

Summary
Trace metals play important roles in biological function, and x‐ray fluorescence microscopy (XFM) provides a way to quantitatively image their distribution within cells. The faithfulness of these measurements is dependent on proper sample preparation. Using mouse embryonic fibroblast NIH/3T3 cells as an example, we compare various approaches to the preparation of adherent mammalian cells for XFM imaging under ambient temperature. Direct side‐by‐side comparison shows that plunge‐freezing‐based cryoimmobilization provides more faithful preservation than conventional chemical fixation for most biologically important elements including P, S, Cl, K, Fe, Cu, Zn and possibly Ca in adherent mammalian cells. Although cells rinsed with fresh media had a great deal of extracellular background signal for Cl and Ca, this approach maintained cells at the best possible physiological status before rapid freezing and it does not interfere with XFM analysis of other elements. If chemical fixation has to be chosen, the combination of 3% paraformaldehyde and 1.5 % glutaraldehyde preserves S, Fe, Cu and Zn better than either fixative alone. When chemically fixed cells were subjected to a variety of dehydration processes, air drying was proved to be more suitable than other drying methods such as graded ethanol dehydration and freeze drying. This first detailed comparison for x‐ray fluorescence microscopy shows how detailed quantitative conclusions can be affected by the choice of cell preparation method.
Keywords: Biological, cryomicroscopy, specimen preparation, x‐ray microanalysis, x‐ray microscopy
Introduction
Synchrotron‐based x‐ray fluorescence microscopy (XFM) has found wide applications in many areas of science, in particular in biomedical science (Paunesku et al., 2006; Fahrni, 2007; Szczerbowska‐Boruchowska, 2008; Majumdar et al., 2012). These applications can be classified into two categories. One involves the mapping of naturally existing elements such as in studies of the roles of Fe, Cu and Zn in essential cellular processes or in disease development (Farquharson et al., 2007; Ortega et al., 2007; Leskovjan et al., 2011; Vogt & Ralle, 2013). The second involves studying how exogenously employed metal‐containing reagents such as anticancer drugs are transported, taken up, compartmentalized and distributed at the level of tissue, cell or even subcellular organelles (Corde et al., 2002; Paunesku et al., 2003; Corezzi et al., 2009; Yuan et al., 2013). Both lines of research have greatly enhanced our understanding of the molecular mechanisms of many cellular processes as well as metal‐related disorders, and assisted the development and assessment of targeted therapeutic and diagnostic compounds.
Several other in situ elemental analysis techniques exist (McRae et al., 2009; Petibois, 2010; Bourassa & Miller, 2012), such as dye or fluorophor‐based fluorescence optical microscopy, electron probe x‐ray microanalysis (EPXMA), proton induced x‐ray emission (PIXE), secondary‐ion mass spectrometry (SIMS) and NanoSIMS. Among these techniques, XFM has some unique characteristics. Due to the high penetration depth of hard x‐rays, various biological samples such as monolayer cells can be directly studied without further sectioning; this not only avoids complex sample preparation procedures as required by EPXMA, but also facilitates elemental quantification and localization within the entirety of a whole cell thickness (Lobinski et al., 2006; Paunesku et al., 2006; Ortega et al., 2009). Another advantage is its exceptional sensitivity. Attomolar amounts of trace elements can be detected in single cell or 10 m tissue sections (Twining et al., 2003). XFM is also known for submicrometer spatial resolution, and simultaneous analysis of a large number of elements. Thus, XFM has been widely regarded as an effective approach for in situ imaging and quantification of trace metals, toxic heavy metals and molecule–metal complexes in whole cells or whole cell‐thick tissue sections (Dillon et al., 2002; Paunesku et al., 2003; Kemner et al., 2004; Yang et al., 2005; Corezzi et al., 2009).
There are many critical factors to be considered while applying XFM to investigate the elemental distribution and quantification of cultured mammalian cells. Sample preparation is one of the most important steps (Perrin et al., 2015). One common preparation approach involves aldehyde‐based chemical fixation followed by dehydration, whereas another involves rapid freezing‐based fixation (cryoimmobilization), followed by imaging in the frozen hydrated state or with dehydrated, room‐temperature specimens. Both approaches have been originally developed and extensively studied in the field of transmission electron microscopy for the preservation of ultrastructure and antigenicity (Sitte et al., 1987; Nicolas, 1991; Monaghan et al., 1998). When these approaches are adapted to sample preparation for XFM studies, it is important to preserve both the total content and also the spatial distribution of biologically important elements.
Aldehyde‐based conventional chemical fixation is in general considered to be suboptimal for the preservation of most biologically important elements, especially for those highly diffusible ions such as K and Cl, because it is slow and selective (Zierold, 1982; Chwiej et al., 2005; Matsuyama et al., 2010; Hackett et al., 2011). It takes time (often seconds or even minutes) for chemical fixatives to reach and react with their counterparts within the entire living cell, where they immobilize only certain macromolecules such as proteins (Gilkey & Staehein, 1986). Many small molecules (such as ions) or macromolecules (such as carbohydrates, lipids and nucleic acids) are not efficiently crosslinked by aldehydes due to the lack of functional free amino groups, which leads them to be subsequently extracted, replaced or lost (Makjanic & Watt, 1999; Chwiej et al., 2005; Hawes, 2015). Furthermore, aldehydes disorganize cellular membranes and alter membrane permeability. This allows free ions and unreactive small molecules to escape from their native sites and to redistribute within the cell or be lost to extracellular space. Loss or redistribution can also happen to bound ions, if the macromolecules to which they were bound were not crosslinked during fixation.
In contrast, cryoimmobilization, which involves instantaneous cooling of cellular water into a crystal‐free solid state (amorphous or vitreous) ice, provides rapid immobilization of both free and bound ions at native sites. Plunge freezing, impact freezing, double propane jet freezing, and high pressure freezing are the most commonly used cryoimmobilization techniques (Moor, 1987; Sitte et al., 1987; McDonald, 2014). With freezing rates above 104 K s–1, these techniques are able to vitrify whole cells or tissues (up to 10 m thickness in plunge freezing and 200 m in high‐pressure freezing) within microseconds or milliseconds (Muller & Moor, 1984; Sartori & Richter, 1993; Studer et al., 2008). At such cooling speeds, the formation of ice crystals is mostly inhibited, leading to reduced structural damage and redistribution of ions and small molecules. (The formation of small ice crystals can be detected via diffraction rings in electron microscopy, see Dubochet et al., 1982, but might not be noticeable in XFM where the present spatial resolution is no better than about 30 nm). Furthermore, cryogenic sample preparation, when combined with cryotransfer and scanning capabilities, is capable of preserving elemental composition, speciation and distribution as close as possible to the native state, and is thus recognized as the most reliable approach for studies of cellular elemental homeostasis in electron (Shuman et al., 1976; Saubermann et al., 1981; Zierold, 1982; Somlyo et al., 1985; Andrews et al., 1987; Saubermann & Heyman, 1987; Andrews et al., 1988; Somlyo et al., 1988; LeFurgey & Ingram, 1990; Zierold, 1991) and x‐ray (Matsuyama et al., 2010; Chen et al., 2014; Perrin et al., 2015) microprobe studies. Even when they are subsequently dehydrated and scanned under room temperature, cryogenically prepared biological samples are still believed to provide more faithful preservation than conventional chemical fixation. This is highly relevant, because the limited availability of cryo‐XFM instruments means that conventional chemical fixation has been and will still remain a valuable and necessary specimen preparation approach even though it might cause modification in elemental content and distribution in biological samples.
So as to better understand the differences among different sample preparation approaches, we decided to conduct side‐by‐side comparisons between chemically fixed and cryoimmbolized biological samples by using the mouse embryonic fibroblast cell line NIH/3T3 as an example of adherent mammalian cells. We examined both highly diffusible ions such as Cl and K, and tightly bound ions such as Fe and Zn. We also summarize our approaches for growing adherent cells on XFM‐compatible substrates, on the effects of various rinsing protocols used before plunge freezing, and how chemically fixed cells should be dehydrated prior to room temperature XFM studies.
Materials and methods
Cell culture
NIH/3T3 mouse embryonic fibroblast cells, originally purchased from American Tissue Culture Collection (www.atcc.org, cat# CRL1658), were routinely grown as described before in complete culture medium (Finney & Jin, 2015; Jin et al., 2015). At about 80% confluence, cells were washed with Dulbecco's phosphate buffer saline without calcium and magnesium (D‐PBS, Invitrogen Life Technologies, Waltham, Massachusetts, USA, cat#14190) and detached by incubation with 0.5 mL 0.05% Trypsin‐EDTA for about 2 min. Detached cells were collected by centrifugation, resuspended in fresh medium and seeded in 6‐well culture plates at the density of 6 × 104 cells per well in 3 mL of culture medium.
Cell growth on silicon nitride windows
For cells to be imaged by XFM, silicon nitride (Si3N4) windows (Norcada Inc., Canada, cat# NX5150D) were washed with distilled water followed by 2 min soaking in 70% ethanol and 2 min soaking in 100% ethanol. Windows were then dried and taped to the bottom of 6‐well culture plate. Si3N4 windows were placed with the flat (nonetched) side up. The 6‐well culture plates with taped Si3N4 windows were sterilized by 20 min UV irradiation, after which 3 mL of cell suspension at the density of 2 × 104 cells per mL was dispensed into each well. Cells were allowed to grow for 48 or 72 h to reach around 60% confluence on the surface of the window. All cell samples prepared for side‐by‐side comparisons were originated from the same batch of cells; all comparisons were independently repeated twice.
Chemical fixation
We compared three chemical fixatives: 2% glutaraldehyde (GA), 4% paraformaldehyde (PFA), and the combination of both aldehydes containing 3% PFA supplemented with 1.5 % GA. Each fixative was freshly prepared on the day of use by diluting 25% EM‐grade GA (Electron Microscopy Sciences, Hatfield, Pennsylvania, USA, cat# 16220) and/or 16% EM‐grade PFA (Electron Microscopy Sciences, cat# 15710) in D‐PBS. After a brief D‐PBS wash, cells on Si3N4 windows were fixed at room temperature either in 4% PFA for 20 min, or in 2% GA for 1 h, or in 3% PFA plus 1.5% GA for 1 h.
Dehydration of chemically fixed cells
Immediately after chemical fixation, cells on Si3N4 windows were sequentially dipped into two tubes of 1 mL fresh D‐PBS buffer to remove excess fixatives, and subjected to different dehydration procedures. For air drying, cells were rinsed with Tris‐glucose buffer (TG buffer: containing 261 mM glucose, 9 mM acetic acid and 10 mM Tris buffer, pH 7.4) twice and then blotted and allowed to dry in the air. For graded ethanol dehydration, cells were sequentially dipped into 30%, 50%, 70%, 90% and 100% ethanol for 10 s each and then dried in the air at room temperature. For freeze drying, chemically fixed cells were rapidly rinsed with TG buffer two times and subjected to plunge freezing and freeze drying as described below.
Plunge freezing (PF) and freeze drying
Si3N4 windows with cells grown on them were carefully detached from the cell culture dish and mounted to the tweezers supplied with a FEI (Hillsboro, Oregon, USA) Vitrobot Mark IV plunge freezer. Cells were then washed respectively with one of the following wash buffers: TG buffer, ammonium acetate buffer (Amac buffer: 100 mM ammonium acetate in H2O, osmolarity 200 mOsm L–1) as has been used in electron microprobe (Wroblewski & Wroblewski, 1993) and synchrotron (Perrin et al., 2015) microanalysis studies, D‐PBS buffer, or complete culture medium for two consecutive times. After all excess liquid on the back of the window (noncell side) and between tweezers was carefully blotted away, the tweezers with window were mounted onto the Vitrobot plunge freezer for rapid freezing following manufacturer's instructions. In brief, the climate chamber was set and stabilized at 25°C with 95% humidity. Windows were blotted in the chamber for 2 s for one single blotting at the blot force setting of 0 mm, and then immediately plunged into liquid ethane cooled by liquid nitrogen. Si3N4 windows were then slowly lifted up from liquid ethane cup and placed vertically into the window storage box (Fig. 1) immersed in liquid nitrogen bath. Plunge frozen cells were either stored in a liquid nitrogen dewar, or freeze dried in the precooled vacuum chamber of an EMS Turbo freeze drier K775 with the following manufacturer‐suggested cycles: 2 h at –120°C, 1 h ramp from –120°C to –80°C, 3 h at –80°C, 1 h ramp from –80°C to –50°C, 2 h at –50°C, 1 h ramp to 25°C, 5 h at 25°C.
Figure 1.

The in house fabricated Si3N4 window storage box. This box has the same outer diameter as most standard cryo‐TEM grid boxes (shown in the far right as a comparison) and fits most cryodevices such as FEI Vitrobot plunge freezer, Instec cryostorage mounting block, and Bionanoprobe sample transferring system. The box at the left has eight storage positions, whereas the box at the right has four storage positions. The window storage box is made of aluminium to minimize possible contamination from other high Z metals. It can be enclosed with a transparent lid secured by a stainless steel screw in the middle. The storage box allows windows to be vertically placed with predetermined orientation and to prevent fragile membranes from touching the aluminium wall during long‐term storage and sample transportation.
XFM elemental analysis
Prior to XFM analysis, frozen hydrated cells were imaged using a (Nikon Instruments, Melville, New York, USA) 50i light microscope with an Instec CLM77K cryostage under cryogenic temperature, whereas dehydrated cells were imaged using a (Leica Microsystems, Buffalo Grove, Illinois, USA) DMXRE microscope at room temperature. The coordinates obtained from light microscopes were recorded and used to locate the areas of interest under XFM. Frozen hydrated cells were scanned at about 100 K temperature using a cryojet fluorescence microprobe (Jin et al., 2015) at the 2‐ID‐D beamline at the Advanced Photon Source (APS), a 7 GeV synchrotron light source at Argonne National Laboratory. Dehydrated cells were scanned at room temperature at the 2‐ID‐E beamline at the APS. Areas of interest were raster‐scanned with 0.8 m step size and 1 s per pixel dwell time, using 10 keV hard x‐rays. The x‐ray beam was focused by a Fresnel zone plate to a spot size of 200–250 nm. The x‐ray induced elemental fluorescence at every scan position was measured by an energy dispersive silicon drift detector (Vortex EM, SII Nanotechnology, Northridge, California, USA). Data were fitted and quantified by comparison to NBS thin film standards 1832 and 1833 (National Institute of Standards and Technology, Gaithersburg, MD, USA). Two‐dimensional elemental maps and regions of interest (ROI) analysis were derived by using MAPS software (Vogt, 2003).
Results and discussion
Growth of adherent mammalian cells on Si3N4 windows
Different substrates have been used for XFM analysis of mammalian cells, including 2 m thick polycarbonate foils/films (Bacquart et al., 2007; Ortega et al., 2007; Kosior et al., 2012), 4 m thick prolene films (Matsuyama et al., 2009), formvar coated TEM grids (Bohic et al., 2001; Paunesku et al., 2003; Harris et al., 2005; Wagner et al., 2005; Chen et al., 2006; Paunesku et al., 2007) and Si3N4 windows (McRae et al., 2006; Finney et al., 2007; Matsuyama et al., 2010; Wolford et al., 2010; Marmorato et al., 2011; Weekley et al., 2011; Arora et al., 2012; Marvin et al., 2012; Yuan et al., 2013; Grubman et al., 2014). A discussion of the relative merits of some substrates is described elsewhere (Carter et al., 2010; Finney & Jin, 2015). Si3N4 windows are by far the most frequently used substrate for adherent cell XFM imaging. We have grown various cell types such as NIH/3T3 cells, ovarian cancer cells, patient primary fibroblast cells and human embryonic stem cells on Si3N4 windows. All cell lines tested can attach on untreated Si3N4 windows and grow robustly, although they are in general more easily detached or aggregated than cells grown on conventional ‘tissue culture treated’ cell culture dishes. To minimize cell detachment and aggregation, window pretreatments are sometimes necessary. In addition to poly‐L‐lysine coating (McRae et al., 2006) or glow discharging (Hagen et al., 2012), we have tried to prewash windows with distilled water followed by 2 min wash in 70% ethanol and 2 min wash in 100% ethanol. We found that prewashed windows provided better support for cell growth.
We have tried several approaches to grow adherent cells on Si3N4 windows (Fig. 2). One possible approach is to use standard office tape (‘Scotch tape’) to affix the windows to the bottom of a cell culture vessel (Finney & Jin, 2015). Upon UV irradiation, cell suspensions can be added into the entire dish containing the windows (Fig. 2A). The cell growth on the windows in this case can be easily monitored using an inverted microscope. If severe cell aggregation occurs, the subsequent treatment can be stopped. As windows are affixed at the bottom of the culture dish with four sides sealed, no cells will migrate and grow on the back side of the window; in addition, this prevents windows from floating around and bumping into each other, which may otherwise cause unnecessary breakage. Another approach is to place the window at the bottom of the culture dish without any affixation (Fig. 2B), and then drop cell suspensions onto the top of the window area only. After cell attachment is well established (roughly 1–2 h after seeding), more media are added to cover the entire culture well. However, windows under this setting might float due to surface tension from the added media and have to be pressed down into the media, thus increasing the chance to have cells detached or windows broken. If immersing the entire window in a culture vessel is not an option due to limited reagent availability, windows can be placed on a sterilized silicone mat in a culture dish (Fig. 2C), after which we dropped 38 L of cell suspension to cover the flat surface of a 5 × 5 mm Si3N4 window while also placing some droplets of cell‐free media or water nearby to maintain humidity. One of the disadvantages of growing cells this way is evaporation; there is about 5–10% water loss within 24 h period. For samples that are sensitive to concentration of ions in media this approach might not be a good option. Another disadvantage is that the cell growth and density on the window cannot be directly observed due to the nontransparent silicon mat. A slight improvement on this method is to use parafilm‐wrapped glass slides instead of a silicon mat. As shown in Figure 2(D), two glass slides are stacked together to provide an elevated surface, then wrapped with parafilm and placed in a culture dish containing, for added moisture, sterilized water at the bottom. Although parafilm is not 100% transparent, cells can be still visualized at lower magnification under this setting.
Figure 2.

Different settings for growing adherent mammalian cells on Si3N4 windows. Si3N4 windows were placed (A) at the bottom of culture dish with all four corners and sides affixed by scotch tape; (B) at the bottom of culture dish without any affixation; (C) on a silicon mat in a Petri dish or (D) on a stack of two glass slides wrapped with parafilm and placed in a culture dish. After sterilization of the window by UV light, 3 mL of cell suspensions were seeded onto the entire cell culture dish for setting A. For settings B, C and D, only a drop (∼38 L) of cell suspension was added onto each window. After cell attachment occurred, 3 mL of fresh culture medium was added to the entire well in setting B, but not in C or D. Cells were allowed to grow and reach appropriate confluence with normal medium change for settings A and B, and only droplet of medium changes for settings C and D. The droplets surrounding the windows in C and D are cell‐free culture media to reduce evaporation.
All cell samples in this study were grown using the ‘Scotch tape’ approach. Although it is tedious to affix the windows to the bottom of the culture dish, this approach seems to have less chance to break the window and causes less cell aggregation. It might be an easier method for first‐time users of fragile Si3N4 windows for growing cells.
Chemical fixation versus cryoimmobilization
The primary objective of the present study is to compare plunge‐freezing‐based cryoimmobilization against aldehyde‐based chemical fixation. Each comparison was repeated twice with similar results. One of such comparisons is shown in Figure 3. All cell samples were passaged at the same time and grown under the same conditions for the same amount of time. The windows were then randomly selected either for plunge freezing followed by freeze drying (PFFD), or for fixation by different chemical fixatives (2% GA, 4%PFA, or 3% PFA plus 1.5% GA, respectively) followed by TG buffer rinse and air drying. Areas containing multiple well‐separated and triangular cells (most likely excluding cells undergoing mitosis) from each treatment were selected and raster‐scanned at the APS 2‐ID‐E x‐ray microprobe. Pixel‐by‐pixel full spectral analysis was analysed to yield elemental concentration images as shown in Figure 3, panel A. The maximum and minimum concentrations of the same elements were adjusted to be the same values across differently prepared samples. Thus, the colour intensities roughly reflect relative abundance of the same element in these dehydrated samples. The content of P, S, Cl, K, Ca, Fe, Cu and Zn in the entire cell [in femtograms per cell (fg cell–1)] was obtained by ROI analysis, and then averaged for all cells in the same sample and plotted as fractions to the average elemental values in PFFD samples (Fig. 3, panel B). In other words, a value of 1 means that the S content in 4% PFA‐fixed cells is equal to that of plunge‐frozen, freeze‐dried PFFD cells.
Figure 3.
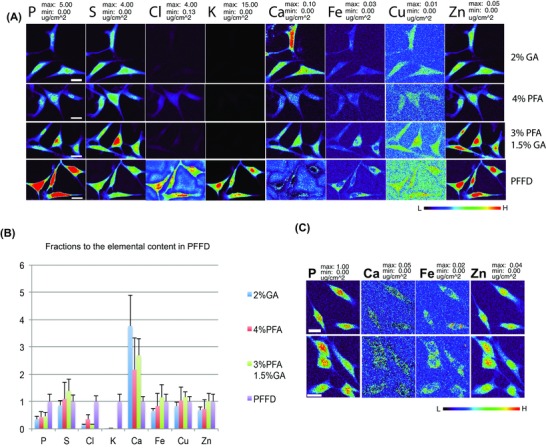
Side‐by‐side comparisons of XFM elemental maps of NIH/3T3 cell samples prepared by different fixation procedures. Panel A: Cells grown on Si3N4 windows were fixed by 2% GA (panel A, 1st row), 4% PFA (panel A, 2nd row), 3% PFA supplemented with 1.5% GA (panel A, 3rd row) or by plunge freezing followed by freeze drying (panel A, 4th row, PFFD). Chemically fixed cells were rinsed with TG buffer and air‐dried. Cells were scanned by XFM microprobe at 2‐ID‐E beamline station. XFM elemental images for each element are presented with the same maximum and minimum values in μg cm–2. Panel B: The total content of P, S, Cl, K, Ca, Fe, Cu and Zn, corresponding to cells shown in panel A, were obtained by ROI analysis and then averaged from all cells in the same sample and calculated as fractions to average elemental content of PFFD sample. Panel C: Two independently grown cell batches were plunge frozen and scanned at 100 K temperature as maintained by a cryojet in the fluorescence microprobe at 2‐ID‐D beamline station (Jin et al., 2015). XFM elemental images for each element are presented with the same maximum and minimum values in μg cm–2. Scale bars: 20 m. Colour scale in false colours spans from low (black) to high (red).
The elemental maps suggested that all fixation methods preserve similar overall cell morphology. Cell‐to‐cell variation of cell size and elemental content was noted both within and across different samples, most likely resulting from unsynchronized cell growth and different physiological conditions of cells at the moment of chemical fixation or freezing. The most striking difference was the severe loss of K and Cl in chemically fixed cells compared to PFFD sample, as indicated by much weaker visual signal in Figure 3, panel A. Only about 28% of Cl and 0.7% of K remained in 4% PFA‐fixed cells, and about 16% of Cl and 0.3 % of K remained in the samples fixed by 2% GTA or 3% PFA plus 1.5% GTA when compared to PFFD samples (Fig. 3, panel B). Dramatic loss and relocation of diffusible ions from their native sites have been reported in chemically fixed biological samples (Stika et al., 1980; Chandra & Morrison, 1992; Matsuyama et al., 2008; Matsuyama et al., 2010). The potassium gradient across cell membranes in living cells is upheld with active ion pumping and ion channels. Exposure to crosslinking chemical fixatives denatures the proteins acting as ion channels and prevents them from keeping the gradient of Cl and K. The longer the cells are incubated in aqueous buffer, the more ions are exchanged between intracellular and extracellular space. This is the most plausible explanation on why there is slightly more Cl and K in cells fixed for a shorter time (20 min) by 4% PFA than cells fixed for a longer time (60 min) in the other two fixatives. In the present study, cells were immediately rinsed and then air‐dried after chemical fixation. If cells were to be kept in aqueous buffer for a longer time and/or washed more extensively after chemical fixation (as required for most postprocessing procedures such as immunochemical labelling), the content of Cl and K would continue to decrease and become undetectable as reported elsewhere (Somlyo et al., 1985; Zierold, 1991; Chwiej et al., 2005; Matsuyama et al., 2008; Matsuyama et al., 2010). In contrast, Cl and K were well preserved in plunge frozen and freeze dried PFFD sample (Fig. 3, panel A, row 4). Both of these highly diffusible elements were homogenously distributed throughout the entire cell as reported previously (Matsuyama et al., 2010).
Another apparent difference was the elevated level of total Ca in chemically fixed cells compared to PFFD cells, especially in cells fixed by GA‐containing solutions. In the batch shown in Figure 3, there was about 54 fg Ca per cell in PFFD samples, but this value increased to about 200 fg cell–1 in 2% GA‐fixed cells, 150 fg cell–1 in cells fixed by 3%PFA supplemented with 1.5% GA, and 120 fg cell–1 in 4% PFA‐fixed cells. In another replica of the same experiment, the increase of Ca content was even more striking, reaching 9‐fold increase in 2% GA‐fixed cells and 2‐fold increase in 4% PFA‐fixed cells. In addition, the distribution of Ca in chemically fixed cells seemed to be different from PFFD prepared cells, with higher Ca signal in the nucleus than in the cytoplasm. To understand which method provides more faithful preservation of Ca content and distribution, we compared them to cells that were plunge frozen followed by imaging in the frozen hydrated state at cryogenic temperature, which we assume preserves elemental distribution as close as possible to the native state. To simplify the comparison, only maps of S, Ca, Fe and Zn are shown in Figure 3, panel C. Average total content of Ca in these cells is around 59 fg per cell with standard deviation of less than 2 fg, which is very close to that calculated from PFFD cells. Thus, we believe the elevated Ca content in chemically fixed cells might reflect some artificial Ca loading from the extracellular space.
The distribution of Ca has been studied in various adherent cells by different techniques. Chandra et al. have used fluorescence light microscopy and SIMS to investigate the distribution of free and total Ca ions in NIH/3T3 and other cell lines. They suggested that Ca ratio in the cytoplasm to the nucleus of NIH/3T3 cells is around 2.9–4.1 (Chandra et al., 1989). In our study, this Ca ratio in frozen hydrated cells (cells in Fig. 3, panel C) is around 3.6, close to the reported value. In contrast, this ratio in PFFD samples is around 6, which might indicate that some Ca ions were redistributed from the nucleus to the cytoplasm during freeze drying. Considering that about one third of intracellular Ca exists as free ions and the nucleus is the cellular region that is most likely to be suboptimally cryofixed, Ca redistribution is plausible. If sizeable ice crystals are formed in the nuclear membrane during plunge freezing, it could lead to the redistribution of Ca or other free ions (Chandra et al., 1989). Of course, an ideal study would employ cells synchronized to be the same phase of the cell cycle, since Ca can be redistributed at various points in the cycle.
In addition to the difference in the content and distribution of K, Cl and Ca, chemically fixed cells also showed lower content of P: only about 35–45% of that in PFFD cells. In contrast, most other tightly bound ions such as S, Fe, Cu and Zn were retained at least at the 65% level compared to PFFD cells, with the combination of both aldehydes leading to a level equal to or greater than that in PFFD cells. Within cells, P is mostly present as a component of proteins, nucleic acids and some small molecules such as ATP. Although aldehyde‐based chemical fixatives are very efficient at crosslinking protein molecules, it is not an efficient fixative for nucleic acids, which could lead to redistribution or loss of some phosphorus in particular in the nucleic acid‐rich nucleus.
The distribution of Fe ions has been reported to be mainly localized in the nuclei in chemically fixed cells, compared to the perinuclear region in plunge frozen cells (Matsuyama et al., 2010). In our study, both chemically fixed and PFFD cells had perinuclear distribution of Fe similar to frozen hydrated cells as shown in Figure 3(C), indicating that chemical fixation procedures used in this study provide a good preservation of Fe distribution. The distribution of Cu was nonhomogeneous but similar in all samples, with more in the cytoplasm than in the nucleus. The distribution of Cu has been reported to follow the distribution of Zn in mitotic cells (McRae et al., 2013), but this was not observed in our study, possibly due to the fact that cells imaged in this study were most likely at interphase based on their triangular shape. The concentration of Zn in cryopreserved nuclei was apparently higher than that in the cytoplasm, so that the Zn concentration map nicely defined the outline of the nucleus. However, the Zn outline of the nucleus in chemically fixed cells was not as distinctive as in PFFD sample, with GA‐fixed cells being the worst and the cells fixed by the combination of both aldehydes the best. Considering that the GA molecule is larger than PFA and takes a longer time to reach the nucleus, this finding is not surprising. Limited by our spatial resolution and the lack of correlative studies to differentiate cell cycle stages, we were unable to conduct any sophisticated colocalization analysis among different elements as previously reported (McRae et al., 2013).
We conclude that plunge freezing‐based cryoimmobilization followed by freeze drying (PFFD) generally provides better preservation of most biologically relevant elements than chemical fixation when cells have to be imaged dehydrated at room temperature. PFFD should be the method of choice whenever possible, especially when the elements of interest are diffusible, or associated with molecules that are difficult to fix chemically; this applies to elements such as K, Cl, Ca and P. However, cryopresrevation followed by freeze drying is not free of artefacts. Various factors such as prolonged rinse before plunge freezing, or insufficient or overaggressive blotting may interfere with cell integrity or cause suboptimal vitrification. Along with many artefacts associated with the freeze drying process, the fidelity of chemical composition might be still compromised to a certain extent. When chemical fixation is chosen, cells should be fixed as promptly as possible and immediately dried upon the completion of fixation. The combination of both aldehydes seemed to provide relatively better preservation on the content and distribution of P, S, Fe, Cu and Zn than either fixative alone.
Cell rinse before plunge freezing
When adherent monolayer cells were used for elemental analysis, a quick wash using buffers containing no high Z (atomic number) elements is generally needed before plunge freezing. This wash is intended to remove most extracellular ions deposited on and around the cell surface, yet to have minimum impact on intracellular elemental composition. Ammonium acetate buffer has been commonly used to rinse cells before plunge freezing. It has neutral pH and can be easily adjusted to have osmolality nearly equivalent to culture media. This buffer sublimes during freeze drying, leaving no extra salt behind. Tris‐based buffer is another one commonly used. It contains only low Z elements and causes minimal interference with the measurement of fluorescence emitted by most metal ions. In this study, we attempted to compare whether cells rinsed with ammonium acetate or Tris‐based buffers before plunge freezing show any difference in the content and intracellular distribution of biologically relevant elements compared to cells rinsed with fresh culture media or D‐PBS buffer.
After two brief rinses with the given buffer or medium, cells were plunge frozen and subsequently freeze dried and imaged by XFM at room temperature. As expected, the culture medium washed cells had higher extracellular signals of P, S, Cl, K and Ca than the cells washed with ammonium acetate or Tris buffers (Fig. 4). The higher extracellular background of these elements is due to their relatively abundance as media components. D‐PBS washed cells have higher extracellular background of P, Cl and K, consistent with the fact that they are the three main constituents of this buffer. There was no significant extracellular background in ammonium acetate and Tris buffer washed cells.
Figure 4.
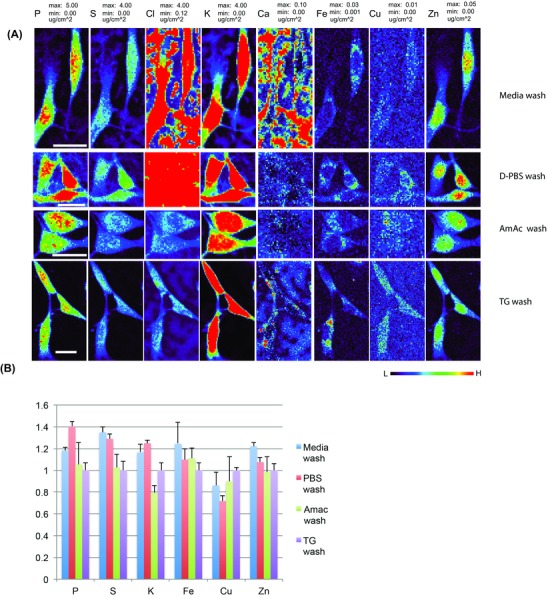
Side‐by‐side comparisons of XFM elemental maps of adherent mouse NIH/3T3 cells rinsed with different buffers before plunge freezing and freeze drying. Cells grown on Si3N4 windows were rinsed with fresh media (media wash, 1st row), D‐PBS (D‐PBS wash, 2nd row), 100 mM ammonium acetate (Amac wash, 3rd row) or Tris‐glucose buffer (TG wash, 4th row), and then plunge frozen and freeze dried. Cells were scanned by XFM microprobe at the 2‐ID‐E beamline station. XFM elemental images for each element are presented with the same maximum and minimum values in μg cm–2 (panel A). The total content of P, S, K, Fe, Cu and Zn, corresponding to cells in panel A, were averaged for all cells in the same sample and calculated as fractions to elemental content in cells washed with TG buffer (panel B). Scale bar: 20 m. Colour scale in false colours spans from low (black) to high (red).
When the total elemental contents (excluding Cl and Ca) were calculated and plotted as fractions to that obtained with TG buffer washed cells (Fig. 4, panel B) the media washed cells had about 40% more S and D‐PBS washed cells had about 40% more P. The contents of K, Fe Cu, and Zn in media or PBS washed cells were within 100% ± 30% of values for TG buffer washed cells. Considering wide cell‐to‐cell variations, none of these differences were considered to be statistically significant. In ammonium acetate washed cells, the contents for all elements showed great consistency with TG buffer washed cells.
The distribution of these biologically relevant elements within cells doesn't seem to be affected by the wash buffer used, as we observed similar elemental distributions to what we saw in PFFD samples (Fig. 3). It is worth noting that the distribution of Ca, which as revealed in PFFD samples (Fig. 3) was predominantly localized in the cytoplasm with little presence in the nucleus, can be still identified in media washed cells, regardless of almost saturated Ca signals in the extracellular space and some cell surface areas. We would also like to point out that mouse fibroblast cells used for this study are quite robust. Neither ammonium acetate nor TG buffer wash led to significant elemental loss, if any, compared to media or PBS washed cells. However, for more delicate cell lines or primary cells, the decision to wash or not wash them before freezing and selection of wash buffers needs to be experimentally established, taking many factors into account. For example, when ammonium acetate buffer was used to wash red blood cells before plunge freezing, much more significant loss of iron was observed than when D‐PBS wash was used (data not shown). Although the media wash or even no wash could cause higher extracellular background for some elements, such preparation approaches have least impact on cell physiological state, which might aid in revealing the contents of some other elements, such as Fe, Cu and Zn, as close as possible to the native state.
Air drying versus freeze drying and ethanol drying
When room temperature scanning of chemically fixed cells is planned, fixation is only the first step in this process. Regardless of how cells are fixed, all cellular water needs to be removed before the sample can be studied using XFM. Dehydration not only reduces beam damage (LeFurgey & Ingram, 1990), but it also ensures that no motion artefacts are generated during scanning. For chemically fixed cells, air drying or graded ethanol drying has been followed (Wagner et al., 2005; Yang et al., 2005; McRae et al., 2006; Finney et al., 2007; Corezzi et al., 2009; Matsuyama et al., 2009; Wolford et al., 2010). In this study, we attempted to compare different dehydration processes side by side and evaluate how such processes affect elemental contents and distributions in chemically fixed adherent cells. NIH/3T3 cells grown from the same batch under the same conditions were first fixed by 4% PFA and then subjected either to air drying, or to ethanol dehydration, or to freeze drying. To subject previously chemically fixed cells to freeze drying, they were first plunge frozen and then freeze dried.
As shown in Figure 5, ethanol dehydrated cells (Fig. 5, row 1), compared to the other two drying processes, had the lowest levels of all analysed elements excluding K. The total contents of S, Ca, Fe and Cu were no more than 25% of that in the air‐dried cells (Fig. 5, panel B). It is possible that some of these ions are lost because ethanol caused protein denaturation or membrane solubilization (Bancroft & Stevens, 1996; Malm et al., 2009). An elevated level of K ions has been also observed in ethanol dehydrated sample by time‐of‐flight secondary ion mass spectrometry analysis (Malm et al., 2009), which was explained by lower efficiency for K ions to be washed away in ethanol dehydrated sample than in freeze dried sample.
Figure 5.
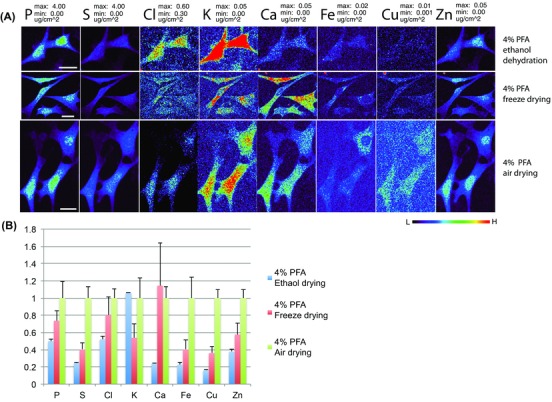
Side‐by‐side comparisons of XFM elemental maps of 4% PFA‐fixed cells subjected to different dehydration and drying procedures. NIH/3T3 cells were fixed by 4% PFA and then subjected to graded ethanol dehydration (4% PFA, ethanol dehydration); plunge freezing and freeze drying (4% PFA, freeze drying); or room temperature air drying (4% PFA, air drying). Cells were scanned by XFM microprobe at 2‐ID‐E beamline station at APS. XFM elemental images for each element are presented with the same maximum and minimum values in μg cm–2 (panel A). The total content of P, S, Cl, K, Ca, Fe, Cu and Zn, corresponding to cells in panel A, were averaged and calculated as fractions to that of air‐dried cells (panel B). Scale bar: 20 m. colour scale in false colours spans from low (black) to high (red).
The total content of all analysed elements in freeze dried samples (Fig. 5, row 2) was lower than in air‐dried samples, with the exception of Ca. This suggests that once the cell membrane has been made porous by chemical fixation, elemental loss through the compromised membrane is possible during plunge freezing and/or freeze drying (Fig. 5, panels A and B). Formation of small holes in cell membranes, ranging from 10 nm to a few micrometres, had been reported in chemically fixed cells but not in cryofixed cells (Malm et al., 2009). Within the limits of our spatial resolution, we could not judge whether such holes were also present in our chemically fixed samples, or whether these holes might be responsible for further elemental loss during plunge freezing and freeze drying. Alternatively, this particular sample may just have had more membrane damage than most chemically fixed samples, so it had preexisting higher content of Ca and lower contents of all other elements before plunge freezing and freeze drying. Thus, the reduced level of these elements was possibly not caused by plunge freezing and/or freeze drying. Although the contents of most elements are lower in freeze dried sample, their distributions seemed to be similar to that of air‐dried samples under current spatial resolution.
In conclusion, graded ethanol dehydration of chemically fixed samples should be avoided to prevent further elemental redistribution. Subjecting chemically fixed cells to plunge freezing followed by freeze drying might not provide added benefits on the preservation of elemental contents and distributions than air drying.
Conclusions
In this work adherent mammalian cells grown on Si3N4 windows have been used as biological specimens, and different sample preparation approaches were employed for x‐ray imaging. The importance of cryoimmobilization and freeze drying has been well documented in EPXMA‐, SIMS and PIXE‐based elemental imaging analysis. Cryopreparation becomes even more necessary when highly diffusible ions, such as Cl, K and Ca, are under study (Chandra & Morrison, 1992; Makjanic & Watt, 1999; Jallot et al., 2004). Some SIMS‐based studies have suggested that only cryogenically prepared cells show well preserved intracellular ionic composition close to native state. The chemical integrity is apparently disrupted in chemically fixed or poorly cryofixed cells, which is often signified by apparently reduced intracellular K:Na ratios and an excessive intracellular loading of calcium (Chandra et al., 1989; Chandra, 2001; Arlinghaus et al., 2006). Cells prepared in this way might well yield visually pleasing elemental concentration maps, but the actual content of those maps might be quite unrepresentative of the living cell prior to fixation.
Cryogenic sample preparation has not yet emerged as the primary means to prepare adherent mammalian cells for XFM studies. Of approximately two dozen recent publications on adherent mammalian cells analysed by XFM, half or so used plunge freezing to prepare their biological specimens (Bohic et al., 2001; Ilinski et al., 2003; Harris et al., 2005; Bacquart et al., 2007; Matsuyama et al., 2010; Kosior et al., 2012; Yuan et al., 2013; Chen et al., 2014; Kashiv et al., 2016). Very recently, high‐pressure freezing followed by freeze substitution and sectioning has been also applied to adherent mammalian cells in order to reveal metal distribution at single organelle level (Kashiv et al., 2016). Nevertheless, the other dozen studies employed chemical fixation either by aldehyde‐based chemicals or organic solvents such as ethanol or methanol (Paunesku et al., 2003; Wagner et al., 2005; Yang et al., 2005; McRae et al., 2006; Finney et al., 2007; Corezzi et al., 2009; Matsuyama et al., 2009; Wolford et al., 2010; Marmorato et al., 2011; Weekley et al., 2011; Marvin et al., 2012; McRae et al., 2013). Although chemical fixation had to be used in some of these studies, we think that lack of extensive comparison between the two methods might also have influenced some researchers to choose chemical fixation based on its convenience. To our knowledge, very few studies had compared the elemental maps of plunge frozen cells to maps of chemically fixed cells in XFM studies (Matsuyama et al., 2010, Perrin et al., 2015). Matsuyama and others scanned the cryoimmobilized cells in the frozen hydrated state, and compared them to the chemically fixed and dehydrated cells scanned in a separated experiment. In our work, we conducted a side‐by‐side comparison between chemically fixed cells and plunge frozen cells, with both samples subsequently dehydrated and scanned by XFM under ambient temperature, with results similar to those reported by Perrin et al., 2015. This comparison revealed significant loss of K and Cl in chemically fixed cells, elevated level and altered distribution of Ca, loss of P to a certain extent and some possible difference in Zn distribution, compared to plunge frozen and freeze dried cells. By choosing different wash buffers to rinse cells before plunge freezing, we conclude that adherent cells might be rinsed with fresh media before plunge freezing, if Ca or other diffusible ions are not the elements of interest. Otherwise, either Tris‐based or ammonium acetate buffers can be used. If chemical fixation has to be used because of additional sample manipulations such as immunocytochemistry, incubation in the aqueous buffers should be kept to the minimum and the addition of small amounts of GA into PFA solution might aid the preservation of elemental composition. Comparison of different dehydration approaches has shown that simple air drying of formaldehyde‐fixed samples provides better preservation than graded ethanol dehydration. This study (as well as comparable work in the literature) shows that cell‐to‐cell variation within the same sample can be extensive with respect to elemental content of single cell. Improved comparisons can be obtained if a more statistically significant number of cells can be imaged. This points the way to rapid scanning approaches (Lombi et al., 2011), and software tools that would enable comparative statistical analyses of many cells at one time (Ward et al., 2013; Wang et al., 2014).
Acknowledgements
We thank Junjing Deng, Young Pyo Hong, Reiner Bleher, and Eric Roth of Northwestern University for helpful discussions. We thank the National Institutes of Health for support under grant R01 GM104530. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract DE‐AC02‐06CH11357.
References
- Andrews, S.B. , Leapman, R.D. , Landis, D.M.D. & Reese, T.S. (1987) Distribution of calcium and potassium in presynaptic nerve terminals from cerebellar cortex. Proc. Nat. Acad. Sci. 84(6), 1713–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S.B. , Leapman, R.D. , Landis, D.M.D. & Reese, T.S. (1988) Activity‐dependent accumulation of calcium in Purkinje cell dendritic spines. Proc. Nat. Acad. Sci. 85(5), 1682–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlinghaus, H.F. , Kriegeskotte, C. , Fartmann, M. , Wittig, A. , Sauerwein, W. & Lipinsky, D. (2006) Mass spectrometric characterization of elements and molecules in cell cultures and tissues. Appl. Surf. Sci. 252(19), 6941–6948. [Google Scholar]
- Arora, H.C. , Jensen, M.P. , Yuan, Y. , Wu, A. , Vogt, S. , Paunesku, T . & Woloschak, G.E. (2012) Nanocarriers enhance Doxorubicin uptake in drug‐resistant ovarian cancer cells. Cancer Res. 72(3), 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacquart, T. , Deves, G. , Carmona, A. , Tucoulou, R. , Bohic, S . & Ortega, R. (2007) Subcellular speciation analysis of trace element oxidation states using synchrotron radiation micro‐X‐ray absorption near‐edge structure. Analytic. Chem. 79(19), 7353–7359. [DOI] [PubMed] [Google Scholar]
- Bancroft, L.W . & Stevens, A. (1996) Theory and Practice of Histological Techniques. New York, Churchill Livingston. [Google Scholar]
- Bohic, S. , Simionovici, A. , Snigirev, A. , Ortega, R. , Devès, G. , Heymann, D . & Schroer, C.G. (2001) Synchrotron hard x‐ray microprobe: fluorescence imaging of single cells. Appl. Phys. Lett. 78(22), 3544–3546. [Google Scholar]
- Bourassa, M.W . & Miller, L.M. (2012) Metal imaging in neurodegenerative diseases. Metallomics 4(8), 721–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, E.A. , Rayner, B.S. , McLeod, A.I. et al (2010) Silicon nitride as a versatile growth substrate for microspectroscopic imaging and mapping of individual cells. Mol Biosyst. 6(7), 1316–1322. [DOI] [PubMed] [Google Scholar]
- Chandra, S. (2001) Studies of cell division (mitosis and cytokinesis) by dynamic secondary ion mass spectrometry ion microscopy: LLC‐PK1 epithelial cells as a model for subcellular isotopic imaging. J. Microsc. 204(Pt 2), 150–165. [DOI] [PubMed] [Google Scholar]
- Chandra, S. , Gross, D. , Ling, Y.C . & Morrison, G.H. (1989) Quantitative imaging of free and total intracellular calcium in cultured cells. Proc. Nat. Acad. Sci. 86(6), 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra, S . & Morrison, G.H. (1992) Sample preparation of animal‐tissues and cell‐cultures for secondary ion mass‐spectrometry (SIMS) microscopy. Biol. Cell 74(1), 31–42. [DOI] [PubMed] [Google Scholar]
- Chen, K.G. , Valencia, J.C. , Lai, B. et al (2006) Melanosomal sequestration of cytotoxic drugs contributes to the intractability of malignant melanomas. Proc. Nat. Acad. Sci. 103(26), 9903–9907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Deng, J. , Yuan, Y. et al (2014) The Bionanoprobe: hard X‐ray fluorescence nanoprobe with cryogenic capabilities. J. Synchrot. Rad. 21(Pt 1), 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwiej, J. , Szczerbowska‐Boruchowska, M. , Lankosz, M. , Wojcik, S. , Falkenberg, G. , Stegowski, Z . & Setkowicz, Z. (2005) Preparation of tissue samples for X‐ray fluorescence microscopy. Spectr. Acta B. 60(12), 1531–1537. [Google Scholar]
- Corde, S. , Biston, M.C. , Elleaume, H. et al (2002) Lack of cell death enhancement after irradiation with monochromatic synchrotron X rays at the K‐shell edge of platinum incorporated in living SQ20B human cells as cis‐diamminedichloroplatinum (II). Rad. Res. 158(6), 763–770. [DOI] [PubMed] [Google Scholar]
- Corezzi, S. , Urbanelli, L. , Cloetens, P. , Emiliani, C. , Helfen, L. , Bohic, S. , Elisei, F . & Fioretto, D. (2009) Synchrotron‐based X‐ray fluorescence imaging of human cells labeled with CdSe quantum dots. Analytic. Biochem. 388(1), 33–39. [DOI] [PubMed] [Google Scholar]
- Dillon, C.T. , Lay, P.A. , Kennedy, B.J. et al (2002) Hard X‐ray microprobe studies of chromium(VI)‐treated V79 Chinese hamster lung cells: intracellular mapping of the biotransformation products of a chromium carcinogen. J. Biol. Inorg. Chem. 7(6), 640–645. [DOI] [PubMed] [Google Scholar]
- Dubochet, J. , Lepault, J. , Freeman, R. , Berriman, J.A . & Homo, J.C. (1982) Electron‐microscopy of frozen water and aqueous‐solutions. J. Microsc. 128(3), 219–237. [Google Scholar]
- Fahrni, C.J. (2007) Biological applications of X‐ray fluorescence microscopy: exploring the subcellular topography and speciation of transition metals. Curr. Opin. Chem. Biol. 11(2), 121–127. [DOI] [PubMed] [Google Scholar]
- Farquharson, M.J. , Geraki, K. , Falkenberg, G. , Leek, R . & Harris, A. (2007) The localisation and micro‐mapping of copper and other trace elements in breast tumours using a synchrotron micro‐XRF system. Appl. Rad. Isotop. 65(2), 183–188. [DOI] [PubMed] [Google Scholar]
- Finney, L. , Mandava, S. , Ursos, L. et al (2007) X‐ray fluorescence microscopy reveals large‐scale relocalization and extracellular translocation of cellular copper during angiogenesis. Proc. Nat. Acad. Sci. 104(7), 2247–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney, L.A . & Jin, Q. (2015) Preparing adherent cells for X‐ray fluorescence imaging by chemical fixation. J. Vis. Exper. 97, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkey, J . & Staehein, L.A. (1986) Advances in ultrarapid freezing for the preservation of cellular ultrastructure. J. Electr. Microsc. Tech. 3, 177–210. [Google Scholar]
- Grubman, A. , James, S.A. , James, J. et al (2014) X‐ray fluorescence imaging reveals subcellular biometal disturbances in a childhood neurodegenerative disorder. Chem. Sci. 5(6), 2503–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett, M.J. , McQuillan, J.A. , El‐Assaad, F. et al (2011) Chemical alterations to murine brain tissue induced by formalin fixation: implications for biospectroscopic imaging and mapping studies of disease pathogenesis. Analyst 136(14), 2941–2952. [DOI] [PubMed] [Google Scholar]
- Hagen, C. , Guttmann, P. , Klupp, B. , Werner, S. , Rehbein, S. , Mettenleiter, T.C. , Schneider, G . & Grünewald, K. (2012) Correlative VIS‐fluorescence and soft X‐ray cryo‐microscopy/tomography of adherent cells. J. Struct. Biol. 177(2), 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, H.H. , Levina, A. , Dillon, C.T. , Mulyani, I. , Lai, B. , Cai, Z . & Lay, P.A. (2005) Time‐dependent uptake, distribution and biotransformation of chromium(VI) in individual and bulk human lung cells: application of synchrotron radiation techniques. J. Biol. Inorg. Chem. 10(2), 105–118. [DOI] [PubMed] [Google Scholar]
- Hawes, P.C. (2015) Preparation of cultured cells using high‐pressure freezing and freeze substitution for subsequent 2D or 3D visualization in the transmission electron microscope. Coronaviruses 1282(Chapter 23), 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilinski, P. , Lai, B. , Cai, Z. et al (2003) The direct mapping of the uptake of platinum anticancer agents in individual human ovarian adenocarcinoma cells using a hard X‐ray microprobe. Cancer Res. 63, 1776–1779. [PubMed] [Google Scholar]
- Jallot, E. , Laquerriere, P. , Grandjean‐Laquerriere, A. , Kilian, L . & Laurent‐Maquin, D. (2004) Characterization of intra‐cellular ionic concentrations of monocytes in contact with bioactive glasses and hydroxyapatite particles. A TEM cryo‐X‐ray analysis of diffusible ions. Instr. Sci. Technol. 32(5), 537–544. [Google Scholar]
- Jin, Q. , Vogt, S. , Lai, B. et al (2015) Ultraviolet germicidal irradiation and its effects on elemental distributions in mouse embryonic fibroblast cells in X‐ray fluorescence microanalysis. PLoS ONE 10(2), e0117437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiv, Y. , Austin, J.R., 2nd , Lai, B. , Rose, V. , Vogt, S . & El‐Muayed, M. (2016) Imaging trace element distributions in single organelles and subcellular features. Sci. Rep. 6, 21437, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemner, K.M. , Kelly, S.D. , Lai, B. et al (2004) Elemental and redox analysis of single bacterial cells by x‐ray microbeam analysis. Science 306(5696), 686–687. [DOI] [PubMed] [Google Scholar]
- Kosior, E. , Bohic, S. , Suhonen, H. et al (2012) Combined use of hard X‐ray phase contrast imaging and X‐ray fluorescence microscopy for sub‐cellular metal quantification. J. Struct. Biol. 177(2), 239–247. [DOI] [PubMed] [Google Scholar]
- LeFurgey, A . & Ingram, P. (1990) Calcium measurements with electron probe X‐ray and electron energy loss analysis. Environ. Health Perspect. 84, 57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskovjan, A.C. , Kretlow, A. , Lanzirotti, A. , Barrea, R. , Vogt, S . & Miller, L.M. (2011) Increased brain iron coincides with early plaque formation in a mouse model of Alzheimer's disease. NeuroImage 55(1), 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobinski, R. , Moulin, C . & Ortega, R. (2006) Imaging and speciation of trace elements in biological environment. Biochimie 88(11), 1591–1604. [DOI] [PubMed] [Google Scholar]
- Lombi, E. , de Jonge, M.D. , Donner, E. , Ryan, C.G . & Paterson, D. (2011) Trends in hard X‐ray fluorescence mapping: environmental applications in the age of fast detectors. Analytic. Bioanalytic. Chem. 400, 1637–1644. [DOI] [PubMed] [Google Scholar]
- Majumdar, S. , Peralta‐Videa, J.R. , Castillo‐Michel, H. , Hong, J. , Rico, C.M . & Gardea‐Torresdey, J.L. (2012) Applications of synchrotron μ‐XRF to study the distribution of biologically important elements in different environmental matrices: a review. Analytic. Chim. Acta. 755, 1–16. [DOI] [PubMed] [Google Scholar]
- Makjanic, J . & Watt, F. (1999) Nuclear microscopy in Alzheimer's disease. Nucl. Instr. Meth. Phys. Res. Sect B. 150(1‐4), 167–172. [Google Scholar]
- Malm, J. , Giannaras, D. , Riehle, M.O. , Gadegaard, N . & Sjovall, P. (2009) Fixation and drying protocols for the preparation of cell samples for time‐of‐flight secondary ion mass spectrometry analysis. Analytic. Chem. 81(17), 7197–7205. [DOI] [PubMed] [Google Scholar]
- Marmorato, P. , Ceccone, G. , Gianoncelli, A. et al (2011) Cellular distribution and degradation of cobalt ferrite nanoparticles in Balb/3T3 mouse fibroblasts. Toxicol. Lett. 207(2), 128–136. [DOI] [PubMed] [Google Scholar]
- Marvin, R.G. , Wolford, J.L. , Kidd, M.J. et al (2012) Fluxes in “free” and total zinc are essential for progression of intraerythrocytic stages of Plasmodium falciparum. Chem. Biol. 19(6), 731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama, M. , Funao, K. , Kawahito, Y. et al (2008) Study of cysteinyl leukotriene‐1 receptor in rat renal ischemia‐reperfusion injury. Transplant. Proc. 40(7), 2149–2151. [DOI] [PubMed] [Google Scholar]
- Matsuyama, S. , Shimura, M. , Fujii, M. et al (2010) Elemental mapping of frozen‐hydrated cells with cryo‐scanning X‐ray fluorescence microscopy. X‐Ray Spectrometr. 39(4), 260–266. [Google Scholar]
- Matsuyama, S. , Shimura, M. , Mimura, H. et al (2009) Trace element mapping of a single cell using a hard x‐ray nanobeam focused by a Kirkpatrick‐Baez mirror system. X‐Ray Spectrometr. 38(2), 89–94. [Google Scholar]
- McDonald, K.L. (2014) Rapid embedding methods into epoxy and LR White resins for morphological and immunological analysis of cryofixed biological specimens. Microsc. Microanal. 20(1), 152–163. [DOI] [PubMed] [Google Scholar]
- McRae, R. , Bagchi, P. , Sumalekshmy, S . & Fahrni, C.J. (2009) In situ imaging of metals in cells and tissues. Chem. Rev. 109(10), 4780–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae, R. , Lai, B . & Fahrni, C.J. (2013) Subcellular redistribution and mitotic inheritance of transition metals in proliferating mouse fibroblast cells. Metallomics 5(1), 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae, R. , Lai, B. , Vogt, S . & Fahrni, C.J. (2006) Correlative microXRF and optical immunofluorescence microscopy of adherent cells labeled with ultrasmall gold particles. J. Struct. Biol. 155(1), 22–29. [DOI] [PubMed] [Google Scholar]
- Monaghan, P. , Perusinghe, N . & Muller, M. (1998) High‐pressure freezing for immunocytochemistry. J. Microsc. 192(Pt 3), 248–258. [DOI] [PubMed] [Google Scholar]
- Moor, H. (1987) Theory and practice of high pressure freezing. Cryotechniques in Biological Electron Microscopy (ed. by Steinbrecht R. & Zierold K.), 175–191. Berlin, Springer‐Verlag. [Google Scholar]
- Muller, M . & Moor, H. (1984) Cryofixation of thick specimens by high pressure freezing. The science of Biological Secimen and Preparation. The Science of Biological Speicmen Preparation (ed. by Muller M., Becker R.P., Boyde A. & Wolosewick J.J.), pp. 131–138. Chicago, IL, AMF O'Hare. [Google Scholar]
- Nicolas, G. (1991) Advantages of fast‐freeze fixation followed by freeze‐substitution for the preservation of cell integrity. J. Electr. Microsc. Tech. 18(4), 395–405. [DOI] [PubMed] [Google Scholar]
- Ortega, R. , Cloetens, P. , Deves, G. , Carmona, A . & Bohic, S. (2007) Iron storage within dopamine neurovesicles revealed by chemical nano‐imaging. PLoS ONE 2(9), e925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega, R. , Devès, G . & Carmona, A. (2009) Bio‐metals imaging and speciation in cells using proton and synchrotron radiation X‐ray microspectroscopy. J. R. Soc. Interf. 6, S649–S658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paunesku, T. , Rajh, T. , Wiederrecht, G. et al (2003) Biology of TiO2‐oligonucleotide nanocomposites. Nat. Mater. 2(5), 343–346. [DOI] [PubMed] [Google Scholar]
- Paunesku, T. , Vogt, S. , Lai, B. et al (2007) Intracellular distribution of TiO2−DNA oligonucleotide nanoconjugates directed to nucleolus and mitochondria indicates sequence specificity. Nano Lett. 7(3), 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paunesku, T. , Vogt, S. , Maser, J. , Lai, B . & Woloschak, G. (2006) X‐ray fluorescence microprobe imaging in biology and medicine. J. Cell. Biochem. 99(6), 1489–1502. [DOI] [PubMed] [Google Scholar]
- Perrin, L. , Carmona, A. , Roudeau, S . & Ortega, R. (2015) Evaluation of sample preparation methods for single cell quantitative elemental imaging using proton or synchrotron radiation focused beams. J. Analytic. Atomic Spectrometr. 30(12), 2525–2532. [Google Scholar]
- Petibois, C. (2010) Imaging methods for elemental, chemical, molecular, and morphological analyses of single cells. Analytic. Bioanalytic. Chem. 397(6), 2051–2065. [DOI] [PubMed] [Google Scholar]
- Sartori, N . & Richter, K. (1993) Vitrifcation depth can be increased more than 10‐fold by high pressure freezing. J. Microsc. 172, 55–61. [Google Scholar]
- Saubermann, A.J. , Echlin, P. , Peters, P.D . & Beeuwkes, R. (1981) Application of scanning electron microscopy to x‐ray analysis of frozen‐hydrated sections. I. Specimen handling techniques. J. Cell Biol. 88(2), 257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saubermann, A.J . & Heyman, R.V. (1987) Quantitative digital x‐ray imaging using frozen hydrated and frozen dried tissue sections. J. Microsc. 146, 169–182. [DOI] [PubMed] [Google Scholar]
- Shuman, H. , Somlyo, A.V . & Somlyo, A.P. (1976) Quantitative electron probe microanalysis of biological thin sections: methods and validity. Ultramicroscopy 1(4), 317–339. [DOI] [PubMed] [Google Scholar]
- Sitte, H. , Edelmann, L . & Neumann, K. (1987) Cryofixation without pretreatment at ambient pressure. Cryotechniques in Biological Electron Microscopy (ed. by Steinbrecht R. & Zierold K.), pp. 87–113. Berlin, Springer‐Verlag. [Google Scholar]
- Somlyo, A.P. , Bond, M . & Somlyo, A.V. (1985) Calcium content of mitochondria and endoplasmic reticulum in liver frozen rapidly in vivo. Nature 314(6012), 622–625. [DOI] [PubMed] [Google Scholar]
- Somlyo, A.V. , Broderick, R. , Shuman, H. , Buhle Jr, E.L . & Somlyo, A.P. (1988) Atrial‐specific granules in situ have high calcium content, are acidic, and maintain anion gradients. Proc. Nat. Acad. Sci. 85(16), 6222–6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stika, K.M. , Bielat, K.L . & Morrison, G.H. (1980) Diffusible ion localization by ion microscopy: a comparison of chemically prepared and fast‐frozen, freeze‐dried, unfixed liver sections. J. Microsc. 118(4), 409–420. [DOI] [PubMed] [Google Scholar]
- Studer, D. , Humbel, B.M . & Chiquet, M. (2008) Electron microscopy of high pressure frozen samples: bridging the gap between cellular ultrastructure and atomic resolution. Histochem. Cell Biol. 130(5), 877–889. [DOI] [PubMed] [Google Scholar]
- Szczerbowska‐Boruchowska, M. (2008) X‐ray fluorescence spectrometry, an analytical tool in neurochemical research. X‐Ray Spectrometr. 37(1), 21–31. [Google Scholar]
- Twining, B.S. , Baines, S.B. , Fisher, N.S. , Maser, J. , Vogt, S. , Jacobsen, C. , Tovar‐Sanchez, A . & Sañudo‐Wilhelmy, S.A. (2003) Quantifying trace elements in individual aquatic protist cells with a synchrotron X‐ray fluorescence microprobe. Analytic. Chem. 75(15), 3806–3816. [DOI] [PubMed] [Google Scholar]
- Vogt, S. (2003) MAPS: a set of software tools for analysis and visualization of 3D X‐ray fluorescence data sets. J. Physiq. IV 104, 635–638. [Google Scholar]
- Vogt, S . & Ralle, M. (2013) Opportunities in multidimensional trace metal imaging: taking copper‐associated disease research to the next level. Analytic. Bioanalytic. Chem. 405(6), 1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, D. , Maser, J. , Lai, B. , Cai, Z. , Barry, C.E. , Honer zu Bentrup, K. , Russell, D.G . & Bermudez, L.E. (2005) Elemental analysis of mycobacterium avium‐, mycobacterium tuberculosis‐, and mycobacterium smegmatis‐containing phagosomes indicates pathogen‐induced microenvironments within the host cell's endosomal system. J. Immunol. 174(3), 1491–1500. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Ward, J. , Leyffer, S. , Jacobsen, W.S.M.C . & Vogt, S. (2014) Unsupervised cell identification on multidimensional X‐ray fluorescence datasets. J. Sync. Rad. 21, 568–579. [DOI] [PubMed] [Google Scholar]
- Ward, J. , Marvin, R.G. , O'Halloran, T.V. , Jacobsen, C . & Vogt, S. (2013) Rapid and accurate analysis of an X‐ray fluorescence microscopy data set through gaussian mixture‐based soft clustering methods. Microsc. Microanal. 19, 1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekley, C.M. , Aitken, J.B. , Vogt, S. et al (2011) Uptake, distribution, and speciation of selenoamino acids by human cancer cells: X‐ray absorption and fluorescence methods. Biochemistry 50(10), 1641–1650. [DOI] [PubMed] [Google Scholar]
- Wolford, J.L. , Chishti, Y. , Jin, Q. , Ward, J. , Chen, L. , Vogt, S . & Finney, L. (2010) Loss of pluripotency in human embryonic stem cells directly correlates with an increase in nuclear zinc. PLoS ONE 5(8), e12308–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewski, J . & Wroblewski, R. (1993) X‐ray microanalysis of cultured mammalian cells X‐ray Microanalysis in Biology: Experimental Techniques and Applications (ed. by Sigee D.C., Morgan A.J., Sumner A.T. & Warley A.) pp. 317–329. Cambridge, UK, Cambridge University Press. [Google Scholar]
- Yang, L. , McRae, R. , Henary, M.M. , Patel, R. , Lai, B. , Vogt, S . & Fahrni, C.J. (2005) Imaging of the intracellular topography of copper with a fluorescent sensor and by synchrotron x‐ray fluorescence microscopy. Proc. Nat. Acad. Sci. 102(32), 11179–11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, Y. , Chen, S. , Paunesku, T. et al (2013) Epidermal growth factor receptor targeted nuclear delivery and high‐resolution whole cell X‐ray imaging of Fe3O4@TiO2 nanoparticles in cancer cells. ACS Nano 7(12), 10502–10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierold, K. (1982) Preparation of biological cryosections for analytical electron microscopy. Ultramicroscopy 10(1‐2), 45–53. [DOI] [PubMed] [Google Scholar]
- Zierold, K. (1991) Cryofixation methods for ion localization in cells by electron‐probe microanalysis: a review. J. Microsc. 161(2), 357–366. [DOI] [PubMed] [Google Scholar]