

Abstract
Acute lymphoblastic leukemia (ALL) remains the leading cause of cancer-related death in children and young adults. Compared to ALL in children, adult ALL has a much lower cure rate. Therefore, it is important to understand the molecular mechanisms underlying high-risk ALL and to develop therapeutic strategies that specifically target genes or pathways in ALL. Here, we explored the IL7R and SH2B3 expression in adult ALL and found that IL7R is significantly higher and Sh2B3 lower expressed in B-ALL compared to normal bone marrow control, and the IL7RhighSH2B3low is associated with high-risk factors, and with high relapse rate and low disease-free survival rate in the patients. We also found that Ikaros deletion was associated with the IL7RhighSH2B3low expression pattern and Ikaros directly binds the IL7R and SH2B3 promoter, and suppresses IL7R and promotes SH2B3 expression. On the other hand, casein kinase inhibitor, which increases Ikaros function, inhibits IL7R and stimulates SH2B3 expression in an Ikaros dependent manner. Our data indicate that IL7RhighSH2B3low expression distinguishes a novel subset of high-risk B-ALL associated with Ikaros dysfunction, and also suggest the therapeutic potential for treatment that combines casein kinase inhibitor, as an Ikaros activator, with drugs that target the IL7R signaling pathway.
Keywords: IL7R, SH2B3, Ikaros, gene expression, acute lymphoblastic leukemia
INTRODUCTION
Acute lymphoblastic leukemia (ALL), the leading cause of cancer-related death in children and young adults (aged 21–39), is less common in adults [1–4] and treatment outcomes are significantly inferior to those in children [1–3, 5, 6]. With front-line therapy, rates of complete remission have ranged between 78% and 93% in recent clinical trials. However, one third of patients with standard-risk ALL and two thirds of high-risk patients relapse, making the cure rate about 40 percent overall in adults [7–9]. The reasons underlying this age-related decline in outcome are not completely understood, but it is observed that a higher incidence of genetic alterations is associated with poor outcome, and genetic alterations such as the BCR–ABL1 fusion, are more common in adults than in children [10, 11]. Nevertheless, when compared to childhood ALL, detailed information on the genetic basis of ALL in adults is lacking. Therefore, it is important to understand the molecular mechanism underlying high-risk adult ALL and to develop therapeutic strategies that specifically target genes or pathways in ALL.
The cell surface interleukin (IL)-7 receptor-α (IL7R) is present in lymphoid progenitor cells, and is required for normal lymphocyte development [12]. IL7R forms heterodimers with IL-2Rγ (common gamma chain) or with the cytokine receptor-like factor 2 (CRLF2) [13, 14] and activates the JAK/STAT5 and the PI3K/Akt/mTOR signaling pathways [15]. IL7R mutations have been identified in malignant and nonmalignant diseases. Somatic mutations in IL7R are detected in 10% of pediatric T-ALL cases and in a few cases of pediatric B-ALL. IL7R gain-of-function mutations are reported to be oncogenic in childhood T-ALL and B-ALL [16, 17]. Overexpression of the IL-7R is also reported to have oncogenic effects [18], however, the mechanism responsible for IL-7R overexpression in ALL it is not fully understood.
The SH2B adaptor protein 3 (SH2B3), also known as lymphocyte adaptor protein (LNK), is a negative regulator of cytokine signaling and plays a critical role in the homeostasis of hematopoietic stem cells and lymphoid progenitors. Loss of function mutations in SH2B3 are reported to play an important role in oncogenesis of ALL [19, 20]. Loss of SH2B3 results in an increase in Janus kinase-signal transduction, activation of transcription signaling and lymphoid cell proliferation, which further promotes leukemia development in a mouse model of NOTCH1-induced ALL [20, 21]. These results demonstrate that SH2B3 functions as a tumor suppressor in the pathogenesis of ALL. However, it is unclear whether low SH2B3 expression is associated with clinical features in ALL.
Two of the three major signaling pathways that are perturbed in high-risk ALLs are 1) loss of function of the lymphoid transcription factors IKZF1 and PAX5, and 2) activating tyrosine kinase lesions [22, 23]. The IL7R/CRLF2 receptor and downstream JAK-STAT pathway plays a critical role in malignancy of B-ALL. SH2B3 regulates pro-B progenitor homeostasis by attenuating IL-7–stimulated JAK/STAT5 signaling via a direct interaction with phosphorylated JAK3. While SH2B3 suppresses IL-7R/JAK/STAT signaling to restrict pro-/pre-B progenitor expansion and leukemia development [20], Ikaros plays an essential role in lymphocyte development and functions as a tumor suppressor in leukemia. Ikaros dysfunction is associated with a high relapse rate and unfavorable outcome in high-risk ALL [24–26]. We reported that restoring Ikaros function by Casein Kinas II (CK2) inhibition has therapeutic efficacy against high-risk leukemia by suppression of expression of Ikaros gene targets [27–30]. Here, we determined SH2B3 and IL7R expression in adult ALL and found that SH2B3 expression is significantly lower and IL7R is significantly higher than in normal bone marrow. Furthermore, the lower SH2B3 expression is associated with high IL7R expression in B-ALL. High IL7R and low SH2B3 expression are associated with high risk factors. High IL7R and low SH2B3 (IL7RhighSH2B3low) was associated with a high relapse rate and low disease-free survival in patients with B-ALL. Our studies show that Ikaros deletion is negatively correlated with SH2B3 expression but positively correlated with IL7R expression in patient samples and that Ikaros directly regulates IL7R and SH2B3 expression. CK2 inhibitor (TBB), which enhances Ikaros function, affected IL7R and SH2B3 expression in an Ikaros-dependent manner. Our findings indicate that IL7RhighSH2B3low expression distinguishes a novel subset of high risk B-ALL associated with Ikaros dysfunction. Our data also suggest the therapeutic potential of therapy that combines CK2 inhibitors to restore Ikaros function with drugs that target IL7R signaling.
RESULTS
IL7R high and SH2B3 low expression in adult ALL
We assessed IL7R and SH2B3 mRNA expression in 63 newly diagnosed adult B-ALL and 32 T-ALL patients. We found that, compared to normal control, SH2B3 expression is significantly lower (Figure 1A) but IL7R expression is significantly higher (Figure 1B) in B-ALL but not in T-ALL patients. Consistent with our data, we also observed the high expression of IL7R and low SH2B3 using a reported microarray expression cohort of ALL patients (Supplementary Figure S1 and S2). We found that IL7R high expression negatively correlated with SH2B3 expression (data not shown). Frequency of high IL7R expression is significantly higher than low IL7R expression among patients with SH2B3 low expression (84.8% vs. 40.0%, P<0.0001). These data indicate that IL7R high and SH2B3 low expression (IL7RhighSH2B3low) is characteristic of a subset of adult ALL.
Figure 1. SH2B3 and IL7R expression in B-ALL patients.
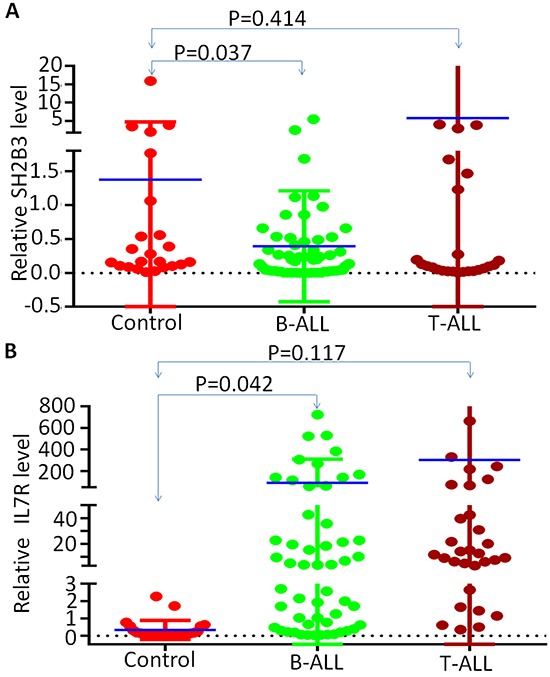
q-PCR was performed to detect IL-7 and SH2B3 in ALL patient samples and normal BM controls. Graphed is the relative expression A. Comparison of SH2B3 expression in B-ALL and T-ALL to normal BM control; B. Comparison of IL7R expression in B-ALL and T-ALL to normal BM control. Median expression is indicated and comparisons were by Mann-Whitney U test.
Association of high IL7R and low SH2B3 expression with characteristics of adult ALL
Patients were divided into high and low IL7R/SH2B3 expression groups (Quartiles 3-4 vs Quartiles 1-2, respectively) and IL7RhighSH2B3low expression was correlated with clinical features in adult B-ALL (Supplementary Table S1). We found that WBC count, a poor outcome marker, was much higher in IL7RhighSH2B3low than the IL7RlowSH2B3high subgroup (median 60.7×109/L vs. 12.9×109/L, P=0.001). Further, WBC ≥30×109/L, a specific marker for poor outcome in B-ALL, was also significantly higher in patients with IL7RhighSH2B3low (71.4 vs. 16.7, P=0.000) (Figure 2A), along with higher leukemic blasts in peripheral blood (median 81.0% vs. 45.0%, P=0.035) (Figure 2A). The percentage of the patients with age ≥35 years old, an important prognostic factor of poor outcome in adult B-ALL, was significantly higher in the IL7RhighSH2B3low than the IL7RlowSH2B3high subgroup (78.6% vs. 44.4%, P=0.018) (Figure 2B). The frequency of splenomegaly and enlarged lymph nodes, which indicates extramedullary infiltration, were found to be higher in the IL7RhighSH2B3low subgroup (60.7 vs. 11.1, P=0.001; 53.6 vs. 22.2, P=0.035, respectively) (Figure 2C). It is worth noting that we observed a significantly higher occurrence of the BCR/ABL1 fusion gene/Ph chromosome in IL7RhighSH2B3low than in the IL7RlowSH2B3high subgroup (75.0 vs. 16.7, P=0.000) (Figure 2D). We did not observe statistical differences in gender, HB, PLT, LDH, leukemic blasts in BM, myeloid markers (CD13, CD33) or the presence of complex karyotypes between the IL7RhighSH2B3low and the IL7RlowSH2B3high subgroups (P>0.05).
Figure 2. Correlation of IL7RhighSH2B3low expression with clinical features in ALL.
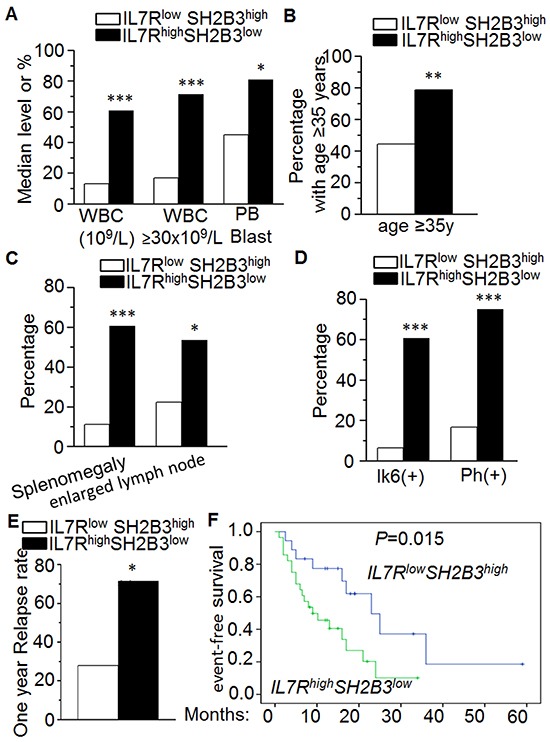
A-B. Comparison of high risk factors [high median WBC (109/L), WBC≥30×109/L, PB blasts] (A) and patient age ≥35 years old (B) in patients with IL7RhighSH2B3lowexpression to those in patients with IL7RlowSH2B3highexpression; C-D. Comparison of percentage of splenomegaly or enlarged lymph node (C) and co-existence of Ik6 (+) or Ph (+) chromosome (D) in these two patients' groups. E-F. Comparison of one-year relapse rate (E) and event-free survival (F) in patients with IL7RhighSH2B3lowexpression to those in patients with IL7RlowSH2B3high expression.
We also compared clinical outcomes of the patients from the two B-ALL subgroups. Patients in the IL7RhighSH2B3low subgroup did not show a higher rate of long duration (≥30 days) from initial chemotherapy to achieve CR when compared with the IL7RlowSH2B3high subgroup (32.1 vs. 16.7, P=0.451). However, we did observe a much higher 1-year relapse rate in the IL7RhighSH2B3low than in the IL7RlowSH2B3high subgroup (71.4 vs. 27.8, P=0.004) (Figure 2E). We also found that the IL7RhighSH2B3low subgroup had shorter event-free survival (EFS) than the IL7RlowSH2B3high subgroup (9 months vs. 23 months, P=0.015) (Figure 2F).
Taken together, these data show that adult B-ALL with IL7RhighSH2B3low gene expression is associated with markers of high-risk leukemia and with an unfavorable outcome. These data also distinguish patients with IL7RhighSH2B3low expression as a novel subset of high-risk leukemia.
Ikaros binds to the promoters of the IL7R and SH2B3 and regulates their expression
Our Ikaros ChIP-seq data show that Ikaros binding peaks in the promoter region of both IL7R and SH2B3 in Nalm6 B-ALL cells (Figure 3A and 3B) as well as in primary B-ALL cells (Supplementary Figure S3). We identified strong Ikaros binding motifs in the promoter regions of the two genes (data not shown). The binding of Ikaros at the promoter regions of IL7R and SH2B3 was further confirmed by qChIP and we found that Ikaros significantly bound to their promoter regions in Nalm6 and Ramos, malignant B cell lines (Figure 3C and 3D) and in primary cells from B-ALL patients (Figure 3E and 3F) but not in Molt 4 and U937 cells (T cell and myeloid leukemias, respectively). We further observed that Ikaros suppresses the promoter activity of IL7R and activates that of SH2B3 by luciferase reporter assay (Figure 4A). These data indicate a direct effect of Ikaros on transcription of IL7R and SH2B3. Moreover, expression of Ikaros suppressed the IL7R mRNA level and increased the SH2B3 mRNA level in Nalm6 cells (Figure 4B). Conversely, Ikaros knockdown induced an increase in IL7R expression and a decrease in SH2B3 expression in Nalm6 cells (Figure 4C, left panel); and also Ikaros mRNA level was obviously knocked down as shown by qPCR on right panel of Figure 4. TBB (CK2 inhibitor) increased the effect of Ikaros on IL7R and SH2B3 promoter activity (Figure 4A). Further treatment of Nalm6 cells with TBB suppressed IL7R expression and increased SH2B3 expression in a dose-dependent manner (Figure 5A). CK2 knockdown with shRNA also suppresses IL7R but increases SH2B3 mRNA levels by qPCR (Figure 5B). Ikaros knockdown with shRNA could block the TBB-induced decrease in IL7R expression and increase in SH2B3 expression (Figure 5C). These data indicate that both IL7R and SH2B3 are direct Ikaros targets in ALL and that Ikaros regulates their expression.
Figure 3. Ikaros binds the promoters of SH2B3 and IL7R.
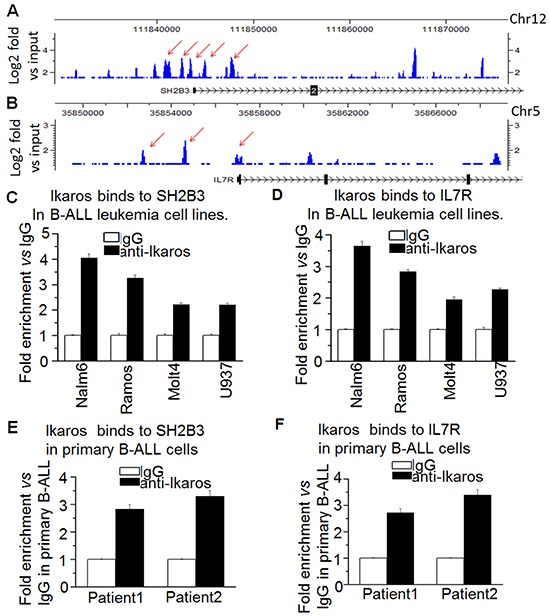
A-B. Ikaros binding peaks at the promoter of SH2B3 (A) and IL7R (B). C-D. qChIP assay to assess Ikaros binding at the promoter of SH2B3 (C) and IL7R (D) in leukemic cell lines. E-F. qChIP assay to assess Ikaros binding at the promoter of SH2B3 (E) and IL7R (F) in primary B-ALL patients' samples.
Figure 4. Ikaros promotes SH2B3 but suppresses IL7R transcription.
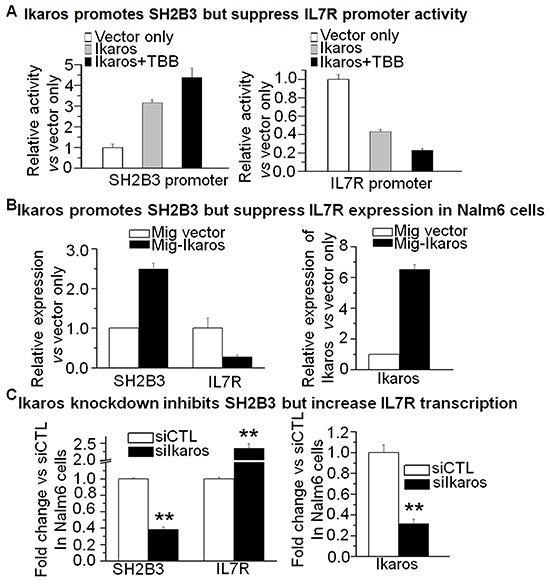
A. The promoter activity of SH2B3 and IL7R promoters by luciferase reporter assay following transfection with Ikaros or control vector in HEK293 cells. B. Expression of SH2B3 and IL7R in Nalm6 cells transduced with vector containing Ikaros as compared to control vector. C. Comparison of SH2B3 and IL7R expression in Nalm6 cells treated with Ikaros shRNA (siIkaros) or scramble shRNA (siCTL). Gene expression is determined by RT-qPCR using total RNA isolated from the cells transfected with scramble shRNA (siCTL) or Ikaros shRNA (siIkaros) for 2 days. compared with siCTL in C: *p <0.05, **p<0.01 compared to siCTL group.
Figure 5. Effect of CK2 inhibitor on expression of SH2B3 and IL7R.
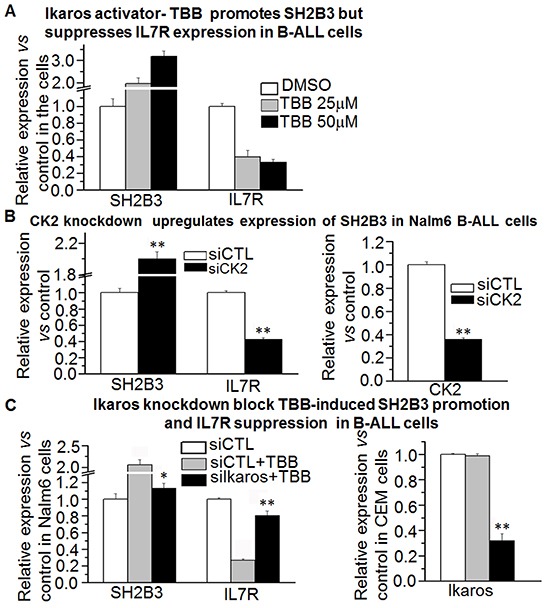
A. The CK2 inhibitor, TBB, promotes SH2B3 but suppresses IL7R expression in B-ALL cells as assessed by q-PCR. B. CK2 knockdown promotes SH2B3 but suppresses IL7R expression in B-ALL cells by q-PCR. C. Ikaros knockdown rescues the TBB-induced change in SH2B3 and IL7R in B-ALL cells. Compared with siCTL+TBB in C, D and E: * P<0.05; ** P<0.01.
IL7R and SH2B3 expression in patients with an Ikaros deletion
We found that Ikaros expression is positively correlated with SH2B3 expression but negatively correlated with IL7R expression in our cohort studies. Deletions that result in expression of Ikaros 6 (Ik6) are the most frequent type of Ikaros deletion. We detected Ik6 in our cohort, and further analyzed the IL7R or SH2B3 expression in patients with Ikaros deletion and patients without Ikaros deletion. Our data indicate significantly higher IL7R expression and lower SH2B3 expression in patients with Ikaros deletion (Ik6+), as compared to those without Ikaros deletion (Ik6-) (Figure 6A and 6B). We also found that the IL7RhighSH2B3low subgroup had a much higher frequency of Ik6+ cases (60.7 vs. 6.3, P<0.0001).
Figure 6. Ikaros deletion results in changes of SH2B3 and IL7R expression in primary B-ALL cells.
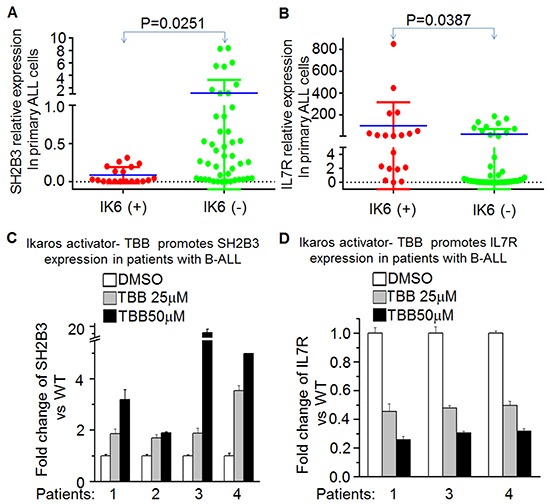
A-B. comparison of SH2B3 (A) and IL7R (B) in patients with or without Ikaros deletion; C-D. Effect of CK2 inhibitor (TBB) on expression of SH2B3 and IL7R in primary B-ALL cells with TBB treatment for 2 days.
These data further support a regulatory effect of Ikaros on both IL7R and SH2B3 in ALL patients and suggested that Ikaros deletion is one of the mechanisms for high IL7R and low SH2B3 expression in the patients. Additionally, TBB can suppress IL7R (Figure 6E) but increase SH2B3 expression (Figure 6F) in primary B-ALL. These data not only indicate the effect of CK2 inhibitor to increase Ikaros function as a transcriptional regulator, but also suggest that Ikaros-induced changes in IL7R/SH2B3 expression are at least partially responsible for the success of CK2 inhibitors in ALL therapy.
The CK2 inhibitor, TBB, promotes SH2B3 expression through chromatin remodeling
CK2 inhibitor functions to increase Ikaros activity. Ikaros regulates transcription of its target genes via chromatin remodeling by inducing epigenetic changes around the transcriptional start site (TSS) [28]. To understand the mechanisms by which CK2 inhibitor affects Ikaros function to promote SH2B3 expression, we tested whether the activation of SH2B3 is achieved through an epigenetic mechanism. Changes in the epigenetic markings around the TSS of SH2B3 were measured by qChIP following TBB treatment. Results show that the TBB induces a strong binding of Ikaros (Figure 7A) and enrichment of histone H3 trimethylation at lysine 4 (H3K4me3) (Figure 7B) in the promoter region of SH2B3 in Nalm6 cells, and also in primary B-ALL cells (Figure 7C and 7D). In addition, we also observed a TBB-induced increase of KDM5B binding in the promoter of the SH2B3 promoter in primary B-ALL cells (Figure 7E).
Figure 7. Ikaros promotes SH2B3 expression via chromatin remodeling.
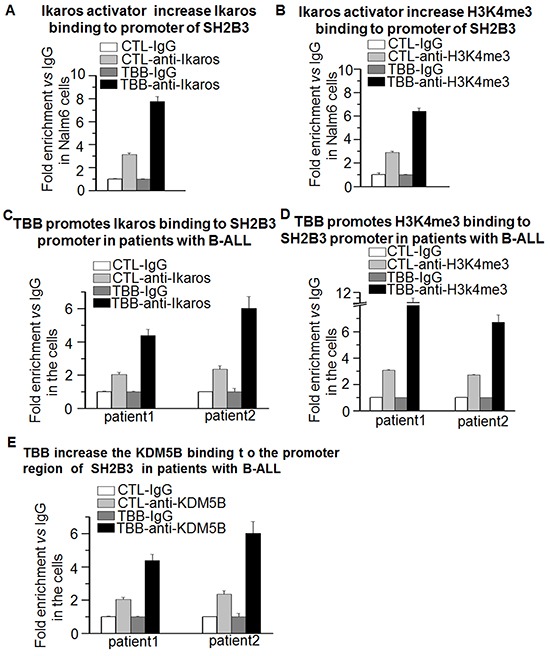
A-D. The CK2 inhibitor, TBB, increases the enrichment of Ikaros (A,C) and H3K4me3 (B,D) in B-ALL cells and patients' samples. The cells were treated with TBB 25μM for 1-2 days and qChIP was performed as described in methods section. E. TBB increases KDM5B binding at the SH2B3 promoter in patient samples.
DISCUSSION
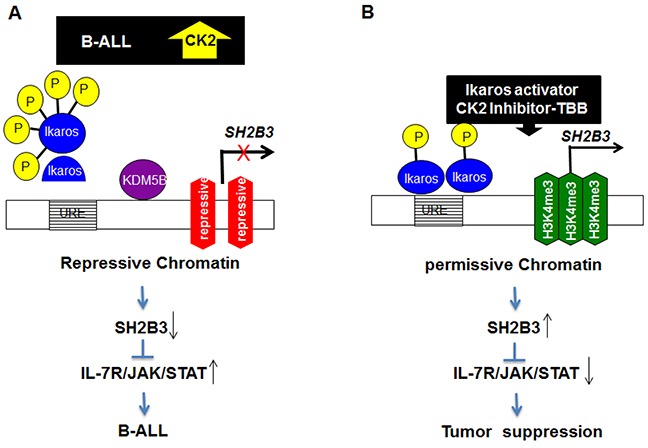
Identifying stage-specific signaling events in malignant transformation and progression and understanding the earliest stages of cellular transformation will provide critical insights in the field of leukemia. These insights will help us to decipher the mechanisms underlying high-risk ALL, delineate a new understanding of B-ALL development and point to potentially drugable pathways for intervention [31, 32]. We identified a novel subclass of high-risk B-ALL that is characterized by IL7RhighSH2B3low expression and associated with Ikaros dysfunction. Ikaros directly binds to and suppresses IL7R and stimulates SH2B3 expression in leukemic cells through chromatin remodeling as shown in the molecular model (Figure 8). Our findings indicate the collaborative effect of Ikaros dysfunction and the IL7R/JAK-STAT/SH2B3 signaling pathway on oncogenesis in high-risk B-ALL. This suggests that the use of CK2 inhibitors to restore Ikaros function combined with the inhibition of IL7R/JAK-STAT could be a potential therapy for treatment of high-risk B-ALL.
Figure 8. Model for the mechanism of Ikaros regulation of SH2B3 expression and IL7R/JAK/STAT signaling.
Most high-risk B-ALL-associated mutations/deletions/translocations identified to date are known or predicted to activate oncogenic cytokine receptor signaling, particularly of JAK-associated pathways [33]. We found that patients with IL7RhighSH2B3low expression have features of high-risk leukemia, unfavorable outcome and a high rate of both Ikaros deletion (60%) and BCR-ABL fusion (75.1%). It is reported that SH2B3 loss-of-function mutations co-occurs with IL7R gain-of-function mutations frequently in Ph-like ALL [22, 34]. Ph-like ALL is characterized with BCL-ABL(−), high CRLF2 rearrangement and high rate of Ikaros deletion. The characteristics of the patients that we identified are high IL7R and low SH2B3 expression, along with a high rate of BCR-ABL (+) and Ikaros deletion (+). This suggests that the patients may be different from both Ph(+) ALL and Ph-like ALL, and may account for a novel subclass of high-risk B-ALL in adult.
IL7R is highly expressed in ALL and IL7R/JAK/STAT signaling plays a critical role in oncogenesis of ALL. SH2B3, in addition to regulating normal and malignant HSC expansion via the TPO/MPL/JAK2 pathway [35–37], also plays a direct role in B cell progenitors. SH2B3 controls pro-B/pre-B homeostasis and aging by regulating IL-7–mediated JAK/STAT signaling in normal and malignant B progenitors. We identified a subclass of high-risk ALL patients characterized as IL7RhighSH2B3low which is consistent with the reported effect of IL7R and SH2B3 in the oncogenesis of B-ALL [19, 21, 34]. It is reported that SH2B3 interacts with JAK3 and IL7R activates JAK3 in B cell lineages [20]. SH2B3 is a negative regulator for IL7R–mediated JAK/SAT signaling that contributes to precursor B-ALL development. Moreover, the elevated activations of STATs such as STAT5 are downstream of IL7R stimulation in ALL [20, 38]. STAT5 is considered indispensable for maintenance of BCR-ABL–positive leukemia [38]. Furthermore, overexpression of oncogenic STAT5 in BM transplant models promotes B-ALL development in mice [39]. Taken together this data suggest that patients with the IL7RhighSH2B3low subclass of high-risk B-ALL will be sensitive to inhibitors of the JAK/STAT pathway. We have reported that CK2 inhibitor restores Ikaros function and shows therapeutic efficacy in high-risk B-ALL [27]. We show here that Ikaros directly binds to IL7R and SH2B3 and that the CK2 inhibitor, TBB, suppresses IL7R but promotes SH2B3 expression in an Ikaros dependent manner. These data indicate that B-ALL in these patients is likely to be sensitive to combination treatment with CK2 and JAK inhibitors.
Our previous studies have shown that Ikaros suppresses gene expression by recruiting repressive histone markers H3K9me3 and H3K27me3 via HDAC1 [27, 28]. We believe Ikaros suppression of IL7R expression is also mediated by induction of the H3K27me3 or H3K9me3 histone modifications to form repressive chromatin in the promoter region. Ikaros can also activate gene expression and the mechanism is not fully determined. We reported that Ikaros suppress KDM5B expression and increases levels of H3K4me3 histone modifications. [30]. We observed that the CK2 inhibitor, TBB, increases KDM5B binding and H3K4me3 histone marks at the SH2B3 promoter suggesting that this could be a mechanism through which Ikaros activates SH2B3 expression.
In summary, we identified a subclass of high-risk B-ALL with IL7RhighSH2B3low expression associated with Ikaros dysfunction. Our data implicate Ikaros/IL7R/SH2B3 signaling in oncogenesis of high-risk leukemia. We also found that CK2 inhibitor acts to increase transcriptional repression of IL7R and transcriptional activation of SH2B3 in an Ikaros dependent manner. This suggests the therapeutic potential of combining CK2 inhibitor with inhibitors of JAK/STAT downstream of IL7R/SH2B3 signaling.
MATERIALS AND METHODS
Patients and samples
BM samples were collected from 95 patients with ALL (63 B-ALL and 32 T-ALL) between June 2009 and June 2015 at the First Affiliated Hospital of Nanjing Medical University and Zhongda Hospital Southeast University Medical School. The ALL diagnosis was made according to the cytogenetic, morphologic, immunophenotypic, and molecular criteria of WHO Diagnosis and Classification of ALL (2008). Written informed consent was provided before enrollment in the study by all patients in accordance with the Declaration of Helsinki. The cohort study was also approved by the Institutional Review Board and the Ethics Committee of the Nanjing Medical University (Nanjing, China).
Cytogenetic and molecular analyses
Conventional cytogenetic analysis was performed at the time of diagnosis using unstimulated short-term cultures according to the recommendations of the International System for Human Cytogenetic Nomenclature (ISCN). At least 20 bone marrow metaphase cells were analyzed for each sample.
Flow cytometry was performed on fresh pretreatment BM samples for immunophenotypic analyses. A cell-surface antigen was defined as positive when fluorescence intensity of at least 20% of cells exceeded fluorescence of negative control as previously described [29, 40].
Cell culture reagents, plasmid construction, and retroviral gene transfer
Cells were incubated at 37°C in a humidified atmosphere of 5% CO2. Nalm6 cells have been previously described [27]. CCRF-CEM (CEM), MOLT-4 and U-937 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). The cell lines werecultured in RPMI 1640 medium (Cellgro) supplemented with 10% fetal bovine serum (Hyclone). HEK 293T cells were cultured in DMEM (Cellgro) supplemented with 10% fetal calf serum and 1% L-glutamine (Cellgro). Primary human B-ALL cells were cultured in RPMI 1640 medium (Cellgro) supplemented with 10% fetal bovine serum (Hyclone). Cells were cultured with or without TBB and collected for total RNA isolation. Human HA-tagged Ikaros (IKZF1) retroviral construct and retroviral production was described previously [27–30].
Luciferase assay
The pGL4.15 luciferase reporters construct for the IL7R and SH2B3 promoters were constructed by insert of the IL7R promoter (−1000bp to 0bp) and SH2B3 promoter (−100 to +300bp). Transient luciferase assays were performed in HEK293T cells using Promega luciferase assay reagents and measured with a luminometer following the manufacturer's instructions. Luciferase activity was calculated as fold change relative to values obtained from pGL vector only control cells and expressed as a percentage of pcDNA 3.1-Ikaros transfection-induced luciferase activity versus that of pcDNA3.1 vector alone. All transfection and reporter assays were performed independently in triplicate at least three times.
Real time-PCR
Total RNA was isolated using the RNeasy Mini Kit (QIAGEN). One μg RNA was reverse transcribed using SuperScriptTM First-Strand Synthesis System for RT-PCR Kit (Invitrogen). q-PCR was performed with qSTAR SYBR Master Mix (OriGene) using a StepOne Plus real-time PCR system (Applied Biosystems). Each experiment was performed in triplicate.
IL7R and SH2B3 expression in patient samples was quantitated as previously described [29, 40]. The expression level of IL7R or SH2B3 was normalized to 18s RNA and expressed as gene expression value of IL7R or SH2B3/18s RNA.
The qPCR for IL7R and SH2B3 expression was performed in Nalm6 cells transfected with Ikaros. The results were normalized to those obtained with 18s RNA and presented as fold induction over vector controls. Primers: 18s RNA, Sense:5′-GTAACCCGTTGAACCCCATT-3′, Antisense: 5′-CCATCCAATCGGTAGTAGCG-3′; IL7R Sense: 5′-AATGAAAAGGCCCCCAAGGTAGTTATCC-3′, Anti-sense: 5′-GTCGTTTCCGCAACAAGTCCTCTTC-3′; SH2B3 Sense: 5′-TCACAGTGCAAGAAGGATACCAA A-3′, Anti-sense: 5′-TGAAAGCCAGCATCGTTCTTAGTC-3′.
Quantitative chromatin immunoprecipitation (qChIP)
qChIP assays were performed by incubation of the chromatin with antibodies against Ikaros and normal rabbit IgG (Abcam) as a control [27–30]. Enrichment of the ChIP sample over input was evaluated by qPCR with three or more replicates, using specific primers in the promoter region of IL7R (forward: 5′-AGGGGA AGGGAGGGGAAGGGAAAG-3‘, reverse: 5′-CCCTT TCCTCCCCTCCCCTCCT-3‘) and SH2B3 (forward: 5′-G GTTTTCTCTGCCTTAAACTCTGAA-3‘, reverse: 5′-GC TGCCAACAGGAAGATTTACTG-3′). The relative concentration of the qPCR product was presented as the fold change of the level of DNA in Ikaros samples in comparison to controls. It is considered the binding is significant if the fold enrichment versus IgG is ≥2.
Ikaros shRNA knockdown
Nalm6 cells were transiently transfected with human Ikaros shRNA constructs in the GFP vector (pGFP-v-RS) (Origene) using the Neon Transfection System (Invitrogen). The 29-mer scrambled shRNA cassette in the pGFP-VRS vector was also used as a control. After transfection for 1 day, Nalm6 cells with transfection efficiency around 80% (green cells) and greater than 95% cell viability were further treated with 20μM TBB or vehicle control (0.01% DMSO) for 2 days and harvested for total RNA isolation. Knockdown of Ikaros was confirmed by measurement of Ikaros mRNA level using qPCR[27, 29]. Primers: IKZF1-F:5′-ggcgcggtgctcctcct-3′, IKZF1-R: 5′-tccgac acgccctacgaca-3′
Statistical analysis
Patients were divided into high and low IL7R or SH2B3 expression groups (Quartiles 3-4 vs Quartiles 1-2, respectively) as determined by SPSS 17.0.
For quantitative parameters, overall differences between the cohorts were evaluated using a Mann – Whitney U-test. For qualitative parameters, overall group differences were analyzed using a χ2 test. All statistical analyses were performed using the SPSS 17.0 and P<0.05 was considered statistically significant.
The experimental data are shown as the mean value with bars representing the standard error of the mean (S.E.M.). Determinations of statistical significance were performed using a Student t-test for comparisons of two groups or using analysis of variance (ANOVA) for comparing multiple groups. The P<0.05 was considered statistically significant.
SUPPLEMENTARY FIGURES AND TABLE
Acknowledgments
This work is supported in part by The National Natural Science Foundation of China (81270613,30973376), Jiangsu Province Key Medical Talents (RC2011077), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (39th), China Postdoctoral Science Foundation (20090461134), Special grade of the financial support from China Postdoctoral Science Foundation (201003598), The Six Great Talent Peak Plan of Jiangsu (2010-WS-024), The Scientific Research Foundation for the Returned Overseas Chinese Scholars, Nanjing Municipal Bureau of Personnel (2009), and Southeast University Basic Research Fund (2242016k40143) (to Z.G.). It was also supported partially by Four Diamond Foundation of Pennsylvania State University, PA, USA (to S.D. and C.S.).
Footnotes
CONFLICTS OF INTEREST
All the authors declare no conflict of interest.
REFERENCES
- 1.Ko RH, Ji L, Barnette P, Bostrom B, Hutchinson R, Raetz E, Seibel NL, Twist CJ, Eckroth E, Sposto R, Gaynon PS, Loh ML. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: a Therapeutic Advances in Childhood Leukemia Consortium study. Journal of clinical oncology. 2010;28:648–54. doi: 10.1200/JCO.2009.22.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raetz EA, Bhatla T. Where do we stand in the treatment of relapsed acute lymphoblastic leukemia? Hematology. 2012;2012:129–36. doi: 10.1182/asheducation-2012.1.129. [DOI] [PubMed] [Google Scholar]
- 3.Bhatla T, Jones CL, Meyer JA, Vitanza NA, Raetz EA, Carroll WL. The biology of relapsed acute lymphoblastic leukemia: opportunities for therapeutic interventions. Journal of pediatric hematology/oncology. 2014;36:413–8. doi: 10.1097/MPH.0000000000000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381:1943–55. doi: 10.1016/S0140-6736(12)62187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burke PW, Douer D. Acute lymphoblastic leukemia in adolescents and young adults. Acta haematologica. 2014;132:264–73. doi: 10.1159/000360204. [DOI] [PubMed] [Google Scholar]
- 6.Curran E, Stock W. How I treat acute lymphoblastic leukemia in older adolescents and young adults. Blood. 2015;125:3702–10. doi: 10.1182/blood-2014-11-551481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, Ma J, Coustan-Smith E, Harvey RC, Willman CL, Mikhail FM, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nature genetics. 2009;41:1243–6. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 9.Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, White D, Hughes TP, Le Beau MM, Pui CH, Relling MV, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–4. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 10.Perez-Andreu V, Roberts KG, Xu H, Smith C, Zhang H, Yang W, Harvey RC, Payne-Turner D2, Devidas M, Cheng IM, Carroll WL, et al. A genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood. 2015;125:680–6. doi: 10.1182/blood-2014-09-595744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stock W. Adolescents and young adults with acute lymphoblastic leukemia. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2010;2010:21–9. doi: 10.1182/asheducation-2010.1.21. [DOI] [PubMed] [Google Scholar]
- 12.Grossmann V, Haferlach C, Weissmann S, Roller A, Schindela S, Poetzinger F, Stadler K, Bellos F, Kern W, Haferlach T, Schnittger S, et al. The molecular profile of adult T-cell acute lymphoblastic leukemia: mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes chromosomes & cancer. 2013;52:410–22. doi: 10.1002/gcc.22039. [DOI] [PubMed] [Google Scholar]
- 13.Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, Park LS, Ziegler SF, Williams DE, Ware CB, Meyer JD, et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. The Journal of experimental medicine. 1994;180:1955–60. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noguchi M, Nakamura Y, Russell SM, Ziegler SF, Tsang M, Cao X, Leonard WJ. Interleukin-2 receptor gamma chain: a functional component of the interleukin-7 receptor. Science. 1993;262:1877–80. doi: 10.1126/science.8266077. [DOI] [PubMed] [Google Scholar]
- 15.Ribeiro D, Melao A, Barata JT. IL-7R-mediated signaling in T-cell acute lymphoblastic leukemia. Advances in biological regulation. 2013;53:211–22. doi: 10.1016/j.jbior.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 16.Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te Kronnie G, Cario G, Cazzaniga G, Kulozik AE, Stanulla M, Schrappe M, et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. The Journal of experimental medicine. 2011;208:901–8. doi: 10.1084/jem.20110580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zenatti PP, Ribeiro D, Li W, Zuurbier L, Silva MC, Paganin M, Tritapoe J, Hixon JA, Silveira AB, Cardoso BA, Sarmento LM, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nature genetics. 2011;43:932–9. doi: 10.1038/ng.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, Borowitz MJ, Camitta BM, Carroll AJ, Devidas M, Pullen DJ, Payne-Turner D, et al. Outcome modeling with CRLF2 IKZF1 JAK and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood. 2012;119:3512–22. doi: 10.1182/blood-2011-11-394221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Garcia A, Ambesi-Impiombato A, Hadler M, Rigo I, LeDuc CA, Kelly K, Jalas C, Paietta E, Racevskis J, Rowe JM, et al. Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood. 2013;122:2425–32. doi: 10.1182/blood-2013-05-500850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng Y, Chikwava K, Wu C, Zhang H, Bhagat A, Pei D, Choi JK, Tong W. LNK/SH2B3 regulates IL-7 receptor signaling in normal and malignant B-progenitors. The Journal of clinical investigation. 2016;126:1267–81. doi: 10.1172/JCI81468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willman CL. SH2B3: a new leukemia predisposition gene. Blood. 2013;122:2293–5. doi: 10.1182/blood-2013-08-519843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, Ma J, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. The New England journal of medicine. 2014;371:1005–15. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Mullighan CG, Harvey RC, Wu G, Chen X, Edmonson M, Buetow KH, Carroll WL, Chen IM, Devidas M, Gerhard DS, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood. 2011;118:3080–7. doi: 10.1182/blood-2011-03-341412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, Maffucci P, Pierce KR, Abbott JK, Voelkerding KV, South ST, et al. Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. The New England journal of medicine. 2016;374:1032–43. doi: 10.1056/NEJMoa1512234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeBoer R, Koval G, Mulkey F, Wetzler M, Devine S, Marcucci G, Stone RM, Larson RA, Bloomfiel d CD, Geyer S, Mullighan CG, et al. Clinical impact of ABL1 kinase domain mutations and IKZF1 deletion in adults under age 60 with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL): molecular analysis of CALGB (Alliance) 10001 and 9665. Leukemia & lymphoma. 2016:1–9. doi: 10.3109/10428194.2016.1144881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, Harvey RC, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. The New England journal of medicine. 2009;360:470–80. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song C, Gowda C, Pan X, Ding Y, Tong Y, Tan BH, Wang H, Muthusami S, Ge Z, Sachdev M, Amin SG, et al. Targeting casein kinase II restores Ikaros tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood. 2015;126:1813–22. doi: 10.1182/blood-2015-06-651505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song C, Pan X, Ge Z, Gowda C, Ding Y, Li H, Li Z, Yochum G, Muschen M, Li Q, Payne KJ, et al. Epigenetic regulation of gene expression by Ikaros HDAC1 and Casein Kinase II in leukemia. Leukemia. 2015 doi: 10.1038/leu.2015.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge Z, Guo X, Li J, Hartman M, Kawasawa YI, Dovat S, Song C. Clinical significance of high c-MYC and low MYCBP2 expression and their association with Ikaros dysfunction in adult acute lymphoblastic leukemia. Oncotarget. 2015;6:42300–11. doi: 10.18632/oncotarget.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Song C, Ding Y, Pan X, Ge Z, Tan BH, Gowda C, Sachdev M, Muthusami S, Ouyang H, Lai L, et al. Transcriptional Regulation of JARID1B/KDM5B Histone Demethylase by Ikaros Histone Deacetylase 1 (HDAC1) and Casein Kinase 2 (CK2) in B-cell Acute Lymphoblastic Leukemia. The Journal of biological chemistry. 2016;291:4004–18. doi: 10.1074/jbc.M115.679332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loghavi S, Kutok JL, Jorgensen JL. B-acute lymphoblastic leukemia/lymphoblastic lymphoma. American journal of clinical pathology. 2015;144:393–410. doi: 10.1309/AJCPAN7BH5DNYWZB. [DOI] [PubMed] [Google Scholar]
- 32.Matnani R, Parekh V, Borate U, Brazelton J, Reddy V, Peker D. Therapy-related B-lymphoblastic leukemia associated with Philadelphia chromosome and MLL rearrangement: Single institution experience and the review of the literature. Pathology international. 2015;65:536–40. doi: 10.1111/pin.12337. [DOI] [PubMed] [Google Scholar]
- 33.Buchner M, Swaminathan S, Chen Z, Muschen M. Mechanisms of pre-B-cell receptor checkpoint control and its oncogenic subversion in acute lymphoblastic leukemia. Immunological reviews. 2015;263:192–209. doi: 10.1111/imr.12235. [DOI] [PubMed] [Google Scholar]
- 34.Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, Chen X, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer cell. 2012;22:153–66. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seita J, Ema H, Ooehara J, Yamazaki S, Tadokoro Y, Yamasaki A, Eto K, Takaki S, Takatsu K, Nakauchi H. Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2349–54. doi: 10.1073/pnas.0606238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bersenev A, Wu C, Balcerek J, Tong W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. The Journal of clinical investigation. 2008;118:2832–44. doi: 10.1172/JCI35808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buza-Vidas N, Antonchuk J, Qian H, Mansson R, Luc S, Zandi S, Anderson K, Takaki S, Nygren JM, Jensen CT, Jacobsen SE. Cytokines regulate postnatal hematopoietic stem cell expansion: opposing roles of thrombopoietin and LNK. Genes & development. 2006;20:2018–23. doi: 10.1101/gad.385606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, Fajmann S, Grebien F, Warsch W, Stengl G, Hennighausen L, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO molecular medicine. 2010;2:98–110. doi: 10.1002/emmm.201000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joliot V, Cormier F, Medyouf H, Alcalde H, Ghysdael J. Constitutive STAT5 activation specifically cooperates with the loss of p53 function in B-cell lymphomagenesis. Oncogene. 2006;25:4573–84. doi: 10.1038/sj.onc.1209480. [DOI] [PubMed] [Google Scholar]
- 40.Guo X, Zhang R, Liu J, Li M, Song C, Dovat S, Li J, Ge Z. Characterization of LEF1 High Expression and Novel Mutations in Adult Acute Lymphoblastic Leukemia. PloS one. 2015;10:e0125429. doi: 10.1371/journal.pone.0125429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.