 μg), 84.5% (FF MDI 9.6 μg), 82.5% (FF MDI 7.2 μg), 40.0% (placebo MDI) and 85.2% (FF DPI 12 μg). Inferential comparisons of the percentage of patients achieving ≥12% improvement in FEV1 were not possible for several comparisons due to the limited number of patients (≤5) receiving each pair of treatments. However, numerical benefits were observed for all active treatments compared to placebo. GFF MDI 36/9.6 μg demonstrated non-inferiority to GFF MDI 72/9.6 μg (Table 3; Additional file 1). At Day 7, GFF MDI 72/9.6 μg and GFF MDI 36/9.6 μg demonstrated superiority to each monocomponent MDI and to open-label FF DPI and tiotropium for morning peak FEV1, peak change in FEV1, and morning pre-dose trough IC (Table 3; Additional file 1). FF MDI 7.2 μg and FF MDI 9.6 μg were both non-inferior to FF DPI, and FF MDI 7.2 μg was non-inferior to FF MDI 9.6 μg in secondary endpoints at Day 7 (Additional file 1).
Table 3.
Secondary efficacy endpoints: Days 1 and 7 – GFF MDI 72/9.6 μg and GFF MDI 36/9.6 μg comparisons (mITT population)
| Comparator | Treatment differences for GFF MDI comparisons | |||||
|---|---|---|---|---|---|---|
| GP MDI 36 μg | Open-label tiotropium 18 μg | FF MDI | Placebo MDI | Open-label FFa DPI 12 μg | ||
| 9.6 μg | 7.2 μg | |||||
| DAY 7 | ||||||
| Change from baseline in morning pre-dose trough FEV1, L | ||||||
| GFF MDI 72/9.6 μg | ||||||
| LSMb difference (SE) | 0.0960 (0.0280)*** | 0.096 (0.0247)† | 0.129 (0.0278)† | 0.120 (0.0271)† | 0.234 (0.0302)† | 0.091 (0.0277)** |
| GFF MDI 36/9.6 μg | ||||||
| LSMb difference (SE) | 0.073 (0.0273)** | 0.073 (0.0245)** | 0.106 (0.0274)† | 0.097 (0.0270)*** | 0.211 (0.0300)† | 0.068 (0.0275)* |
| Peak change from baseline in FEV1, L | ||||||
| GFF MDI 72/9.6 μg | ||||||
| LSMb difference (SE) | 0.125 (0.0282)† | 0.140 (0.0248)† | 0.101 (0.0279)*** | 0.108 (0.0271)† | 0.342 (0.0300)† | 0.082 (0.0278)** |
| GFF MDI 36/9.6 μg | ||||||
| LSMb difference (SE) | 0.127 (0.0273)† | 0.141 (0.0245)† | 0.103 (0.0273)*** | 0.110 (0.0268)† | 0.344 (0.0298)† | 0.083 (0.0276)** |
| Change from baseline in morning pre-dose trough IC, L | ||||||
| GFF MDI 72/9.6 μg | ||||||
| LSMb difference (SE) | 0.083 (0.0445) | 0.090 (0.0399)* | 0.156 (0.0452)*** | 0.110 (0.0436)* | 0.255 (0.0483)† | 0.096 (0.0447)* |
| GFF MDI 36/9.6 μg | ||||||
| LSMb difference (SE) | 0.098 (0.0445)* | 0.105 (0.0387)** | 0.172 (0.0433)† | 0.126 (0.0428)** | 0.271 (0.0471)† | 0.111 (0.0434)* |
| Peak change from baseline in IC, L | ||||||
| GFF MDI 72/9.6 μg | ||||||
| LSMb difference (SE) | 0.078 (0.0532) | 0.095 (0.0470)* | 0.050 (0.0529) | 0.033 (0.0513) | 0.265 (0.0572)† | 0.016 (0.0527) |
| GFF MDI 36/9.6 μg | ||||||
| LSMb difference (SE) | 0.107 (0.0513)* | 0.124 (0.0461)** | 0.078 (0.0513) | 0.062 (0.0503) | 0.293 (0.0559)† | 0.045 (0.0518) |
| DAY 1 | ||||||
| Peak change from baseline in FEV1, L | ||||||
| GFF MDI 72/9.6 μg | ||||||
| LSMb difference (SE) | 0.081 (0.0309)** | 0.104 (0.0268)† | 0.062 (0.0307)* | 0.060 (0.0297)* | 0.265 (0.0328)† | 0.072 (0.0306)* |
| GFF MDI 36/9.6 μg | ||||||
| LSMb difference (SE) | 0.068 (0.300)* | 0.090 (0.0266)*** | 0.048 (0.0300) | 0.046 (0.0293) | 0.251 (0.0326)† | 0.058 (0.0303) |
| Peak change from baseline in IC, L | ||||||
| GFF MDI 72/9.6 μg | ||||||
| LSMb difference (SE) | 0.065 (0.0567) | 0.149 (0.0493)** | 0.134 (0.0564)* | 0.144 (0.0547)** | 0.412 (0.0607)† | 0.121 (0.0561)* |
| GFF MDI 36/9.6 μg | ||||||
| LSMb difference (SE) | −0.019 (0.0555) | 0.065 (0.0491) | 0.050 (0.0554) | 0.060 (0.0542) | 0.328 (0.0602)† | 0.037 (0.0557) |
| Time to onset of action, hazard ratioc | ||||||
| GFF MDI 72/9.6 μg | ||||||
| HR | 1.399* | 1.754*** | 0.980 | 1.150 | 3.475† | 0.971 |
| 95% CI | 1.038, 1.884 | 1.300, 2.367 | 0.746, 1.289 | 0.904, 1.465 | 2.095, 5.765 | 0.713, 1.321 |
| GFF MDI 36/9.6 μg | ||||||
| HR | 1.323 | 1.695*** | 0.888 | 1.062 | 3.358† | 0.878 |
| 95% CI | 0.936, 1.870 | 1.275, 2.253 | 0.671, 1.175 | 0.0806, 1.400 | 2.091, 5.391 | 0.660, 1.169 |
* p < 0.05; ** p < 0.01; *** p < 0.001; † p ≤ 0.0001
aForadil® Aerolizer®; bLSM allows for any imbalances in baseline covariates that relate to responses to be adjusted for in order to avoid bias in treatment effect estimates; ca hazard ratio of 1.399 signifies a 39.9% higher probability of onset of action at any time point post-dose
CI confidence interval, DPI dry powder inhaler, FEV 1 forced expiratory volume in 1 s, FF formoterol fumarate, GFF glycopyrrolate/formoterol fumarate, GP glycopyrrolate, HR hazard ratio, IC inspiratory capacity, LSM least squares mean, MDI metered dose inhaler, mITT modified intent-to-treat, SE standard error
Exploratory endpoint
All active treatments were superior to placebo (p < 0.0001) for change from baseline FVC (calculated as AUC0–12) on Day 7 (Additional file 1: Figure S2). Treatment comparisons are shown in the Additional file 1.
Safety and tolerability
Most AEs were of mild (32.0%) or moderate (29.5%) intensity. Treatment-emergent AEs (TEAEs) reported in more than two patients receiving treatment are displayed in Table 4. The most commonly reported TEAEs were: dry mouth, headache, tremor, cough and dysphonia (Table 4). Dry mouth was reported more frequently by patients receiving GP MDI, GFF MDI, and open-label tiotropium compared to the other groups, while headache and tremor were reported more frequently by patients receiving GFF MDI. No patient in any treatment period reported paradoxical bronchospasm. The incidence of TEAEs was similar for the two doses of GFF MDI (31.7% vs 27.9%).
Table 4.
Summary of adverse events (safety population)
| GFF MDI | GP MDI 36 μg (N = 41) | Open-label tiotropium 18 μg (N = 58) | FF MDI | Placebo MDI (N = 52) | Open-label FFa DPI 12 μg (N = 55) | |||
|---|---|---|---|---|---|---|---|---|
| 72/9.6 μg (N = 41) | 36/9.6 μg (N = 43) | 9.6 μg (N = 64) | 7.2 μg (N = 64) | |||||
| Patients with at least one AE, n (%) | 17 (41.5) | 18 (41.9) | 11 (26.8) | 22 (37.9) | 24 (37.5) | 16 (25.0) | 9 (17.3) | 17 (30.9) |
| Patients with AE related to study treatment, n (%) | 13 (31.7) | 12 (27.9) | 7 (17.1) | 7 (12.1) | 7 (10.9) | 4 (6.3) | 2 (3.8) | 7 (12.7) |
| Patients with SAE, n (%) | 0 | 1 (2.3) | 0 | 2 (3.4) | 1 (1.6) | 2 (3.1) | 0 | 0 |
| Patients with SAE related to study treatment, n (%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Patients with AE leading to early withdrawal, n (%) | 1 (2.4) | 0 | 1 (2.4) | 1 (1.7) | 4 (6.3) | 3 (4.7) | 1 (1.9) | 0 |
| Patients with SAE leading to early withdrawal, n (%) | 0 | 0 | 0 | 0 | 0 | 2 (3.1) | 0 | 0 |
| TEAEs reported in ≥2 patients in any treatment group | ||||||||
| Dry mouth | 8 (19.5) | 3 (7.0) | 5 (12.2) | 4 (6.9) | 3 (4.7) | 2 (3.1) | 1 (1.9) | 2 (3.6) |
| Headache | 3 (7.3) | 4 (9.3) | 1 (2.4) | 1 (1.7) | 1 (1.6) | 0 | 1 (1.9) | 2 (3.6) |
| Tremor | 1 (2.4) | 5 (11.6) | 0 | 0 | 0 | 0 | 0 | 0 |
| Cough | 0 | 2 (4.7) | 0 | 1 (1.7) | 0 | 0 | 0 | 0 |
| Dysphonia | 1 (2.4) | 2 (4.7) | 0 | 0 | 0 | 0 | 0 | 0 |
% = 100 × n/N: n = no. of patients in the preferred term category for treatment group
aForadil® Aerolizer®
AE adverse event, DPI dry powder inhaler, FF formoterol fumarate, GFF glycopyrrolate/formoterol fumarate, GP glycopyrrolate, MDI metered dose inhaler, SAE serious adverse event, TEAE treatment-emergent adverse event
Six SAEs were reported in five patients, none of which was related to study drug (one patient in the GFF MDI 36/9.6 μg group [ruptured appendix]; two patients in the open-label tiotropium group [inhaled foreign body; abdominal aortic aneurysm]; one patient in the FF MDI 9.6 μg group [gastritis]; and two patients in the FF MDI 7.2 μg group [COPD exacerbation; atypical chest pain leading to early withdrawal]). No deaths were reported in the study.
There were no notable changes in hematology or chemistry laboratory values and no clinically significant abnormalities in vital signs, ECG, or physical examination.
Pharmacokinetics
Following chronic administration of GFF MDI 36/9.6 μg, the geometric LSM of glycopyrrolate was approximately 9% (AUC0–12) and 14% (maximum observed plasma concentration [Cmax]) lower than those observed following GP MDI 36 μg (Fig. 3a). In addition, the geometric LSM for formoterol was approximately 7% (AUC0–12) and 14% (Cmax) lower than those observed when FF MDI 9.6 μg was administered alone (Fig. 3b).
Fig. 3.
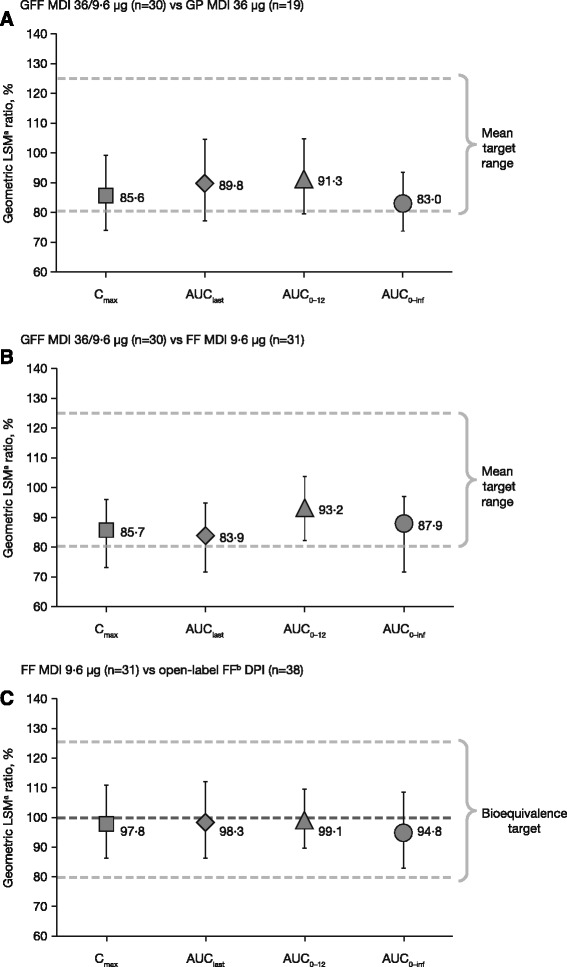
Ratio of geometric LSMs and 90% CIs. a GFF MDI 36/9.6 μg versus GP MDI 36 μg (b) GFF MDI 36/9.6 μg versus FF MDI 9.6 μg (c) FF MDI 9.6 μg versus FF DPI (PK-mITT population). aLSM allows for any imbalances in baseline covariates that relate to responses to be adjusted for in order to avoid bias in treatment effect estimates. bForadil® Aerolizer® . AUC0–inf, area under the curve from time 0 to infinity; AUC0–12, area under the curve from 0 to 12 h post-dose; CI, confidence interval; Cmax, maximum observed plasma concentration; DPI, dry powder inhaler; FF, formoterol fumarate; GFF, glycopyrrolate/formoterol fumarate; GP, glycopyrrolate; LSM, least squares mean; MDI, metered dose inhaler; PK-mITT, pharmacokinetic modified intent-to-treat
Results from an analysis of variance (ANOVA) of the dose-normalized exposure parameters of formoterol between the FF MDI 9.6 μg and the FF DPI showed that 90% CIs for the ratios of LSM for the exposure parameters AUC0–12 and Cmax were within the 80–125% interval, demonstrating that monocomponent FF MDI 9.6 μg was bioequivalent to the FF DPI formulation (equivalent to an FF 10 μg dose), with dose normalization. Furthermore, ANOVA results based on non-dose-normalized PK parameters also demonstrated equivalence between FF MDI and FF DPI (Fig. 3c).
Discussion
This 7-day Phase IIb study of GFF MDI 72/9.6 μg and 36/9.6 μg BID is the first study to investigate the FDC GFF MDI formulated using Co-Suspension delivery technology in patients with COPD. Previous studies have investigated the dose-response of the individual components delivered via MDI using Co-Suspension delivery technology [9, 13]. GFF MDI 72/9.6 μg and 36/9.6 μg BID led to statistically significant and clinically relevant improvement in the primary endpoint FEV1 AUC0–12 at Day 7 compared with the monocomponent MDIs and placebo MDI in patients with moderate-to-very severe COPD and were well tolerated (p ≤ 0.0001).
It has become established that the combination of a LAMA and LABA provides benefits in lung function in patients with COPD over LAMA or LABA monotherapy in those patients who are not adequately controlled by a single long-acting bronchodilator. Within this therapeutic approach, LAMA/LABA FDCs glycopyrrolate/indacaterol, tiotropium/olodaterol, aclidinium/formoterol, and umeclidinium/vilanterol have previously demonstrated lung function benefits over monocomponents [14–19]. This study was part of a Phase IIb program including dose-ranging studies of GFF MDI and monocomponents to define the optimal doses of the GFF MDI FDC to take forward to Phase III studies, and utilized non-inferiority testing, based on the targeted 100 mL minimally clinically significant difference, as a well-defined and reproducible treatment effect for trough FEV1 to define therapeutic effect [12]. In this study, non-inferiority was confirmed between GFF MDI 72/9.6 μg and 36/9.6 μg, incorporating GP doses at the higher end of the dose range, and both doses showed statistically significantly greater FEV1 AUC0–12 at Day 7 versus open-label tiotropium DPI and FF DPI. Notably, GFF MDI demonstrated superiority to placebo and statistically significant (GFF MDI 72/9.6 μg) and numerically greater (GFF MDI 36/9.6 μg) improvements in IC compared with open-label tiotropium DPI. In patients with COPD, changes in IC reflect changes in hyperinflation and have shown a higher correlation to patient-focused outcomes, such as dyspnea with exercise, than other standard spirometric measurements [20]. Additional information was gained concerning the monocomponents, whereby the doses of the two monocomponents, GP and FF, demonstrated non-inferiority to the open-label active comparators such that GP MDI 36 μg BID was non-inferior to open-label tiotropium DPI and both doses of FF MDI demonstrated non-inferiority to open-label FF delivered via DPI.
The PK component of the study characterized the systemic exposure of glycopyrrolate and formoterol delivered as an FDC compared with individual components delivered using Co-Suspension delivery technology. The findings support the absence of a significant drug-drug interaction for formoterol and glycopyrrolate following administration of GFF MDI relative to the individual MDI formulations. It was also shown that formoterol exposure (AUC0–12) increases in a dose-proportional manner when delivered via the MDI and that FF MDI 9.6 μg delivered by MDI using Co-Suspension delivery technology was bioequivalent to FF delivered using DPI, which taken together endorse the dose of FF MDI 9.6 μg in the FDC.
All treatments were generally well tolerated in this study. The tolerability and safety profiles observed for GFF MDI 36/9.6 μg and 72/9.6 μg BID were consistent with the patient population and drug classes [21]. The most commonly reported TEAEs (more than two patients in any treatment group) were dry mouth, headache, tremor, cough and dysphonia in descending order of incidence.
Whilst the sample size of this first study of GFF delivered by MDI using novel Co-Suspension delivery technology was calculated to provide reasonable information to characterize the response, we recognize that the study was limited by a relatively small population and enrolled a lower than anticipated number of patients with very severe COPD. In addition, the study was only conducted over 7 days. However, information from this study guided study design and sample size for subsequent studies.
GFF MDI will potentially widen treatment options for patients with COPD by providing LAMA/LABA therapy in an MDI when other LAMA/LABA FDCs are available as DPI and soft-mist inhaler devices. Unlike other devices, MDIs are not breath-actuated, consequently, MDIs may be able to offer patients with quite severe airflow limitation, who may be unable to breathe deeply enough to release the medication from a DPI, a more practical and reliable method of delivery of their medication. The PKs of GFF MDI support twice-daily administration and the improvements in lung function demonstrated here after BID dosing with GFF MDI as maintenance dual-bronchodilator therapy may provide benefits over QD dosing, preventing excessive worsening of symptoms during the night or towards the morning in patients with this pattern of symptoms [7]. The results of a patient evaluation survey conducted by Partridge et al., and similarly in the ASSESS study, show that patients with COPD begin to experience worsening symptoms in the evening through the night-time, with the worst symptoms, affecting activity and productivity, occurring in the morning [22, 23]. This is also an area for future investigation of the effects of GFF MDI on COPD symptom burden.
Conclusions
Co-Suspension delivery technology allows formulation of FDCs of different drug classes, at different concentrations, in a single MDI. GFF MDI 72/9.6 μg and 36/9.6 μg BID were associated with a similarly greater magnitude of effect on FEV1 AUC0–12 at Day 7 compared with the monocomponent MDIs and placebo MDI in patients with moderate-to-very severe COPD, which is likely to be the maximal therapeutic effect. Additional studies are required to establish the optimal doses of GP and FF for combination in the FDC GFF MDI.
Acknowledgements
The authors would like to thank all of the patients and their families, the team of investigators, research nurses and operations staff involved in this study. The authors would also like to thank Catherine Stanton, of Complete Medical Communications, who provided medical writing support under the direction of the authors, funded by AstraZeneca. The authors would also like to thank Mervyn Thomas for his work in the statistical design and planning of this study and Everest Clinical Research, who conducted the statistical analyses of the study data.
Funding
Pearl Therapeutics Inc., a member of the AstraZeneca Group.
Availability of data and material
All relevant data generated or analyzed during this study are included in this published article (and its additional information files).
Authors’ contributions
KFR, FJM, JFD, LMF, GTF, SD, CR, CO, PD, ES, TF, MG, SSt, SSp made substantial contributions to the conception or design of the work reported. EMK, DQ, CF, SSp participated in the acquisition of reported data. SD, CR, CO, PD participated in the analysis of reported data. KFR, FJM, JFD, LMF, GTF, EMK, DQ, CF, SSt, SSp, SD, CR, CO, PD participated in the interpretation of reported data. All authors contributed to the writing of the report and participated in the review and interpretation of the data. All authors read and approved the final report before submission.
Authors’ information
[as per title page].
Co-Suspension™ is a trademark of the AstraZeneca group of companies.
List of principal investigators
JoAnne Marjason, Robyn Huttenmeister, Maria Quartararo, Peter Clyne, Paul Seale, Philip Thompson, Philip Bardin, Dean Quinn, Andrew Veale, Margaret Wilsher, Graham Mills, Michael Chia, Charles M. Fogarty, Selwyn Spangenthal, Edward M. Kerwin, Leonard Dunn.
Competing interests
CR is an employee of Pearl Therapeutics, Inc., a member of the AstraZeneca Group. LMF has received grants, personal fees and non-financial support from Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Laboratori Guidotti, Merck Sharp & Dohme, Menarini, Novartis and Takeda. He has received personal fees and non-financial support from Boston Scientific, Mundipharma and Pearl Therapeutics Inc. He has received personal fees from Bayer, Kyorin and Zambon. He has received grants from Biofutura Italia, Dompè, Malesci, Pfizer and Vree Health Italia. EMK has served on advisory boards, speaker panels, or received travel reimbursement from Amphastar, Boehringer Ingelheim, Forest, Mylan, Novartis, Pearl Therapeutics Inc. (a member of the AstraZeneca Group), Sunovion, Teva and Theravance. He has conducted multicenter clinical trials for ~40 pharmaceutical companies. CF is a consultant and investigator for Pearl Therapeutics, Inc. SSp has no potential conflicts of interest to disclose. KFR has received grants from Boehringer Ingelheim, the German Federal Ministry of Education and Research (BMBF) and Novartis. He has received personal fees from AstraZeneca, Boehringer Ingelheim, Chiesi, Intermune, Novartis and Takeda. GTF has received grants and personal fees from AstraZeneca, Boehringer Ingelheim Novartis, Pearl Therapeutics Inc., Sunovian and Theravance. He has received grants from Forest. He has received personal fees GlaxoSmithKline, Meda, Mylan and Verona. FJM has received personal fees from Adept, Afferent, Amgen, AstraZeneca, Axon, Axon Communication, Boehringer Ingelheim, Clarion, ConCert, Forest, Genentech, GlaxoSmithKline, Ikaria/Bellerophon, Informa, Janssen, Kadmon, Lucid, Methodist Hospital, Novartis, Nycomed/Takeda, Pearl Therapeutics Inc., Pfizer, Prime, Roche, Sunovion, Theravance, Unity Biotechnology, Veracyte and WebMD. He has received non-financial support from Biogen/Stromedix, Boehringer Ingelheim, Centocor and Gilead and grants from the National Institutes of Health. He has received personal fees for delivering CME programs for Academic CME, American Thoracic Society, Annenberg, California Society for Allergy and Immunology, CME Incite, Falco, Haymarket Communications, Integritas, InThought, Miller Medical, National Association for Continuing Education, Paradigm, Peer Voice, Potomac, UpToDate and Western Society of Allergy and Immunology. He has received royalty fees from Informa. He has spoken on behalf of AstraZeneca and Nycomed/Takeda. He is currently a member of the GOLD Scientific Committee. JFD has received personal fees from AstraZeneca, GlaxoSmithKline, Novartis, Sunovion and Teva. PD, ES, CO, SSt, TF, MG and SD are employees of Pearl Therapeutics, Inc., a member of the AstraZeneca Group.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study was conducted in accordance with International Conference on Harmonization guidelines, the Declaration of Helsinki, and the US Code of Federal Regulations. The protocol, its amendments and patient informed consent form were approved by an Independent Ethics Committee or Institutional Review Board.
Abbreviations
- AEs
Adverse events
- AUC
Area under curve
- BID
Twice daily
- CI
Confidence interval
- COPD
Chronic obstructive pulmonary disease
- CT
Computed tomography
- DPI
Dry powder inhaler
- ECG
Electrocardiogram
- FDC
Fixed-dose combination
- FEV1 AUC0–12
Forced expiratory volume in 1 s area under the curve from 0 to 12 h
- FEV1
Forced expiratory volume in 1 s
- FF
Formoterol fumarate
- FVC
Forced vital capacity
- GFF MDI
Glycopyrrolate/formoterol fumarate MDI
- GP
Glycopyrrolate
- HFA
Hydrofluoroalkane
- HR
Hazard ratio
- IC
Inspiratory capacity
- ITT
Intent-to-treat
- LABA
Long-acting beta agonist
- LAMA
Long-acting muscarinic antagonist
- LRTI
Lower respiratory tract infection
- LSM
Least squares mean
- MDI
Metered dose inhaler
- mITT
Modified intent-to treat
- NA
Not available
- PK
Pharmacokinetic
- QD
Once-daily
- SAEs
Serious adverse events
- SD
Standard deviation
- SE
Standard error
- TEAEs
Treatment-emergent AEs
Additional file
Study design; Additional Fig. 1. Study design schematic; Additional Fig. 2. Mean change from baseline in PEFR over time by treatment on Day 7 (mITT population); Additional Fig. 3. Mean change from baseline FVC over time by treatment on Day 7 (mITT population); Additional Table 1. Secondary efficacy endpoints: Days 1 and 7 – FF 9.6 μg and FF 7.2 μg comparisons (mITT population). (DOCX 466 kb)
References
- 1.Cazzola M, Page CP, Calzetta L, Matera MG. Pharmacology and therapeutics of bronchodilators. Pharmacol Rev. 2012;64:450–504. doi: 10.1124/pr.111.004580. [DOI] [PubMed] [Google Scholar]
- 2.Cooper CB, Barjaktarevic I. A new algorithm for the management of COPD. Lancet Respir Med. 2015;3:266–268. doi: 10.1016/S2213-2600(15)00091-0. [DOI] [PubMed] [Google Scholar]
- 3.Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Diseases (GOLD) 2016 2016. [http://www.goldcopd.org]. Accessed 16 June 2016.
- 4.Yu AP, Guérin A, De Leon DP, Ramakrishnan K, Wu EQ, Mocarski M, et al. Clinical and economic outcomes of multiple versus single long-acting inhalers in COPD. Respir Med. 2011;105:1861–1871. doi: 10.1016/j.rmed.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Yu AP, Guerin A, Ponce de Leon D, Ramakrishnan K, Wu EQ, Mocarski M, et al. Therapy persistence and adherence in patients with chronic obstructive pulmonary disease: multiple versus single long-acting maintenance inhalers. J Med Econ. 2011;14:486–496. doi: 10.3111/13696998.2011.594123. [DOI] [PubMed] [Google Scholar]
- 6.Calzetta L, Rogliani P, Matera MG, Cazzola M. A systematic review with meta-analysis of dual bronchodilation with LAMA/LABA for the treatment of stable COPD. Chest. 2016;149:1181–1196. doi: 10.1016/j.chest.2016.02.646. [DOI] [PubMed] [Google Scholar]
- 7.Beier J, Kirsten AM, Mróz R, Segarra R, Chuecos F, Caracta C, et al. Efficacy and safety of aclidinium bromide compared with placebo and tiotropium in patients with moderate-to-severe chronic obstructive pulmonary disease: results from a 6-week, randomized, controlled Phase IIIb study. COPD. 2013;10:511–522. doi: 10.3109/15412555.2013.814626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vehring R, Lechuga-Ballesteros D, Joshi V, Noga B, Dwivedi SK. Cosuspensions of microcrystals and engineered microparticles for uniform and efficient delivery of respiratory therapeutics from pressurized metered dose inhalers. Langmuir. 2012;28:15015–15023. doi: 10.1021/la302281n. [DOI] [PubMed] [Google Scholar]
- 9.Fabbri LM, Kerwin EM, Spangenthal S, Ferguson GT, Rodriguez-Roisin R, Pearle J, et al. Dose-response to inhaled glycopyrrolate delivered with a novel Co-Suspension™ Delivery Technology metered dose inhaler (MDI) in patients with moderate-to-severe COPD. Respir Res. 2016;17:109. doi: 10.1186/s12931-016-0426-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quinn D, Seale JP, Reisner C, Fischer T, Golden M, Fernandez C, et al. A randomized study of formoterol fumarate in a porous particle metered-dose inhaler in patients with moderate-to-severe COPD. Respir Med. 2014;108:1327–1335. doi: 10.1016/j.rmed.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Rennard S, Fogarty C, Reisner C, Fernandez C, Fischer T, Golden M, et al. Randomized study of the safety, pharmacokinetics, and bronchodilatory efficacy of a proprietary glycopyrronium metered-dose inhaler in study patients with chronic obstructive pulmonary disease. BMC Pulm Med. 2014;14:118. doi: 10.1186/1471-2466-14-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donohue JF. Minimal clinically important differences in COPD lung function. COPD. 2005;2:111–124. doi: 10.1081/COPD-200053377. [DOI] [PubMed] [Google Scholar]
- 13.Sethi S, Fogarty C, Hanania NA, et al. Efficacy of formoterol fumarate delivered by metered dose inhaler using Co-Suspension™ Delivery Technology versus Foradil® Aerolizer® in moderate-to-severe COPD: a randomized, dose-ranging study. Chronic Obstr Pulm Dis (Miami). 2017;4(1): In press. doi:http://doi.org/10.15326/jcopdf.4.1.2016.0158. [DOI] [PMC free article] [PubMed]
- 14.Mahler DA, Kerwin E, Ayers T, FowlerTaylor A, Maitra S, Thach C, et al. FLIGHT1 and FLIGHT2: efficacy and safety of QVA149 (indacaterol/glycopyrrolate) versus its monocomponents and placebo in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192:1068–1079. doi: 10.1164/rccm.201505-1048OC. [DOI] [PubMed] [Google Scholar]
- 15.Cazzola M, Calzetta L, Matera MG. β2 -adrenoceptor agonists: current and future direction. Br J Pharmacol. 2011;163:4–17. doi: 10.1111/j.1476-5381.2011.01216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cazzola M, Rogliani P, Matera MG. Aclidinium bromide/formoterol fumarate fixed-dose combination for the treatment of chronic obstructive pulmonary disease. Expert Opin Pharmacother. 2013;14:775–781. doi: 10.1517/14656566.2013.776539. [DOI] [PubMed] [Google Scholar]
- 17.Decramer M, Anzueto A, Kerwin E, Kaelin T, Richard N, Crater G, et al. Efficacy and safety of umeclidinium plus vilanterol versus tiotropium, vilanterol, or umeclidinium monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from two multicentre, blinded, randomised controlled trials. Lancet Respir Med. 2014;2:472–486. doi: 10.1016/S2213-2600(14)70065-7. [DOI] [PubMed] [Google Scholar]
- 18.Donohue JF, Maleki-Yazdi MR, Kilbride S, Mehta R, Kalberg C, Church A. Efficacy and safety of once-daily umeclidinium/vilanterol 62.5/25 mcg in COPD. Respir Med. 2013;107:1538–1546. doi: 10.1016/j.rmed.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 19.Buhl R, Maltais F, Abrahams R, Bjermer L, Derom E, Ferguson G, et al. Tiotropium and olodaterol fixed-dose combination versus mono-components in COPD (GOLD 2–4) Eur Respir J. 2015;45:969–979. doi: 10.1183/09031936.00136014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Celli B, ZuWallack R, Wang S, Kesten S. Improvement in resting inspiratory capacity and hyperinflation with tiotropium in COPD patients with increased static lung volumes. Chest. 2003;124:1743–1748. doi: 10.1378/chest.124.5.1743. [DOI] [PubMed] [Google Scholar]
- 21.Cohen JS, Miles MC, Donohue JF, Ohar JA. Dual therapy strategies for COPD: the scientific rationale for LAMA + LABA. Int J Chron Obstruct Pulmon Dis. 2016;11:785–797. doi: 10.2147/COPD.S54513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Partridge MR, Karlsson N, Small IR. Patient insight into the impact of chronic obstructive pulmonary disease in the morning: an internet survey. Curr Med Res Opin. 2009;25:2043–2048. doi: 10.1185/03007990903103006. [DOI] [PubMed] [Google Scholar]
- 23.Miravitlles M, Worth H, Soler Cataluna JJ, Price D, De Benedetto F, Roche N, et al. Observational study to characterise 24-hour COPD symptoms and their relationship with patient-reported outcomes: results from the ASSESS study. Respir Res. 2014;15:122. doi: 10.1186/s12931-014-0122-1. [DOI] [PMC free article] [PubMed] [Google Scholar]