

ABSTRACT
Bisphenol A (BPA), used in the manufacture of products based on polycarbonate plastics and epoxy resins, is well known as an endocrine‐disrupting monomer. In the current study, BPA increased cytotoxicity in hBMSCs in a dose‐ and time‐dependent manner, concomitantly with increased lipid peroxidation. Increased cell death in BPA‐treated cells was markedly blocked by pretreatment with the superoxide dismutase mimetic MnTBAP and MnTMPyP, but not by catalase, glutathione, the glutathione peroxidase mimetic ebselen, the NOS inhibitor NAME, or the xanthine oxidase inhibitor allopurinol. Furthermore, the decline in nuclear β‐catenin and cyclin D1 levels in hBMSCs exposed to BPA was reversed by MnTBAP treatment. Finally, treatment of hBMSCs with the GSK3β inhibitor LiCl2 increased nuclear β‐catenin levels and significantly attenuated cytotoxicity compared with BPA treatment. Our current results in hBMSCs exposed to BPA suggest that BPA causes a disturbance in β‐catenin signaling via a superoxide anion overload. © 2016 The Authors Environmental Toxicology Published by Wiley Periodicals, Inc. Environ Toxicol 32: 344–352, 2017.
Keywords: bisphenol A, superoxide dismutase, MnTBAP, human bone mesenchymal stem cells, β‐catenin, GSK3β
INTRODUCTION
Reactive oxygen species (ROS), which include superoxide anions ( ), hydrogen peroxide (H2O2), and hydroxyl radicals (HO−), are physiologically generated during aerobic oxygen metabolism. These species are detoxified by the cellular antioxidant defense system, which includes superoxide dismutase (SOD), catalase, glutathione, and nicotinamide adenine dinucleotide phosphate (NADPH). Oxidative stress caused by an imbalance in the intracellular redox state leads to cellular damage that includes the oxidation of proteins, unsaturated lipids, and nucleic acid bases, thereby promoting cell death in eukaryotic cells (Sies, 1993; Perry et al., 2001; Barnham et al., 2004). Several chemicals and environmental contaminants cause cytotoxicity by oxidative stress, which can result from excess ROS or a reduced cellular ability to neutralize these species (Ho et al., 1998; Aydogan et al., 2008; Hassan et al., 2012).
Bisphenol A [BPA; 2,2‐bis‐(4‐hydroxyphenol)‐propane] is a common monomer used in a variety of consumer products worldwide. It has been reported to be an environmental contaminant that leaches out from polycarbonate plastic products, such as beverage containers, food packaging, and dental sealant (Yamamoto and Yasuhara, 1999; Calafat et al., 2008). Because of the prevalent usage of these kinds of manufacturing products based on polycarbonate plastics and epoxy resins, people are easily exposed to BPA. BPA acts as an endocrine disrupter that competitively binds to estrogen, androgen, and thyroid receptors, thereby blocking their physiological action and triggering diverse disorders connected with abnormalities of chromosomes and reproductive systems (Sohoni and Sumpter, 1998; Moriyama et al., 2002; Lahnsteiner et al., 2005; Bonefeld‐Jorgensen et al., 2007; Richter et al., 2007; Wetherill et al., 2007; Schug et al., 2011).
Adult cross‐sectional studies have shown a highly positive correlation between total urinary BPA levels and oxidative stress. The higher the total urinary BPA concentration, the higher the levels of the oxidative stress markers malondialdehyde (MDA) and 8‐hydroxydeoxyguanosine, suggesting that BPA exposure is closely linked to oxidative stress in humans, and that persistent BPA exposure may cause serious health problems (Hong et al., 2009; Yang et al., 2009; Yi et al., 2011). Moreover, several lines of evidence suggest that BPA‐induced cytotoxicity modulated by oxidative stress occurs in both cell culture and in animals. Increased oxidative insults in various tissues have been observed in response to various doses and durations of BPA exposure through an unregulated balance of pro‐oxidants/antioxidants in cultured cells and rodents (Tyl et al., 2002; Kabuto et al., 2003; Oh and Lim, 2008; Hassan et al., 2012).
The Wnt pathway is well established to play a crucial role in normal organogenesis during embryonic development, and to contribute to cellular processes in several systems, including cell survival, cell proliferation, and cell fate specification (Bakre et al., 2007). Recently, some studies in diverse cell types demonstrated that Wnt/β‐catenin signaling mediated by ROS turns off the cell survival program and promotes cell death via an altered β‐catenin behavior under sustained oxidative stress conditions (Essers et al., 2005; Lin et al., 2008; Kawamoto et al., 2012).
Bone mesenchymal stem cells (BMSCs) are multipotent and, under appropriate environmental cues, can differentiate into diverse lineages of mesenchymal origin, including osteogenic, chondrogenic, myogenic, and adipogenic lineages (pittenger et al., 1999; Jacobs et al., 2013). For this reason, maintaining the cellular integrity of BMSCs is very important for tissue repair and regeneration in the body.
Although studies on the deleterious actions of BPA have been extensively performed in various cells, such as gametes, neuronal cells, and hepatocytes, BPA‐induced cell death of BMSCs remains to be determined. For this reason, we investigated BPA‐induced cytotoxicity focusing on oxidative stress and β‐catenin signaling in hBMSCs.
MATERIALS AND METHODS
Materials
BPA, catalase, ebselen, LiCl2, allopurinol, and glutathione‐ethyl ester, and MnTMPyP were purchased from Sigma (St Louis, MO); MnTBAP was purchased from Biomol (Butler Pike, PA); L‐NAME was purchased from Tocris (Ellisville, MO); and cell culture ingredients were obtained from Gibco BRL (Gaithersburg, MD).
hBMSC Isolation and Culture
hBMSCs were isolated from human bone marrow aspirated during hip joint arthroplasties after informed consent was obtained in accordance with the Asan Medical Center Internal Review Board policy (Seoul, Korea). Briefly, human bone marrow was collected with a heparin‐coated syringe from patients undergoing hip replacement surgery by aspiration from the proximal femur while the bone marrow was prepared for the insertion of the femoral implant. Bone marrow was separated into cell fractions by mixing with Ficoll‐Hypaque solution (Lymphoprep; Axis‐Shield PoC AS, Oslo, Norway) and centrifuging at 2000 rpm for 30 min at room temperature. Using a sterile Pasteur pipette, the leukocyte bands from the tubes were recovered and pooled into a new conical tube. The cells were diluted with alpha Minimum Essential Medium (α‐MEM) and pelleted by centrifugation at 1500 rpm for 10 min at 4°C. The pellet was washed and resuspended in α‐MEM supplemented with 10 μg/mL penicillin, 10 μg/mL streptomycin, 0.2 μg/mL amphotericin B, and 10% fetal bovine serum (Invitrogen, Grand Island, NY). The cells were seeded onto a 100‐mm culture dish and placed in a humidified 37°C, 5% CO2 incubator. After 3–4 days, nonadherent cells were removed and the cell layer was washed two to three times with Hank's Balanced Salt Solution. The culture media was renewed every 3 days. After the cells reached confluence, they were subcultured to the next passage using Trypsin‐EDTA (Invitrogen).
Cell Death Assay
Cell death was assessed by measuring lactate dehydrogenase (LDH) released into the culture medium using a LDH assay kit (Roche Diagnostics Corporation, Indianapolis, IN) according to the manufacturer's instructions. Culture medium was collected 18 h after drug treatment. Complete cell death was accomplished by treating cells with 2 mM H2O2 for 18 h. The results were normalized such that sham‐treated cultures and those showing complete cell death were counted as 0 and 100%, respectively.
MDA Determination
MDA levels were measured in cell lysates using the Bioxytech MDA 586 kit (Oxis Research, Portland, OR) in accordance with the manufacturer's instructions. MDA data were normalized to sample protein concentrations.
Preparation of Nuclear Fraction
Nuclear proteins were prepared using a Subcellular Proteome Extraction Kit (Calbiochem, Darmstadt, Germany). In brief, cells were lysed in 1 mL of Extraction Buffer I (20 mM HEPES [pH 7.9], 20 mM KCl, and 30% sucrose) and 5 μL of Protease Inhibitor Cocktail (0.5 mM PMSF, 100 μg/mL aprotinin, 5 μg/mL leupeptin, and 5 μg/mL pepstatin; Calbiochem), and then centrifuged at 3000 rpm for 10 min. The pellets obtained were agitated with 1 mL of Extraction Buffer II (20 mM HEPES [pH 7.9], 20 mM KCl, 30% sucrose, and 0.5% NP‐40) and 5 μL of Protease Inhibitor Cocktail for 30 min. After centrifugation at 7000 rpm for 10 min, the pellets were agitated with 0.5 mL of Extraction Buffer III (20 mM HEPES [pH 7.9], 100 mM NaCl, 20% glycerol, and 1 mM DTT), 1.5 μL of benzonase (350 U), and 5 μL of Protease Inhibitor Cocktail. The final nuclear supernatant was obtained by centrifugation at 9000 rpm for 10 min.
Western Blot Analysis
Protein samples (50 µg) were electrophoretically separated on 10% polyacrylamide gels, transferred to nitrocellulose membranes (Amersham Bioscience, Buckinghamshire, UK), and incubated overnight with primary antibody in a blocking buffer at 4°C. The next day, the membranes were washed in a washing buffer and incubated with horseradish peroxidase‐conjugated secondary antibody for 1 h at room temperature. The optical density of each band was measured using the SCION program (NIH Image Engineering, Bethesda, MD). Anti‐β‐catenin, anti‐cyclin D1, and anti‐β‐actin antibodies were obtained from Cell Signaling Tech (Danvers, MA).
Statistical Analyses
All experiments were performed a minimum of three times independently. Data are expressed as mean ± standard deviation (SD). Statistical comparisons of results from multiple groups were analyzed using one‐way analysis of variance, followed by an S‐N‐K (Student‐Newman‐Keuls) test for post hoc analysis. A value of p < 0.05 was considered statistically significant.
RESULTS
BPA Induces Cytotoxicity in hBMSCs in a Time‐ and Dose‐Dependent Manner
To elucidate the time‐ and dose‐dependent cytotoxic effect of BPA on hBMSCs, we measured the cell death of hBMSCs by quantifying extracellular LDH levels. First, we verified that BPA treatment for 18 h at concentrations ranging from 0 to 500 μM accelerated cytotoxicity in hBMSCs in a dose‐dependent manner; 250 μM BPA produced about 50% cell death (52.15 ± 3.15%) and 500 μM BPA was 100% cytotoxic in hBMSCs [Fig. 1(A)]. Next, to prove the time‐dependent cytotoxicity of BPA in hBMSCs, we treated hBMSCs with 250 μM BPA for 0–18 h, finding that 250 μM BPA for 6 or more hours (63.13 ± 1.54% to 98.14 ± 1.20%) aggravated cell death in hBMSCs [Fig. 1(B)]. Based on these results showing a time‐ and dose‐dependent effect of BPA, we used a cytotoxic experiment paradigm whereby hBMSCs cultures were treated with 250 μM BPA for 18 h, unless otherwise indicated.
Figure 1.
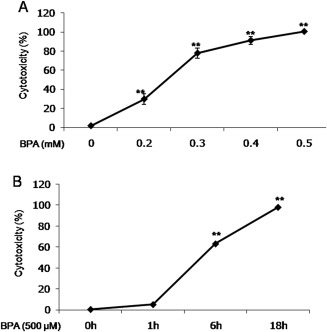
BPA induces cytotoxicity in hBMSCs in a time‐ and dose‐dependent manner. A: The dose‐dependent effect of BPA on cytotoxicity in hBMSCs. B: The time‐dependent effect of BPA on cytotoxicity in hBMSCs. Data are presented as the means ± SD. **denote differences at p < 0.01.
BPA Induces Cytotoxicity in hBMSCs via Superoxide Anion Generation
To identify the relationship between BPA‐induced cytotoxicity in hBMSCs and oxidative stress, we measured the cellular levels of MDA, an oxidative stress marker. MDA was at negligible levels in hBMSCs treated with 100 μM BPA, but profoundly increased in hBMSCs treated with 250 μM BPA for 18 h [Fig. 2(A)]. Given the BPA‐induced increase in MDA levels in hBMSCs, we investigated the ROS type and ROS‐producing factor involved in BPA‐induced cytotoxicity. BPA‐induced cytotoxicity was suppressed to about 20% (21.79 ± 5.35% to 19.37 ± 8.80%) in hBMSCs preincubated for 2 h with the SOD mimetic MnTBAP (30–50 μM), but not with catalase (100–300 Units; 92.35 ± 4.71%), the glutathione peroxidase mimetic ebselen (1–5 μM; 87.70 ± 5.65%), glutathione (1–3 mM), or the NOS inhibitor NAME (50–100 mM; 96.37 ± 6.50% to 98.20 ± 8.62%) [Fig. 2(B)]. In addition, BPA toxicity was significantly reduced by pretreatment with a cell‐permeable SOD mimetic MnTMPyP or the NADPH oxidase inhibitor diphenyleneiodonium (DPI), but not xanthine oxidase inhibitor, Allopurinol (0.3–1 mM; 98.80 ± 8.83% to 97.97 ± 9.37%); BPA cytotoxicity was reduced to about 20% in hBMSCs preincubated for 2 h with MnTMPyP (30–100 μM), and also between 65 and 80% (66.42 ± 6.26% to 82.45 ± 8.04%) in hBMSCs preincubated for 2 h with DPI (5–10 μM) [Fig. 2(C)]. These findings suggest that BPA‐induced cytotoxicity in hBMSCs occurs through superoxide anion generation.
Figure 2.
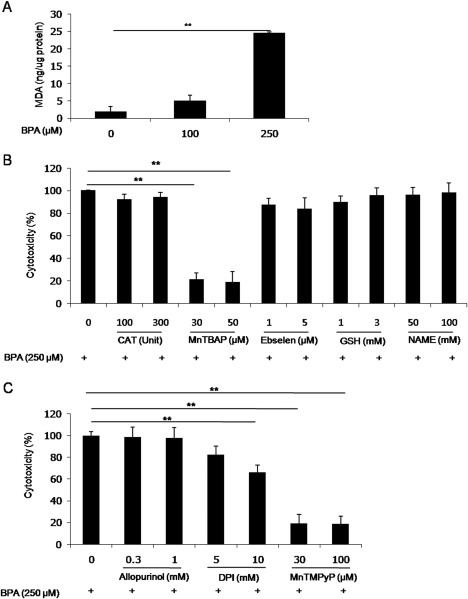
BPA induces cytotoxicity in hBMSCs via superoxide anion generation. A: Quantitative analysis of cellular MDA levels. B: Photomicrographs showing the suppressive effects of antioxidant mimetics on cytotoxicity in hBMSCs. C: Photomicrographs showing the suppressive effects of MnTMPyP, Allopurinol, and DPI on cytotoxicity in hBMSCs. Data are presented as the means ± SD. **denote differences at p < 0.01.
BPA Reduces the Accumulation of Nuclear β‐Catenin and Expression of the β‐Catenin/LEF Pathway‐Dependent Target Gene Cyclin D1 Through Increased Superoxide Anion Levels
Because β‐catenin has recently been linked to oxidative stress and BPA caused oxidative stress‐mediated cytotoxicity of hBMSCs in this study, we measured β‐catenin levels in nuclear fractions of hBMSCs treated with 250 μM BPA for 6 h. We found that treatment of hBMSCs for 6 h with 100–250 μM BPA decreased nuclear β‐catenin levels in a dose‐dependent manner; 100 or 250 μM BPA reduced nuclear β‐catenin levels to 0.75 fold (0.747 ± 0.05 fold) or 0.65 fold (0.64 ± 0.10 fold) compared with basal levels, respectively [Fig. 3(A)]. Furthermore, the expression levels of cyclin D1, which is transcriptionally regulated by the β‐catenin/LEF complex, were markedly reduced by 250 μM BPA for 2–6 h in a time‐dependent manner; cyclin D1 protein expression completely disappeared after 2 h in response to 250 μM BPA [Fig. 3(A)]. These results suggest that the transcriptional activity of the β‐catenin/LEF complex is disrupted by BPA exposure in hBMSCs. In addition, reduced nuclear β‐catenin levels in hBMSCs exposed to 250 μM BPA was conversely increased following a 2‐h pretreatment with 30 μM MnTBAP [Fig. 3(B)], indicating that reduced nuclear β‐catenin accumulation was regulated by BPA‐provoked superoxide anion.
Figure 3.
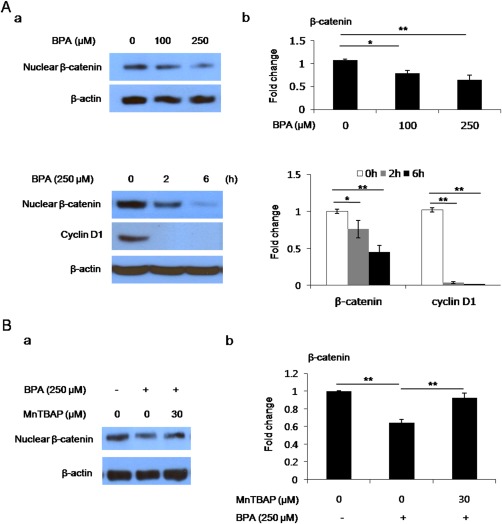
BPA reduces the accumulation of nuclear β‐catenin and expression of the β‐catenin/LEF pathway‐dependent target gene cyclin D1 through increased superoxide anion. A: Western blotting for nuclear β‐catenin and cyclin D1 in BPA‐treated hBMSCs. (a‐left): Photomicrographs showing nuclear β‐catenin and cyclin D1 immunoreactivity. (b‐right): Quantitative analysis of western blots (normalized to β‐actin). B: Western blotting for nuclear β‐catenin following pretreatment with MnTBAP in BPA‐treated hBMSCs. (a‐left): Photomicrographs showing nuclear β‐catenin immunoreactivity. (b‐right): Quantitative analysis of western blots (normalized to β‐actin). Data are presented as the means ± SD (n = 8 animals). **denote differences at p < 0.01. [Color figure can be viewed at wileyonlinelibrary.com.]
The BPA‐Induced Deficit in β‐Catenin Signaling and Cytotoxicity Are Blocked by GSK3β Inhibitor in hBMSCs
We wondered whether BPA‐induced cytotoxicity of hBMSCs was directly suppressed by the transcriptional activation of β‐catenin signaling. To verify that the reduction in nuclear β‐catenin accumulation was restored by GSK3β inhibitor in hBMSCs exposed to BPA, we preincubated hBMSCs with 0–5 mM LiCl2 for 1 h, and then treated the cells with 250 μM BPA. Nuclear β‐catenin levels in hBMSCs treated with 250 μM BPA increased to 1.5–2.5 fold (1.62 ± 0.15 fold to 2.57 ± 0.39 fold) in response to 1–5 mM of LiCl2 compared with the levels seen with 0 mM LiCl2 [Fig. 4(A)]. To investigate the mitigative effect of GSK3β inhibitor on BPA‐induced cytotoxicity of hBMSCs, we preincubated hBMSCs with 0–5 mM LiCl2 for 1 h, followed by 250 μM BPA for 18 h. The cytotoxicity of hBMSCs treated with 250 μM BPA was significantly suppressed to 55% (55.32 ± 6.25%) by pretreatment with 1–5 mM LiCl2 [Fig. 4(B)]. These results suggest that BPA‐induced cytotoxicity is at least partly relieved by β‐catenin transcriptional activation modulated by GSK3β inhibition.
Figure 4.
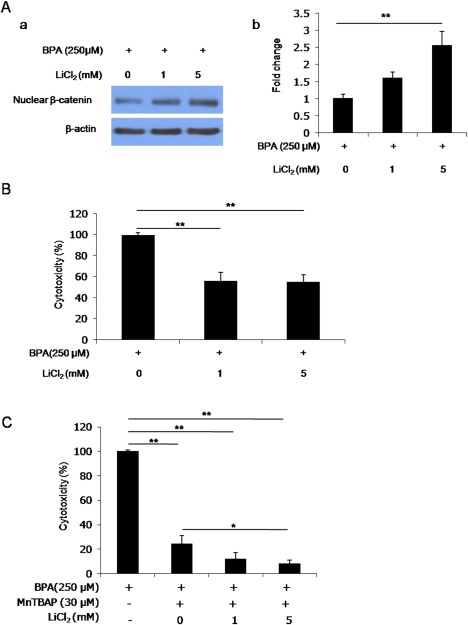
BPA‐induced deficits in β‐catenin signaling and cytotoxicity are alleviated by GSK3β inhibitor in hBMSCs. A: Western blotting for nuclear β‐catenin by pretreatment with LiCl2 in BPA‐treated hBMSCs. (a) Photomicrographs showing nuclear β‐catenin immunoreactivity. (b) Quantitative analysis of western blots (normalized to β‐actin). B: Photomicrographs showing the suppressive effects of LiCl2 on cytotoxicity in hBMSCs. C: Photomicrographs showing the suppressive effects of LiCl2 and MnTBAP on cytotoxicity in hBMSCs. Data are presented as the means ± SD. * and ** denote differences at p < 0.05 and p < 0.01, respectively. [Color figure can be viewed at wileyonlinelibrary.com.]
Given that BPA‐induced cytotoxicity of hBMSCs was not completely blocked by MnTBAP or LiCl2 pretreatment alone, we pretreated the cells with both of these factors for 2 h before treatment with 250 μM BPA. Interestingly, the cytotoxicity in hBMSCs exposed to BPA was almost completely blocked by co‐pretreatment with both MnTBAP (30 μM) and LiCl2 (1–5 mM) [Fig. 4(C)]. In particular, co‐pretreatment with both 30 μM MnTBAP and 5 mM LiCl2 showed more significant inhibition of cell death than 30 μM MnTBAP alone. This result suggests that BPA‐induced cytotoxicity in hBMSCs is blocked by both superoxide anion clearance and β‐catenin signaling activation more synergistically than by either alone.
DISCUSSION
Several endocrine‐disrupting chemicals such as BPA are known to perturb intrinsic hormone regulation, which causes damage to body organs. BPA, which is leached out from plastic products based on polycarbonate plastics and epoxy resins, has shown cytotoxic effects in various cells (Ooe et al., 2005; Crain et al., 2007; Hassan et al., 2012). Here, we first measured the cytotoxicity of hBMSCs treated with BPA in a time‐ and dose‐dependent manner. Cell death was increased in hBMSCs exposed to 0 to 500 μM BPA for 18 h in a dose‐dependent fashion, and was progressively aggravated in cells treated with 500 μM BPA for 6 h or more (Fig. 1). This result suggests the possibility of BPA cytotoxicity in hBMSCs.
Much evidence has indicated that cellular damage is closely linked to oxidative stress in BPA‐exposed cells or tissues. BPA has been shown to induce oxidative stress by reducing antioxidant capacity in various tissues, including the epididymal sperm, liver, kidney, and testes of rodents (Chitra et al., 2003; Kabuto et al., 2003). Increased thiobarbituric acid levels and decreased SOD activity in conjunction with a DNA insult were found in BPA‐treated male germ cells (Wu et al., 2013). In addition, a decline in the expression levels and activity of diverse antioxidant enzymes, including SOD, catalase, glutathione peroxidase, and glutathione reductase, was seen in rat liver following oral administration of BPA (Hassan et al., 2012).
As mentioned above, BPA can induce oxidative stress‐mediated cytotoxicity in various cell types, but the defective balance of the intracellular redox state by BPA is differentially modulated in a cell type‐specific manner. For this reason, we investigated whether BPA treatment of hBMSCs caused oxidative stress, and which type of ROS and ROS‐releasing factor was participating. The dose‐dependent increase in the levels of MDA, the oxidative stress marker, was consistently observed in hBMSCs exposed to BPA (Fig. 2). We found that BPA toxicity in hBMSCs was markedly alleviated by pretreatment with the SOD mimetic MnTBAP and MnTMPyP, but not by catalase, the glutathione peroxidase mimetic ebselen, the thiol compound glutathione, and the NOS inhibitor NAME (Fig. 2). This result indicates that increased superoxide anion ( ) production, rather than hydrogen peroxide (H2O2), nitric oxide (NO), and a depleted thiol group of cysteine, predominantly causes oxidative stress‐induced cytotoxicity in hBMSCs exposed to BPA, consistent with previous studies that showed that BPA toxicity was modulated by oxidative stress through decreased expression and enzymatic activity of SOD (Hassan et al., 2012; Wu et al., 2013). The superoxide anion can be detrimentally generated from some oxidases, such as NADPH oxidase and xanthine oxidase, as well as mitochondria (Muller et al., 2007). Increased BPA toxicity of hBMSCs was found to be alleviated by pretreatment with DPI in a dose‐dependent manner, but not by the xanthine oxidase inhibitor allopurinol; 10 μM of DPI treatment significantly decreased BPA toxicity, although 5 μM of DPI nonsignificantly decreased the toxic effect. Ooe et al. (2005) demonstrated that BPA causes intracellular ROS accumulation through damaged mitochondrial complex I function by dysregulated mitochondrial DJ‐1 in Neuro2a and GC1 cells. Moreover, superoxide anion and proton (H+) are generated via the transfer of electrons from NADPH to molecular oxygen by the NADPH oxidase complex, and this enzyme participates in the overproduction of superoxide anion as part of a widespread oxidative stress‐related pathology, such as those of cardiovascular and neurodegenerative diseases (Bedard and Krause, 2007; Farrow et al., 2013). The above‐mentioned studies and our results, at least in part, provide the evidence for an underlying mechanism for BPA toxicity in hBMSCs, in which the deleterious generation of superoxide anion in hBMSCs exposed to BPA is derived from the mitochondrial electron transfer chain (most notably from complex I and III) and a membrane‐bound NADPH oxidase complex.
A growing body of recent evidence has suggested that Wnt/β‐catenin signaling is modulated by ROS and that oxidative stress alters the association of β‐catenin with its nuclear partners, which include LEF, forkhead box transcription factors (FoxOs), hypoxia‐inducible factor (HIF‐1), and p53, and then decides cell fate, such as cell survival or apoptosis, depending on the type, intensity, and duration of the oxidative stress (Lin et al., 2008; Ladelfa et al., 2011; Tao et al., 2013). To explain the relationship between BPA toxicity in hBMSCs and the Wnt/β‐catenin pathway, we measured the accumulation of nuclear β‐catenin and its transcriptional target gene. Nuclear levels of β‐catenin and cyclin D1, a transcriptional target gene of the Wnt/β‐catenin/LEF pathway, were significantly reduced in a dose‐ and time‐dependent manner in hBMSCs treated with BPA [Fig. 3(A)]. In contrast, pretreatment of hBMSCs exposed to BPA with MnTBAP, a scavenging superoxide anion, increased nuclear β‐catenin accumulation compared with BPA treatment [Fig. 3(B)], suggesting a deficit in β‐catenin signaling modulated by superoxide anion in BPA‐treated hBMSCs. Lin et al. (2008) have reported that high glucose administration increased superoxide anion production and apoptosis by enhancing GSK3β activity and suppressing Wnt5a/β‐catenin signaling in mesangial cells (Lin et al., 2008). Activation of Wnt/β‐catenin signaling protected against hepatocyte apoptosis by inhibiting the oxidative stress‐mediated proapoptotic function of FoxO3 (Tao et al., 2013). Several studies suggested that oxidative stress facilitates a p53‐dependent apoptotic program via negative regulation of the survival functions of FoxO and HIF (An et al., 1998; Chen et al., 2003; Hoogeboom and Burgering, 2009). The results of previously published articles are consistent with and support this notion, namely that BPA causes a deficit in β‐catenin signaling regulated by the deleterious accumulation of superoxide anion in hBMSCs exposed to BPA. This process promotes cell death, although the type of partner associated with β‐catenin by BPA exposure is unknown.
Finally, our investigation of whether BPA‐induced cytotoxicity in hBMSCs is directly suppressed by activating β‐catenin signaling allowed us to verify that this cytotoxicity is ameliorated by GSK3β inhibitor in BPA‐treated hBMSCs. Inhibition of GSK3β increased nuclear β‐catenin levels and reduced BPA cytotoxicity to 55%, which, however, was not nearly as much as MnTBAP pretreatment [Fig. 4(A,B)]. However, the protective effect of both suppressing GSK activity and scavenging superoxide anion was greater than that of either alone [Fig. 4(C)]. This shows that BPA toxicity is synergistically mitigated via both neutralizing superoxide anion and activating β‐catenin signaling in hBMSCs.
Taken together, BPA triggers a deficit in β‐catenin signaling caused by intracellular superoxide anion ( ) overload, thereby promoting cell death in hBMSCs. Due to the therapeutic potential for tissue repair and regeneration, understanding BPA‐toxicity in hBMSCs is very important. Accordingly, our results may provide a basis for elucidating the underlying mechanism for BPA toxicity in hBMSCs and aid strategies targeting BPA‐induced damage of hBMSCs.
CONFLICT OF INTEREST
The authors declare that there are no conflict of interest.
ACKNOWLEDGEMENTS
This work was supported by a Korea Research Foundation grant (MRC, 2010‐0029355) funded by the Korean Government (MEST) and the Korea Research Institute of Bioscience and Biotechnology and the Korea Research Council of Fundamental Science and Technology (KRIBB/KRCF Research Initiative Program; NAP).
Contributor Information
Yea‐Hyun Leem, Email: leemyy@empas.com.
Jae‐Suk Chang, Email: jschang@amc.seoul.kr.
REFERENCES
- An WG, Kanekal M, Simon MC, Maltepe E, Blagoslonny MV, Neckers LM. 1998. Stabilization of wild‐type p53 by hypoxia‐inducible factor 1alpha. Nature 392:405–408. [DOI] [PubMed] [Google Scholar]
- Aydogan M, Korkmaz A, Barlas N, Kolankaya D. 2008. The effect of vitamin C on bisphenol A, nonylphenol and octylphenol induced brain damages of male rats. Toxicology 249:35–39. [DOI] [PubMed] [Google Scholar]
- Bakre MM, Hoi A, Mong JC, Koh YY, Wong KY, Stanton LW. 2007. Generation of mulitipotential mesendodermal progenitors from mouse embryonic stem cells via sustained Wnt pathway activation. J Biol Chem 282:31703–31712. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Masters CL, Bush AI. 2004. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 3:205–214. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. 2007. The NOX family of ROS‐generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev 87:245–313. [DOI] [PubMed] [Google Scholar]
- Bonefeld‐Jorgensen EC, Long M, Hofmeister MV, Vinggaard AM. 2007. Endocrine‐disrupting potential of bisphenol A, bisphenol A dimethacrylate, 4‐n‐nonylphenol, and 4‐n‐octylphenol in vitro: New data and a brief review. Environ Health Perspect 115:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL. 2008. Exposure of US population to bisphenol A and 4‐tertiary‐octylphenol: 2003‐2004. Environ Health Perspect 116:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Li M, Luo J, Gu W. 2003. Direct interactions between HIF‐1 alpha and Mdm2 modulate p53 function. J Biol Chem 278:13595–13598. [DOI] [PubMed] [Google Scholar]
- Chitra KC, Latchoumicandance C, Mathur PP. 2003. Induction of oxidative stress by bisphenol A in the epididymal sperm of rats. Toxicology 14:119–127. [DOI] [PubMed] [Google Scholar]
- Crain DA, Eriksen M, Iguchi T, Jobling S, Laufer H, LeBlanc GA Jr, Guillette LJ. 2007. An ecological assessment of bisphenol‐A: Evidence from comparative biology. Reprod Toxicol 24:225–239. [DOI] [PubMed] [Google Scholar]
- Essers MA, De Vries‐Smts LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. 2005. Functional interaction between β‐catenin and FOXO in oxidative stress signaling. Science 308:1181–1184. [DOI] [PubMed] [Google Scholar]
- Farrow MA, Chumbler NM, Lapierre LA, Franklin JL, Rutherford SA, Goldenring JR, Lacy DB. 2013. Clostridium difficile toxin B‐induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc Natl Acad Sci U S A 110:18674–18679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan ZK, Elobeid MA, Virk P, Omer SA, ElAmin M, Daghestani MH, AlOlayan EM. 2012. Bisphenol A induces hepatotoxicity through oxidative stress in rat model. Oxid Med Cell Longev 2012:194829 DOI: 10.1155/2012/194529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YS, Magnenat JL, Gargano M, Cao J. 1998. The nature of antioxidant defense mechanisms: A lesson from transgenic studies. Environ Health Perspect 106:1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YC, Park EY, Park MS, Ko JA, Oh SY, Kim H, Lee KH, Leem JH, Ha EH. 2009. Community level exposure to chemicals and oxidative stress in adult population. Toxicol Lett 184:139–144. [DOI] [PubMed] [Google Scholar]
- Hoogeboom D, Burgering BM. 2009. Should I stay or should I go: beta‐catenin decides under stress. Biochim Biophys Acta 1798:63–74. [DOI] [PubMed] [Google Scholar]
- Jacobs SA, Roobrouck VD, Verfaillie CM, Gool SWV. 2013. Immunological characteristics of human mesenchymal stem cells and multipotent adult progenitor cells. Immunol Cell Biol 91:32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabuto H, Hasuike S, Minagawa N, Shishibori T. 2003. Effects of bisphenol A on the metabolisms of active oxygen species in mouse tissues. Environ Res 93:31–35. [DOI] [PubMed] [Google Scholar]
- Kawamoto EM, Gleichmann M, Yshii LM, de Sa Lima L, Mattson MP, Scavone C. 2012. Effects of activation of canonical Wnt signaling by the Wnt‐3a protein on the susceptibility of PC12 cells to oxidative and apoptotic insults. Braz J Med Biol Res 45:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladelfa MF, Toledo MF, Laiseca JE, Monte M. 2011. Interaction of p53 with Tumor Suppressive and oncogenic signaling pathways to control cellular reactive species production. Antioxid Redox Signal 15:1749–1761. [DOI] [PubMed] [Google Scholar]
- Lahnsteiner F, Berger B, Kletzl M, Weisman T. 2005. Effects of bisphenol A on maturation and quality of semen and eggs on the brown trout Salmotrutta F. fario. Aquat Toxicol 75:213–224. [DOI] [PubMed] [Google Scholar]
- Lin CL, Wang JY, Ko JY, Surendran K, Huang YT, Kuo YH, Wang FS. 2008. Superoxide destabilization of β‐catenin augments apoptosis of high‐glucose‐stressed mesangial cells. Endocrinology 146:2934–2942. [DOI] [PubMed] [Google Scholar]
- Moriyama K, Tagami T, Akamizu T, Usui T, Saijo M, Kanamoto N, Hataya Y, Shimatsu A, Kuzuya H, Nakao K. 2002. Thyroid hormone action is disrupted by bisphenol A as an antagonist. J Clin Endocrinol Metab 87:5185–90. [DOI] [PubMed] [Google Scholar]
- Muller FL, Lustgarten MS, Jang Y, Richardson A, Remmen HV. 2007. Trends in oxidative aging theories. Free Radic Biol Med 43:477–503. [DOI] [PubMed] [Google Scholar]
- Oh PS, Lim KT. 2008. Blocking of intracellular ROS production by phytoglycoprotein (30kDa) causes anti‐proliferation in bisphenol A‐stimulated Chang liver cells. J Appl Toxicol 28:749–758. [DOI] [PubMed] [Google Scholar]
- Ooe H, Taira T, Iguchi‐Ariga SMM, Ariga H. 2005. Induction of reactive oxygen species by bisphenol A and abrogation of bisphenol A‐induced cell injury by DJ‐1. Toxicol Sci 88:114–126. [DOI] [PubMed] [Google Scholar]
- Perry G, Nunomura A, Siedlak SL, Harris PLR, Zhu X, Castellani RJ, Aliev G, Smith MA. 2001. Oxidant and antioxidant responses in Alzheimer disease. Recent Res Dev Biophys Biochem 1:35–41. [Google Scholar]
- Pittenger MF, Mackay AM, Beck SC, Jaiswai RK, Douglas R, Mosca JD. 1999. Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147. [DOI] [PubMed] [Google Scholar]
- Richter CA, Birnbaum LS, Farabollini F, Newbold RR, Rubin BS, Talsness CE, Vandenbergh JG, Walser‐Kuntz DR, vom Saal FS. 2007. In vivo effects of bisphenol A in laboratory rodent studies. Reprod Toxicol 24:199–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug T, Janesick A, Blumberg B, Heindela JJ. 2011. Endocrine disrupting chemicals and disease susceptibility. J Steroid Biochem Mol Biol 127:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H. 1993. Strategies of antioxidant defense. Eur J Biochem 215:213–219. [DOI] [PubMed] [Google Scholar]
- Sohoni P, Sumpter JP. 1998. Several environmental oestrogens are also anti‐androgens. J Endocrinol 158:327–339. [DOI] [PubMed] [Google Scholar]
- Tao G, Lehwald N, Jang KY, Baek J, Xu B, Bishr Omary M, Sylvester KG. 2013. Wnt/β‐catenin signaling protects mouse liver against oxidative stress‐induced apoptosis through the inhibition of Forkhead transcription factor FoxO3. J Biol Chem 288:17214–17224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyl RW, Myers CB, Marr MC, Thomas BF, Keimowitz AR, Brine DR, Veselica MM, Fail DA, Chang TY, Seely JC, Joiner RC, Butala JH, Dimond SS, Cagen SZ, Shiotsuka RN, Stropp GD, Waechter JM. 2002. Three‐generation reproductive toxicity study of dietary bisphenol A in CD Sprague‐Dawley rats. Toxicol Sci 68:121–146. [DOI] [PubMed] [Google Scholar]
- Wetherill YB, Akingbemi BT, Kanno J, McLachlan JA, Nadal A, Sonnenschein C, Watson CS, Zoeller RT, Belcher SM. 2007. In vitro molecular mechanisms of bisphenol A action. Reprod Toxicol 24:178–98. [DOI] [PubMed] [Google Scholar]
- Wu HJ, Liu C, Duan WX, Xu SC, He MD, Chen CH, Wang Y, Zhou Z, Yu ZP, Zhang L, Chen Y. 2013. Melatonin ameliorates bisphenol A‐induced DNA damage in the germ cells of adult male rats. Mutat Res 752:57–67. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Yasuhara A. 1999. Quantities of bisphenol A leached from plastic waste sample. Chemosphere 38:2569–2576. [DOI] [PubMed] [Google Scholar]
- Yang YJ, Hong YC, Oh SY, Park MS, Kim H, Leem JH, Ha EH. 2009. Bisphenol A exposure is associated with oxidative stress and inflammation in postmenopausal women. Environ Res 109:797–801. [DOI] [PubMed] [Google Scholar]
- Yi B, Kasai H, Lee HS, Kang Y, Park JY, Yang M. 2011. Inhibition by wheat sprout (Triticumaestivum) juice of bisphenol A‐induced oxidative stress in young women. Mutat Res 724:64–68. [DOI] [PubMed] [Google Scholar]