

Abstract
Four new Y-type actinomycin analogues named Y6–Y9 (1–4) were isolated and characterized from the scale up fermentation of the Streptomyces sp. strain Gö-GS12, as well as actinomycin Zp (5) that was, for the first time, isolated as a natural product. Structures of the new compounds were elucidated by the cumulative analyses of NMR spectroscopy and HRMS. The 4-hydroxythreonine on the β-ring of 1 uniquely undergoes both a rearrangement by a two-fold acyl shift and an additional ring closure with the amino group of the phenoxazinone chromophore, and the α-rings of 4 and 5 contain a rare 5-methyl proline. Compounds 2–5 showed potent antibacterial activities against Gram-positive bacteria that correlated with cytotoxicity against representative human cell lines. The combination of a β-ring rearrangement and additional ring closure in 1 rendered this actinomycin significantly less potent relative to the non-rearranged comparator actinomycin Y5 and other actinomycins.
TOC image
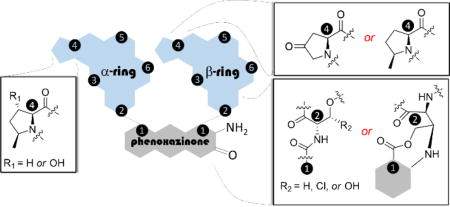
Actinomycins, which have been isolated from many species of Streptomyces, are well known chromopeptides consisting of two cyclic pentapeptide lactones – termed the α- and β-rings – attached to a central phenoxazinone chromophore via amide bonds (Figure 1A).1 Presently more than 30 unique actinomycin natural products have been described that are grouped into C-, F-, X-, Z-, G- and Y-type actinomycins. Numerous other, structurally distinct analogues have also been obtained from precursor-directed biosynthesis and synthetic efforts.2–6 Most actinomycins have very potent antitumor and antibacterial activities due to their ability to bind and intercalate double-stranded DNA.7 More specifically, the planar phenoxazinone chromophore of the model actinomycin D has been demonstrated to intercalate between guanine/cytosine base pairs, while several amino acid residues in both peptide rings interact with and bind to the minor groove of DNA.8 As a result of the strong interaction with DNA, actinomycin D, also known as dactinomycin and commercialized under the trade name of Cosmegen®, was the first antibiotic natural product shown to also have anti-cancer properties and remains FDA-approved for the treatment of less common and difficult-to-treat cancers such as Wilms tumor, rhabdomycosarcoma, and Ewing’s sarcoma.9,10
Figure 1.
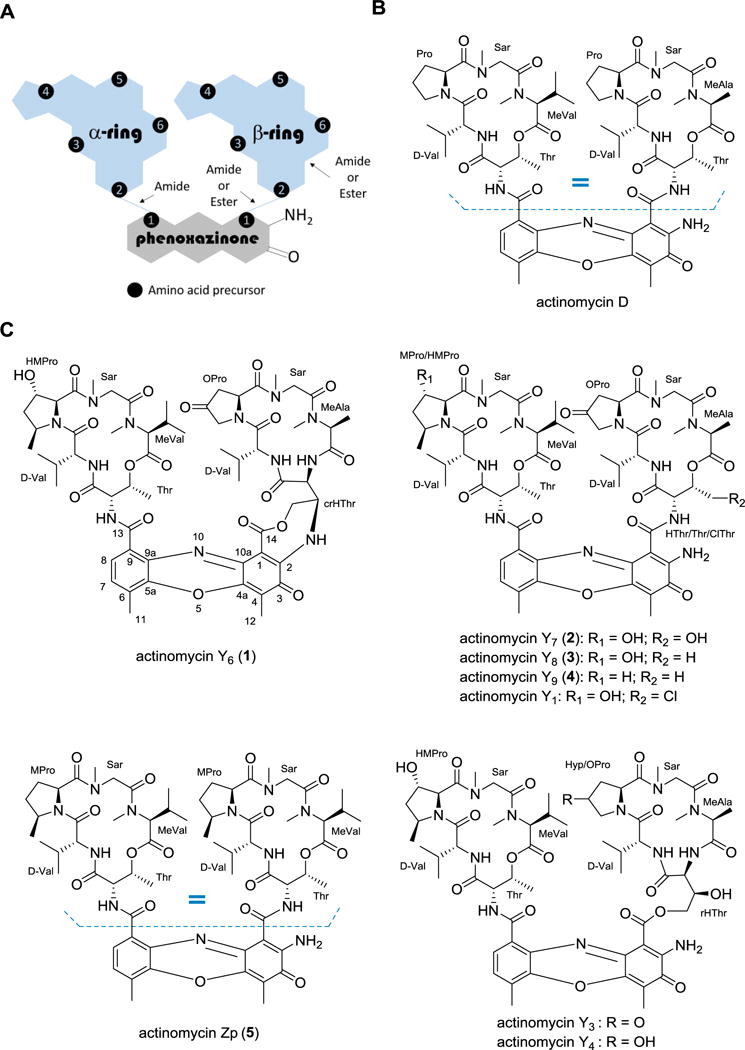
Structures of actinomycins. (A) Cartoon depiction of the general structure of all actinomycins highlighting key connectivity variations found in the β-ring. (B) Structure of the clinically approved actinomycin D, an iso-actinomycin that is isolated from several unique strains of actinomycetes. (C) Actinomycins isolated and structurally characterized from Streptomyces sp. strain Gö-GS12.
The chromophore (2-amino-4,6-dimethylphenoxazine-3-one-1,9-dicarboxylic acid) is identical in all reported actinomycins and serves as the bridge for the attachment of the two cyclic pentapeptides, which can vary in amino acid composition and results in the structural diversity of the family. The Pro in both peptidolactone rings is one such variable amino acid, as it can undergo hydroxylation, oxidation and/or methylation.4–6 In a given actinomycin, the amino acid composition of the depsipeptide α- and β-rings can be identical, which leads to the so-called, pseudo-symmetrical iso-actinomycins such as seen in actinomycin D (Figure 1B), or can differ between the two rings, leading to aniso-actinomycins. An example of the latter is the β-ring modification of the threonine (Thr) residue, which can be hydroxylated (HThr) or chlorinated (ClThr) as observed in G-type6 and Y-type3 actinomycins; rearranged through a two-fold acyl shift of HThr to generate a β-ring pentapeptide lactam that is bonded to the phenoxazinone chromophore through an ester bond, exemplified by actinomycins G6, Y3, and Y4; or the side chain bonded to the chromophore by the formation of an O- or N-phenoxazinone-fused L-Thr bridge, exemplified by actinomycin G5 and Y5, respectfully (Figure 1C). Intriguingly, actinomycin Y5, which was discovered and reported by the Grond group in 2009 along with actinomycins Y1–Y4, was the first identified ‘divergent’ actinomycin that was active as an antibacterial agent but did not have detectable cytotoxicity.3
Owing to our general interest in peptide antibiotics and our efforts to discover novel antibacterial agents, here we report the isolation, structural elucidation and bioactivity assay of five new actinomycin natural products (1–5) that are coproduced with three known congeners Y1, Y3 and Y4, from the strain Streptomyces sp. strain Gö-GS12, the previously reported Y-type actinomycin producer (Figure 1C).3 Distinct from other actinomycins, 1 contains a rearranged β-ring as well as the additional N-phenoxazinone-fused ring with HThr that was previously noted in actinomycin Y5. Compound 5 was the only iso-actinomycin isolated from this strain and contained an N-methyl-L-valine (MeVal) instead of N-methyl-L-alanine (MeAla). The antimicrobial activity of the isolated actinomycins against various Gram-positive, Gram-negative and fungal strains was determined and shown to correlate with their cytotoxicity against representative human cell lines. We also identified Y3 as the degradation product of 1 under acidic conditions, suggesting a potential biosynthetic relationship between actinomycins containing HThr and those with the additional N-phenoxazinone-fused ring.
RESULTS AND DISCUSSION
A 100 mL seed culture of Streptomyces sp. strain Gö-GS12 was grown for three days and used to inoculate 5 L of production media. After fermentation for ten days, followed by extraction, fractionation, and resolution of the components within the crude extract mixture, eight actinomycins were isolated and characterized including five new congeners: actinomycins Y6 (1, yield: 0.4 mg/L), Y7 (2, yield: 2 mg/L), Y8 (3, yield: 2 mg/L), Y9 (4, yield: 3 mg/L) and Zp (5, yield: 2 mg/L), and three known actinomycins Y1 (yield: 6 mg/L), Y3 (yield: 3 mg/L) and Y4 (yield: 2 mg/L).
Compound 1 was obtained as a red amorphous powder, and the molecular formula was established as C61H80N12O18 on the basis of (+)-HRESIMS (Figure S1). The UV/VIS and NMR spectroscopic data for 1 in CD3OD displayed typical features of an actinomycin compound, including two pentapeptidolactone residues and the phenoxazinone chromophore (Tables 1 and 2; Figures S2–S4). The assignment of the amino acids within the α-ring as Thr, valine (Val), 3-hydroxy-cis-5-methylproline (HMPro), sarcosine (Sar) and MeVal; and within the β-ring as cyclic rearranged hydroxythreonine (crHThr), Val, 4-oxoproline (OPro), Sar and MeAla was derived primarily from a thorough analysis of the 2D NMR data (COSY, TOCSY, HSQC and HMBC) (Figures S5–S8). The amino acid composition of both rings is identical with those of actinomycin Y3, a β-ring rearranged product isolated from the same strain in 2009.3 However, the 18 amu difference between compound 1 and actinomycin Y3 indicated a loss of H2O in 1. The significant differences in the chemical shifts for the rearranged hydroxythreonine residue (rHThr) in the β-ring suggested the ring closure in 1 occurs through a dehydration between C-3 of rHThr and the amino group of the chromophore, which was further confirmed by COSY correlations between H-2crHThr (δH 4.26) and H-3crHThr (δC 5.69), and between H-3crHThr (δH 5.69)/H2-4crHThr (δH 4.96, 5.23); and key HMBC correlations from H-2crHThr to C-1crHThr (δC 165.5), from H-3crHThr to 71.5 (δC C-4crHThr), and from H-4crHThr to C-2crHThr (δC 54.6) and C-3crHThr (δC 64.0) (Figure 2). The CD spectra of 1 showed a strong cotton effect at approximately 210 nm (Figure S9) that – considering the origin from the same strain as the other known y-type actinomycins – suggested the absolute configurations of the amino acids in 1 are identical with actinomycin Y3.3 Subsequently, the absolute stereochemistry of 1 was analyzed by total hydrolysis using 6 N HCl, followed by Marfey’s analysis as described previously.11 The presence of D-Val, Sar, L-Thr and L-MeVal was confirmed by comparison with authentic standards (Figure S10). Thus, the structure of compound 1 was established as a new member of the actinomycin family and subsequently named actinomycin Y6.
Table 1.
13C NMR data (100 MHz) of actinomycin Y6–Y9 and Zp (1–5)
| no. | pentapeptidolactone (α-ring, δC) | no. | pentapeptidolactone (β-ring, δC) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 2b | 3b | 4b | 5b | 1a | 2b | 3b | 4b | 5b | ||
| Thr 1 | 171.9 | 169.0 | 168.4 | 168.7 | 168.4 | HThr/1 | 165.5 | 168.2 | 167.8 | 168.7 | 168.9 |
| 2 | 56.7 | 55.0 | 54.5 | 54.6 | 55.1 | Thr 2 | 54.6 | 52.2 | 54.7 | 54.9 | 54.7 |
| 3 | 74.6 | 74.6 | 74.5 | 74.7 | 75.0 | 3 | 64.0 | 77.1 | 75.2 | 75.4 | 75.0 |
| 4 | 17.3 | 17.0 | 17.0 | 17.1 | 17.4 | 4 | 71.5 | 59.6 | 17.4 | 17.4 | 17.8 |
| Val 1 | 177.2 | 174.2 | 173.9 | 173.9 | 173.4 | Val 1 | 175.6 | 173.8 | 173.9 | 173.2 | 173.4 |
| 2 | 61.2 | 59.1 | 59.0 | 59.1 | 59.3 | 2 | 58.3 | 57.0 | 56.9 | 57.0 | 59.2 |
| 3 | 33.5 | 31.9 | 31.9 | 32.0 | 32.0 | 3 | 32.9 | 31.9 | 31.9 | 32.3 | 32.0 |
| 4 | 19.9 | 19.3 | 19.3 | 19.3 | 19.2 | 4 | 19.8 | 19.3 | 19.2 | 19.2 | 19.2 |
| 5 | 19.6 | 18.9 | 19.2 | 19.0 | 19.0 | 5 | 19.2 | 18.8 | 18.7 | 18.7 | 19.0 |
| HMPro/1 | 172.5 | 170.7 | 171.0 | 173.6 | 173.4 | OPro/1 | 174.9 | 172.6 | 172.7 | 172.7 | 173.4 |
| MPro 2 | 69.4 | 68.3 | 68.5 | 58.2 | 58.0 | MPro 2 | 56.1 | 54.6 | 54.4 | 54.3 | 58.2 |
| 3 | 74.8 | 75.5 | 75.8 | 29.4 | 29.4 | 3 | 42.3 | 41.8 | 41.7 | 41.7 | 29.6 |
| 4 | 40.5 | 41.0 | 41.5 | 31.9 | 31.8 | 4 | 209.5 | 208.6 | 208.8 | 208.9 | 31.8 |
| 5 | 55.4 | 53.4 | 54.0 | 55.4 | 55.4 | 5 | 54.1 | 52.8 | 52.7 | 52.8 | 55.2 |
| 6 | 18.9 | 18.8 | 18.9 | 18.8 | 18.8 | 18.8 | |||||
| Sar 1 | 168.5 | 166.0 | 166.1 | 166.3 | 166.4 | Sar 1 | 168.5 | 165.8 | 165.7 | 165.6 | 166.5 |
| 2 | 52.8 | 51.4 | 51.4 | 51.4 | 51.4 | 2 | 52.8 | 51.1 | 51.2 | 51.2 | 51.4 |
| NMe | 35.7 | 35.0 | 35.0 | 34.8 | 34.8 | NMe | 35.4 | 34.7 | 34.6 | 34.6 | 34.8 |
| MeVal 1 | 169.7 | 167.4 | 167.4 | 167.4 | 167.6 | MeAla/1 | 170.6 | 169.0 | 168.9 | 168.8 | 167.5 |
| 2 | 72.1 | 71.2 | 71.2 | 71.3 | 71.3 | MeVal 2 | 61.2 | 60.0 | 60.5 | 60.5 | 71.3 |
| 3 | 28.4 | 27.0 | 26.9 | 26.8 | 26.7 | 3 | 13.7 | 13.4 | 13.5 | 13.4 | 26.7 |
| 4 | 21.8 | 21.6 | 21.6 | 21.6 | 21.5 | 4 | 21.6 | ||||
| 5 | 19.6 | 19.0 | 19.0 | 19.0 | 19.1 | 5 | 19.1 | ||||
| NMe | 39.6 | 39.3 | 39.4 | 39.2 | 39.2 | NMe | 37.3 | 37.0 | 37.2 | 37.2 | 39.3 |
| chromophore (δC) | |||||||||||
| 1 | 88.4 | 98.9 | 100.8 | 101.8 | 102.0 | 8 | 127.1 | 125.9 | 125.8 | 126.2 | 125.7 |
| 2 | 153.5 | 148.3 | 147.3 | 147.3 | 147.3 | 9 | 132.8 | 132.7 | 132.7 | 132.2 | 132.9 |
| 3 | 177.4 | 178.2 | 178.6 | 179.0 | 179.1 | 9a | 128.7 | 128.7 | 128.9 | 129.1 | 129.1 |
| 4 | 116.6 | 113.5 | 113.8 | 113.5 | 113.3 | 10a | 147.2 | 146.1 | 146.0 | 145.9 | 145.7 |
| 4a | 148.9 | 145.2 | 145.0 | 144.9 | 144.9 | 11 | 15.0 | 14.9 | 15.0 | 15.0 | 15.0 |
| 5a | 142.4 | 140.4 | 140.5 | 140.4 | 140.4 | 12 | 8.3 | 7.7 | 7.8 | 7.7 | 7.7 |
| 6 | 129.9 | 127.6 | 127.6 | 127.7 | 127.4 | 13 | 169.1 | 166.0 | 166.0 | 165.8 | 166.1 |
| 7 | 132.7 | 130.4 | 130.3 | 130.1 | 130.2 | 14 | 167.9 | 168.6 | 167.8 | 168.8 | 166.3 |
measured in CD3OD;
measured in CDCl3
Table 2.
1H NMR data (500/400 MHz, J in Hz) of actinomycin Y6–Y9 and Zp (1–5)
| no. | 1a | 2b | 3b | 4b | 5b |
|---|---|---|---|---|---|
| pentapeptidolactone (α-ring, δH) | |||||
| Thr 2 | 4.81 | 4.61 brs | 4.49 | 4.59 m | 4.51 brs |
| 3 | 5.36 (m) | 5.23 m | 5.20 m | 5.14 m | 5.20 m |
| 4 | 1.28 d (6.0) | 1.15 d (6.0) | 1.12 | 1.11 d (6.2) | 1.24 d (5.3) |
| NH | 7.17 brs | 6.92 d (7.0) | 7.14 d (7.1) | 7.05 d (6.4) | |
| Val 2 | 3.75 | 3.43 | 3.40 m | 3.51 | 3.48 |
| 3 | 2.05 m | 2.10 m | 2.05 m | 2.07 m | 2.11 m |
| 4 | 1.11 d (6.6) | 1.14 d (6.6) | 1.12 | 1.08 d (6.6) | 1.10 |
| 5 | 0.93 d (6.4) | 0.94 d (6.4) | 0.91 | 0.91 d (6.8) | 0.90 d (6.8) |
| NH | 7.87 d (5.3) | 7.82 d (4.6) | 7.64 d (5.0) | 7.61 | 8.39 d (5.7) |
| HMPro/2 | 6.30 brs | 5.87 brs | 5.92 brs | 6.00 d (9.0) | 6.10 d (9.0) |
| MPro 3 | 4.28 brs | 4.19 brs | 4.11 m | 1.75, 2.74 m | 1.27, 1.73 m |
| 4 | 1.98, 2.13 m | 2.12, 2.19 m | 2.10, 2.20 m | 1.99 m | 1.92, 2.23 m |
| 5 | 4.24 brs | 4.76 m | 4.69 m | 4.35 m | 4.29 m |
| 6 | 1.48 d (6.0) | 1.52 d (6.0) | 1.51 d (5.9) | 1.47 d (6.0) | 1.46 d (6.0) |
| Sar 2 | 4.11 d(12.5) | 3.65 d (12.5) | 3.65 d (17.7) | 3.62 | 3.58 d (11.5) |
| 4.80 d (12.8) | 4.72 d (18.0) | 4.75 d (17.6) | 4.64 | 4.68 | |
| NMe | 2.58 s | 2.90 s | 2.88 s | 2.86 s | 2.87 s |
| MeVal 2 | 3.09 | 2.69 | 2.66 | 2.64 | 2.65 brs |
| 3 | 2.63 m | 2.67 m | 2.64 m | 2.65 m | 2.68 |
| 4 | 1.01 d (6.4) | 0.96 d (6.4) | 0.94 d (5.9) | 0.93 d (6.0) | 0.95 d (5.8) |
| 5 | 0.82 d (6.9) | 0.75 d (6.9) | 0.72 d (6.1) | 0.71 d (5.8) | 0.72 d (6.1) |
| NMe | 3.06 s | 2.91 s | 2.92 s | 2.90 s | 2.90 s |
| pentapeptidolactone (β-ring, δH) | |||||
| HThr/2 | 4.26 d (2.8) | 4.96 brs | 4.90 dd (6.6, 2.6) | 4.48 m | 4.65 m |
| Thr 3 | 5.69 brs | 5.15 brs | 5.22 m | 5.15 m | 5.20 m |
| 4 | 4.96 d (17.9), 5.23 d (12.2) | 3.31, 3.70 m | 1.28 d (6.2) | 1.25 d (6.2) | 1.24 d (5.3) |
| NH | 8.15 d (6.1) | 7.72 d (6.4) | 7.63 d (6.0) | 7.61 d (6.4) | |
| Val 2 | 4.08 | 3.84 | 3.83 m | 3.78 m | 3.48 |
| 3 | 2.23 m | 2.22 m | 2.17 m | 2.24 m | 2.11 m |
| 4 | 0.90 d (6.2) | 0.94 d (6.2) | 0.91 | 0.88 d (7.0) | 1.10 |
| 5 | 1.20 d (6.5) | 1.18 d (6.5) | 1.15 d (6.6) | 1.16 d (6.6) | 0.88 d (6.6) |
| NH | 8.78 d (5.3) | 8.35 d (5.1) | 8.27 d (6.2) | 8.38 d (5.6) | 8.20 d (6.0) |
| OPro/2 | 6.69 d (8.0) | 6.54 d (11.0) | 6.60 d (10.4) | 6.57 d (10.7) | 6.03 d (9.0) |
| MPro 3 | 2.41, 3.42 m | 2.33, 3.93 m | 2.31, 3.97 m | 2.31, 3.79 m | 1.27, 1.73 m |
| 4 | 1.92, 2.23 m | ||||
| 5 | 3.90 d (19.0) | 4.00 d (19.0) | 3.97 d (19.0) | 3.95 d (19.0) | 4.40 m |
| 4.42 d (19.1) | 4.55 d (19.1) | 4.53 d (6.6) | 4.58 m | ||
| 6 | 1.46 d (6.0) | ||||
| Sar 2 | 4.00 d (5.4) | 3.65 d (5.4) | 3.61 d (17.6) | 3.62 | 3.60 d (11.5) |
| 4.66 d (17.9) | 4.49 d (17.9) | 4.52 | 4.49 | 4.68 | |
| NMe | 2.88 s | 2.88 s | 2.86 s | 2.86 s | 2.87 s |
| MeAla/2 | 3.75 m | 3.26 m | 3.23 m | 3.23 m | 2.65 brs |
| MeVal 3 | 1.43 d (6.8) | 1.36 d (6.8) | 1.36 d (6.8) | 1.36 d (6.8) | 2.68 |
| 4 | 0.95 d (5.8) | ||||
| 5 | 0.72 d (6.1) | ||||
| NMe | 2.93 s | 2.92 s | 2.94 s | 2.92 s | 2.91 s |
| chromophore (δH) | |||||
| 7 | 7.48 d (8.0) | 7.36 d (7.6) | 7.34 d (7.8) | 7.34 d (7.7) | 7.32 d (7.7) |
| 8 | 7.43 d (7.8) | 7.55 d (7.8) | 7.54 d(7.7) | 7.61 d (7.7) | 7.58 d (7.7) |
| 11 | 2.56 s | 2.53 s | 2.52 s | 2.53 s | 2.52 s |
| 12 | 2.24 s | 2.12 s | 2.20 s | 2.21 s | 2.22 s |
measured in CD3OD;
measured in CDCl3
Figure 2.
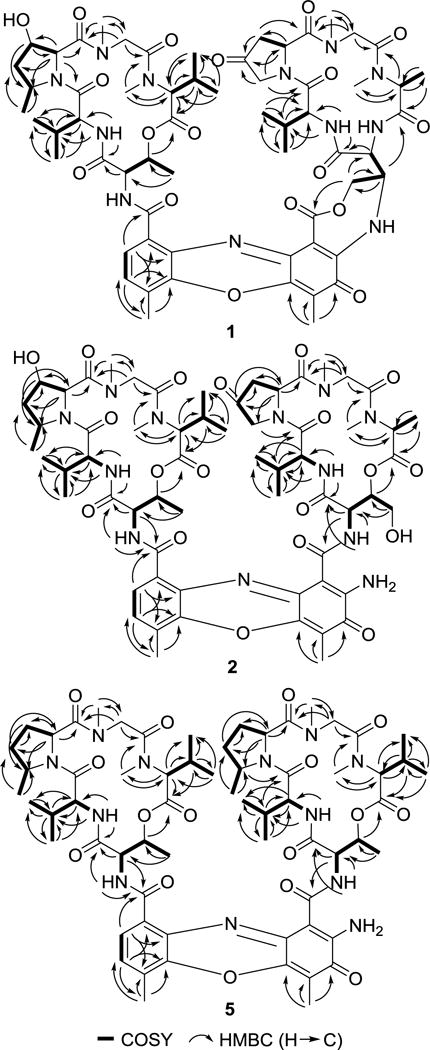
1H,1H-COSY (▬) and selected HMBC (→) correlations of actinomycins Y6 (1), Y7 (2) and Zp (5)
While purifying 1, we noticed that this compound is not stable under acidic conditions and produced a new compound with a UV/Vis spectrum comparable to other common actinomycins (Figure S11). A similar phenomenon was also observed with Y5, but was not interrogated further.3 Thus we collected and characterized the structure of the degradation product. NMR analyses revealed the chromophore-β-peptide ring underwent hydrolysis with 0.1% of TFA (pH ~ 4) to generate non-fused Y3. This result further confirmed the presence of the bridging macrocycle in 1, and also provided a possible clue to the biosynthetic origin of the third ring: the ring closure is potentially formed by nucleophilic attack of the amino group of the chromophore to C-4 of HThr concomitant with dehydration. For comparison it was previously proposed that macrocycle formation occurred via a nucleophilic attack of the amino group of the chromophore to C-4 of ClThr.3 Contrastingly, actinomycin G5, which has an oxygen bridge between the chromophore and the threonine moiety on the β-ring, was proposed to be formed via a nucleophilic attack of the 4-OH of HThr on C-2 of the chromophore.6 The interconversion between ring open and closed form was clearly observable by comparisons of the UV/Vis spectra (Figures S11), which could be monitored in real-time and could be used as a diagnostic feature to identify ring-fused actinomycins.
The molecular formula of compound 2, also obtained as red amorphous powder, was determined to be C61H82N12O19 on the basis of (+)-HRESIMS (Figure S12). The UV/VIS and NMR spectroscopic data for 2 revealed characteristic features of other actinomycins (Tables 1 and 2; Figures S13–S18) and were quite similar to that of actinomycin Y1.3 When compared to Y1, the two high-field shifted proton signals of the HThr in the β-ring showed a much higher chemical shift at the C-4 position. Additionally, no chloride atom was observed in 2, which indicates a hydroxy substitution at the C-4 position of HThr. Further COSY, TOCSY and HMBC correlations were in full agreement with HThr in the β-ring (Figure 2), and the overall structure of compound 2 was assigned as a new analog of the actinomycin series and designated as actinomycin Y7.
Compound 3 was assigned the molecular formula C61H82N12O18 on the basis of (+)-HRESIMS (Figure S19), which was suggestive of a deoxygenated derivative of 2. This deduction was corroborated by the NMR data (Tables 1 and 2; Figures S20–S25), in which the diagnostic methyl signal at C-4 of Thr in the β-ring [δC 17.4, δH 1.25 (d, J = 6.2)], showed a COSY correlation with H-3 and HMBC correlations with C-2 and C-3, indicating the absence of the 4-OH in the HThr residue of 2 (Figure 2). The relative configuration of 3 was assigned to be identical with that of 2 based on the NOESY spectrum (Figure S26) and their similar NMR pattern and designated as actinomycin Y8.
Compound 4 was assigned the molecular formula C61H82N12O17 on the basis of (+)-HRESIMS (Figure S27), which was suggestive of a deoxygenated derivative of 3. Analysis of the 1H NMR spectrum revealed the oxygenated methine group (δH 5.92) of HMPro in the α-ring of 3 was absent and a signal for a methylene (δH 1.75 and 2.74) appeared in 4 (Figure S28). The corresponding methylene signal (δC 29.4) was also observed in the 13C NMR spectra and confirmed by HSQC (Figure S29 and S30). Analysis of the key COSY and HMBC correlations and comparison of the data with that of 3 indicated that there was a MPro residue in the α-ring (Figure 2; Figures S31 and S32). In combination with the other 2D NMR data (Figure S33 and S34), the data were consistent with structure of compound 4 as shown in Figure 1 and named actinomycin Y9.
Compound 5 was assigned the molecular formula C64H90N12O16 on the basis of (+)-HRESIMS (Figure S35). The NMR data showed the existence of the phenoxazinone chromophore and two identical pentapeptidolactones that contained Val, MPro, Sar, MeVal and Thr moieties (Tables 1 and 2; Figures S36 and S37). Compound 5 differed with actinomycin D by an additional methylation at C-3 of proline, which was confirmed through COSY, HSQC, HMBC and NOESY experiments (Figure S38–S41). Upon comparison to the published data of known actinomycins, 5 was identical to the semisynthetic actinomycin Zp.12 Thus the iso-actinomycin Zp was, for the first time, isolated as a natural product and is the only iso-actinomycin identified from this strain. Interestingly, with the possible exception of C-type actinomycins, this is the only published example of a strain that produces an iso-actinomycin along with several aniso-actinomycins, all of which give comparable titers suggesting low selectivity upon coupling of the α- and β-rings.
Along with compounds 1–5, three previously reported compounds were isolated and identified as actinomycins Y1, Y3 and Y4. The structures were confirmed by comparing our spectroscopic data to the literature.3 Previously identified actinomycins Y2 and Y5 were not detected using the fermentation and purification conditions described here. In total, however, 10 total actinomycins have now been isolated and characterized from Streptomyces sp. strain Gö-GS12.
The activities of the isolated actinomycins were evaluated against common Gram-positive, Gram-negative and fungal strains using actinomycin D as a positive control (Table 3). Except for Y3 and Y4, the isolated actinomycins showed potent inhibition against representative Gram-positive strains. Compound 4 showed the highest antibacterial activity, followed by 3 and 5. Compound 2 had significantly less activity than 3, 4 and 5, while 1 was even less active with a 5–50-fold decrease in activity relative to 2.
Table 3.
In vitro antimicrobial activities of 1–5 and actinomycins Y1, Y3, Y4 and D
| Compounds | MIC (μg/mL)
|
||||||
|---|---|---|---|---|---|---|---|
| S. aureus | M. luteus | B. subtilis | M. aurum | S. enterica | E. coli | S. cerevisae | |
| 1 | 2.37 | 0.59 | 0.59 | 0.95 | >9.46 | >9.46 | 9.46 |
| 2 | 0.47 | 0.12 | 0.12 | 0.93 | >9.33 | >9.33 | >9.33 |
| 3 | 0.031 | 0.003 | 0.012 | 0.95 | >9.45 | >9.45 | 9.45 |
| 4 | 0.012 | 0.003 | 0.003 | 0.96 | >9.57 | >9.57 | 9.57 |
| 5 | 0.047 | 0.006 | 0.006 | 0.94 | >9.36 | 9.36 | >9.36 |
| Y1 | 0.023 | 0.003 | 0.002 | 0.58 | >9.20 | >9.20 | 4.60 |
| Y3 | >9.33 | 4.67 | >9.33 | >9.33 | >9.33 | >9.33 | >9.33 |
| Y4 | >9.32 | 4.66 | >9.32 | >9.32 | >9.32 | >9.32 | >9.32 |
| D | 0.006 | 0.003 | 0.002 | 0.60 | 9.57 | 9.57 | 4.79 |
The results of antibacterial screening provided insight into structure-activity relationship of actinomycins. Compound 5 differs from actinomycin D only in the 5-methylproline moiety of both the α- and β-rings, which results in a 1.6-8-fold decrease in activity, indicating that the modification of the Pro has a slight negative impact on antibacterial activity. When compared with 4, the decreased activity of 3 against Staphylococcus aureus and Bacillus subtilis indicates that the introduction of an extra hydroxy group to the α-ring Pro is also detrimental to the antibacterial activity. Modifications to the Thr moiety of the β-ring also significantly lowered the activities, since 2 was 10-fold less active than 3, which differed solely by the presence/absence of a hydroxy group. Another important factor in the antibacterial activity is the connective chemical functionality of the β-ring. In standard actinomycins such as actinomycin D, the connection between Thr and MeVal is through an ester bond (lactone), and the peptide is attached to the chromophore via an amide (Figure 1). This is not the case for 1, actinomycins Y3 and Y4, all of which have undergone an acyl rearrangement that essentially swaps the functional connections to a lactam and ester, respectively. This renders actinomycins Y3 and Y4 essentially inactive, and 1 with a lowered antibacterial activity. In addition, actinomycin G6 which also undergoes an acyl rearrangement of β-ring also has a significant decrease in antimicrobial activity. Co-crystallization of bioactive actinomycin analogues indicated that the carbonyl oxygen atom and amide hydrogen of threonine on β-ring strongly interact with amino group N-2 and ring nitrogen N-3 of guanine residues via hydrogen bonds, respectively. Furthermore, The N-2 amino group of phenoxazone core also interacts with DNA backbone between GC steps by forming an additional hydrogen bond.13–16 The β-ring rearrangement results in blockage of the amide group and N-2 amino group of phenoxazone, thus is possibly responsible for the loss of their ability of DNA binding leading to abolishment of antimicrobial activities against tested strains. Modeling and energy minimization of actinomycin Y3 and 1 with respect to actinomycin D, whose crystal structure has been solved bound to a GpC site of a dsDNA oligonucleotide13, suggests an essentially unchanged α-ring conformation with modest perturbation of the β-ring conformation due to the acyl rearrangement (Y3) that is even more pronounced upon fusion of the β-ring peptidolactone and the phenoxazinone (1) (Figure S42). Thus, the loss of β-ring-DNA binding interactions is a logical explanation for the overall loss in antibacterial activity.
On the other hand, considering 1 is minimally 9-fold more potent than actinomycin Y3, the fusion of the β-ring peptidolactone to the phenoxazinone appears to counter the decrease in activity due to the acyl rearrangement, thus implying that there is a gain-of-function upon fusion that is not readily apparent based solely on inspection of the structural model. Finally, although the activity of the actinomycins was quite variable with other Gram-positive bacteria, it is noteworthy that the anti-mycobacterial activity using Mycobacterium aurum as an indicator strain was independent of any structural modification, suggesting a possible distinct mechanism of action in the genera.
As expected, the actinomycins displayed a similar structural-activity relationship regarding the cytotoxicity against selected human cancer cell lines (Table 4). In general the actinomycins with higher antibacterial potency were more toxic to mammalian cells (PC3 and A549). This trend included 1, the only congener that contains the additional N-phenoxazinone fused peptidolactone component that is also found in actinomycin Y5. Contrastingly, actinomycin Y5 was shown to maintain modest antibacterial activity against S. aureus and B. subtilis based on zones of inhibition yet succumb to a much greater reduction in cytotoxicity: while compound 1 was less potent (278–355-fold change in EC50) compared to actinomycin D against the two human cancer cell lines tested here, actinomycin Y5 was reportedly >5000-fold less potent than actinomycin D against three distinct human tumor cell lines. Thus, neither 1 nor any of the newly discovered actinomycins appears to have a divergent antibacterial vs. cytotoxic activity as previously reported with actinomycin Y5. The exception to this, however, is with respect to antibacterial activity against M. aurum, wherein both 1 and 2 have MIC’s that are comparable to actinomycin D yet are markedly less toxic against human cell lines. The divergence in antimycobacterial versus cytotoxic activity is an interesting discovery, the reason for – and the spectrum of – this observed effect is part of ongoing investigations.
Table 4.
Viability of PC3 (prostate), A549 (lung) human cancer cell lines at 10 μM treatment for 1–5, actinomycins Y1 and D after 48 h.
| Compounds | EC50 (nM)
|
|
|---|---|---|
| PC3 Cells | A549 Cells | |
| 1 | 76.70 | 71.48 |
| 2 | 51.18 | 119.30 |
| 3 | 2.77 | 7.67 |
| 4 | 0.47 | 1.54 |
| 5 | 0.16 | 0.74 |
| Y1 | 0.319 | 0.135 |
| D | 0.276 | 0.201 |
CONCLUSION
With the goal of expanding the family of actinomycins by discovering novel members, we conducted a thorough examination of the metabolic profile of Streptomyces sp. strain Gö-GS12, and several actinomycins were characterized by multiple spectroscopic methods. We successfully identified four new members of actinomycin family Y6–Y9 (1–4) which differed in the asymmetry of the peptidolactones compared to other aniso-actinomycins isolated from Streptomyces sp. strain Gö-GS12. Furthermore, actinomycin Zp (5) – for the first time – was isolated as a natural product. Intriguingly, 5 is the only iso-actinomycin produced by this strain and, with the possible exception of the C-type actinomycins, this appears to be the first report of the isolation of iso- and aniso-actinomycins from the same strain. The current actinomycin catalogue can now be summarized as shown in Table 5.
Table 5.
| α-ring | β-ring | |
|---|---|---|
| (1) Actinomycin D as core structure | ||
| Actinomycin D | Thr-D-Val-Pro-Sar-MeVal | Thr-D-Val-Pro-Sar-MeVal |
| (2) N-demethyl actinomycins | ||
| Actinomycin D0 | Thr-D-Val-Pro-Sar-MeVal | Thr-D-Val-Pro-Gly-MeVal |
| N,N′-Didemethyl- actinomycin D | Thr-D-Val-Pro-Gly-MeVal | Thr-D-Val-Pro-Gly-MeVal |
| (2) C-type actinomycins | ||
| Actinomycin C2 | Thr-D-Val-Pro-Sar-MeVal | Thr-D-aIle-Pro-Sar-MeVal |
| Actinomycin C2a | Thr-d-aIle-Pro-Sar-MeVal | Thr-D-Val-Pro-Sar-MeVal |
| Actinomycin C3 | Thr-D-aIle-Pro-Sar-MeVal | Thr-D-aIle-Pro-Sar-MeVal |
| (3) F-type actinomycins | ||
| Actinomycin F8 | Thr-D-Val-Sar-Sar-MeVal | Thr-D-Val-Sar-Sar-MeVal |
| Actinomycin F9 | Thr-D-Val-Sar-Sar-MeVal Thr-D-Val-Pro-Sar-MeVal | Thr-D-Val-Pro-Sar-MeVal Thr-D-Val-Sar-Sar-MeVal |
| (4) X-type actinomycins | ||
| Actinomycin X0α | Thr-D-Val-Sar-Sar-MeVal | Thr-D-Val-Hyp-Sar-MeVal |
| Actinomycin X0β | Thr-D-Val-Pro-Sar-MeVal | Thr-D-Val-Hyp-Sar-MeVal |
| Actinomycin X0δ | Thr-D-Val-Pro-Sar-MeVal | Thr-D-Val-aHyp-Sar-MeVal |
| Actinomycin X1a | Thr-D-Val-Sar-Sar-MeVal | Thr-D-Val-OPro-Sar-MeVal |
| Actinomycin X2 | Thr-D-Val-Pro-Sar-MeVal | Thr-D-Val-OPro-Sar-MeVal |
| (5) Z-type actinomycins | ||
| Actinomycin Z1 | Thr-D-Val-HMPro-Sar-MeVal | HThr-D-Val-MOPro-Sar-MeAla |
| Actinomycin Z2 | Thr-D-Val-HMPro-Sar-MeVal | Thr-D-Val-MOPro-Sar-MeAla |
| Actinomycin Z3 | Thr-D-Val-HMPro-Sar-MeVal | ClThr-D-Val-MOPro-Sar-MeAla |
| Actinomycin Z4 | Thr-D-Val-MPro-Sar-MeVal | Thr-D-Val-MOPro-Sar-MeAla |
| Actinomycin Z5 | Thr-D-Val-MPro-Sar-MeVal | ClThr-D-Val-MOPro-Sar-MeAla |
| Actinomycin Zp | Thr-D-Val-MPro-Sar-MeVal | Thr-D-Val-MPro-Sar-MeVal |
| (6) G-type actinomycins | ||
| Actinomycin G1 | Thr-D-Val-Pro-Sar-MeVal | HThr-D-Val-HMPro-Sar-MeAla |
| Actinomycin G2 | Thr-D-Val-HMPro-Sar-MeVal | ClThr-D-Val-Pro-Sar-MeAla |
| Actinomycin G3 | Thr-D-Val-HMPro-Sar-MeVal | HThr-d-Val-Pro-Sar-MeAla |
| Actinomycin G4 | Thr-D-Val-HMPro-Sar-MeVal | Thr-D-Val-Pro-Sar-MeAla |
| Actinomycin G5 | Thr-D-Val-HMPro-Sar-MeVal | cHThr-D-Val-Pro-Sar-MeAla |
| Actinomycin G6 | Thr-D-Val-HMPro-Sar-MeVal | rHThr-D-Val-Pro-Sar-MeAla |
| (7) novel Y-type actinomycins | ||
| Actinomycin Y1 | Thr-D-Val-HMPro-Sar-MeVal | ClThr-D-Val-OPro-Sar-MeAla |
| Actinomycin Y2 | Thr-D-Val-HMPro-Sar-MeVal | ClThr-D-Val-Hyp-Sar-MeAla |
| Actinomycin Y3 | Thr-D-Val-HMPro-Sar-MeVal | rHThr-D-Val-OPro-Sar-MeAla |
| Actinomycin Y4 | Thr-D-Val-HMPro-Sar-MeVal | rHThr-D-Val-Hyp-Sar-MeAla |
| Actinomycin Y5 | Thr-D-Val-HMPro-Sar-MeVal | cThr-D-Val-OPro-Sar-MeAla |
| Actinomycin Y6 | Thr-D-Val-HMPro-Sar-MeVal | crThr-D-Val-OPro-Sar-MeAla |
| Actinomycin Y7 | Thr-D-Val-HMPro-Sar-MeVal | HThr-D-Val-OPro-Sar-MeAla |
| Actinomycin Y8 | Thr-D-Val-HMPro-Sar-MeVal | Thr-D-Val-OPro-Sar-MeAla |
| Actinomycin Y9 | Thr-D-Val-MPro-Sar-MeVal | Thr-D-Val- OPro -Sar-MeAla |
Differences to actinomycin D are shown in bold letters. All amino acids are L-configuration except when indicated otherwise. Abbreviations: MeVal=N-methylvaline, MeAla=N-methyl-L-alanine, aIle=allo-isoleucine, Sar=sarcosine, MPro=cis-5-methylproline, HMPro=trans-3-hydroxy-cis-5-methylproline, Hyp=trans-4-hydroxyproline, OPro=4- oxoproline, MOPro=cis-5-methyl-4-oxoproline, aHyp=cis-4-hydroxyproline, HThr=4-hydroxythreonine, ClThr=4-chlorothreonine, cThr/cHThr=cyclic Thr or HThr, i.e. forming an additional ring closure to the chromophore, rHThr= rearrangement of the HThr (β-ring) connectivity.
This table was updated based on previous overview on naturally occurring actinomycins (ref. 3).
The numbers in red indicated compounds identified in this study
Comparison of the UV/VIS spectra (λmax 238, 429 and 441 nm) of the actinomycins has revealed two diagnostic features that will likely aide in future discovery efforts. An additional absorption at 309 nm was found in the phenoxazinone-fused L-Thr congener 1, and a red shift at 452 nm was observed for the acyl rearranged and inactive actinomycins Y3 and Y4 (Figure S2). These noted differences in physicochemical properties are consistent with conformational changes of the β-ring that may contribute to the binding of actinomycins to DNA. It is interesting to note that the least toxic actinomycins against human cancer cell lines, 1 and 2, retained activity against M. aurum that was comparable to actinomycin D, suggesting that the divergent activity has the potential to be exploited for developing an anti-mycobacterial agent. The rearrangement of the β-ring and formation of the additional ring also has interesting ramifications regarding actinomycin biosynthesis. The heterocyclic ring was previously proposed to be formed via nucleophilic substitution of the hydroxy group of HThr,3,6 and the fact that 1 undergoes ring opening through hydration under mild acidic conditions supports this hypothesis. However, whether the ring closure is catalyzed by a putative dehydrogenase or happens nonenzymatically6 remains an unanswered question.
EXPERIMENTAL SECTION
General Experimental Procedures
UV spectra were recorded on an Ultraspec 8000 spectrometer (GE, Pittsburgh, PA, USA). CD spectra were obtained on a Jasco J-810 spectropolarimeter (Jasco, Easton, MD). All NMR data were recorded at 500 MHz or 400 MHz for 1H and 100 and 125 MHz for 13C with Varian Inova NMR spectrometers (Agilent, Santa Clara, CA). LC-MS was conducted with an Agilent 6120 Quadrupole MSD mass spectrometer (Agilent Technologies, Santa Clara, CA) equipped with an Agilent 1200 Series Quaternary LC system and an Eclipse XDB-C18 column (150 × 4.6 mm, 5 μm). HR-ESI-MS spectra were recorded on an AB SCIEX Triple TOF 5600 System (AB Sciex, Framingham, MA, USA). Analytic HPLC was performed with Waters Alliance 2695 separation module (Milford, MA) equipped with a Waters 2998 diode array detector and an analytical Apollo C18 column (250 mm } 4.6 mm, 5 μm). Semipreparative HPLC was performed with a Waters 600 controller and pump (Milford, MA) equipped with a 996 diode array detector, 717plus autosampler, and an Apollo C18 column (250 × 10 mm, 5 μm) purchased from Grace (Deerfield, IL). All solvents used were of ACS grade and purchased from Pharmco-AAPER (Brookfield, CT). Sephadex LH-20 (25 ~ 100 μm) was purchased from GE Healthcare (Little Chalfont, United Kingdom). TLC silica gel plates (60 F254) were purchased from EMD Chemicals Inc. (Darmstadt, Germany). Celite (TM) was purchased from Fisher Scientific (Pittsburgh, PA, United States).
Fermentation, Extraction, Isolation and Purification
Streptomyces sp. strain Gö-GS12 was provided by Prof. Stephanie Grond from Eberhard-Karls-Universität Tübingen, Germany. The strain was cultivated in two 250 mL Erlenmeyer flasks, each containing 50 mL of oatmeal medium (20g/L) enhanced with trace elements solution (CaCl2·2H2O 3g/L, Fe(III)-citrate 1 g/L, MnSO4 0.2 g/L, ZnCl2 0.1 g/L, CuSO4·5 H2O 25 mg/L, Na2B4O7 ·10H2O 20 mg/L, CoCl2 4 mg/L, Na2MoO4 mg/L·2 H2O 10 mg/L). After three days of incubation at 28 °C with 200 rpm agitation, the cultures were used to inoculate 50 flasks (250 mL), each containing 100 mL of oatmeal medium. The fermentation was continued for ten days at 28 °C with 200 rpm agitation.
All culture flasks were combined, mixed with 300 g of celite follwed by filteration under vacum to seperate the mycelium and water phase. The mycelial cake-celite portion was extracted with acetone (3 × 500 mL) by sonication, filtered off and the organic phase was evaporated to afford 5g of a dark red crude extract. The supernatant was extracted with CH2Cl2 (3 × 1 L). The organic phase was dried in vacuo to afford 3 g of dark red crude extract.
Crude extract from mycelium and supernatant was combined and subjected to a silica gel colunm chromatography using a gradient of CHCl3-MeOH (100:0 ~ 10:90) to yield four fractions, I–IV. Fraction I was applied to Sephadex LH-20 column chromatography and then semi-preparative HPLC (CH3CN/H2O, 0.01% TFA; flow rate: 3.6 mL/min) to yield 5 (10 mg). Compounds actinomycin Y1 (30 mg), 3 (10 mg) and 4 (15 mg) were isolated from fractions II and III by Sephadex LH-20 and semi-preparative HPLC (CH3CN/H2O, 0.1% TFA; flow rate: 3.6 mL/min). Similarly, 1 (2 mg), 2 (10 mg) and actinomycins Y3 (15 mg) and Y4 (10 mg) were purified from fraction IV. Since 1 was unstable under acidic condition, 1/10 amount of TFA was used during HPLC purification, and solvent was rapidly dried in vacuo.
Actinomycin Y6 (1)
red amorphous powder; UV (MeOH) λmax (log ε) 234 (sh, 9.13), 306 (1.22), 429 (4.30) nm; CD (MeOH) λmax ([θ]) 212 (−103,949), 244 (7,851), 306 (−14,000), 375 (1,261); 13C and 1H NMR data, see Tables 1 and 2; (+)-HR-ESI-MS: m/z 1269.5770 [M + H]+ (calcd for C61H81N12O18, 1269.5792).
Actinomycin Y7 (2)
red amorphous powder; UV (MeOH) λmax (log ε) 241 (sh, 7.91), 441 (3.73) nm; CD (MeOH) λmax ([θ]) 209 (−94,694), 243 (9,034), 271 (−16,047), 306 (1,664), 378 (−5,637); 13C and 1H NMR data, see Tables 1 and 2; (+)-HR-ESI-MS: m/z 1287.5918 [M + H]+ (calcd for C61H83N12O19, 1287.5897); m/z 1309.5748 [M + Na]+ (calcd for C61H82N12O19Na, 1309.5717).
Actinomycin Y8 (3)
red amorphous powder; UV (MeOH) λmax (log ε) 240 (sh, 9.37), 440 (4.34) nm; CD (MeOH) λmax ([θ]) 215 (−125,568), 242 (26,967), 267 (−56,168), 379 (−20,196); 13C and 1H NMR data, see Tables 1 and 2; (+)-HR-ESI-MS: m/z 1271.5968 [M + H]+ (calcd for C61H83N12O18, 1271.5948); m/z 1293.5788 [M + Na]+ (calcd for C61H82N12O18Na, 1293.5768).
Actinomycin Y9 (4)
red amorphous powder; UV (MeOH) λmax (log ε) 238 (sh, 8.58), 440 (3.95) nm; CD (MeOH) λmax ([θ]) 210 (−67,274), 242 (11,893), 272 (−21,000), 381 (−7,255); 13C and 1H NMR data, see Tables 1 and 2; (+)-HR-ESI-MS: m/z 1255.5935 [M + H]+ (calcd for C61H83N12O17, 1255.5999), m/z 1277.5749 [M + Na]+ (calcd for C61H82N12O17Na, 1277.5819).
Actinomycin Zp (5)
red amorphous powder; UV (MeOH) λmax (log ε) 242 (sh, 9.44), 442 (4.13) nm; CD (MeOH) λmax ([θ]) 212 (−52,199), 241 (11,713), 269 (−16,359), 380 (−5,068); 13C and 1H NMR data, see Tables 1 and 2; (+)-HR-ESI-MS: m/z 1283.6575 [M + H]+ (calcd for C64H91N12O16, 1283.6676), m/z 1305.6400 [M + Na]+ (calcd for C64H90N12O16Na, 1305.6495).
Antimicrobial bioactivities
The protocol used for the determination of the minimum inhibitory concentration (MIC) was as that described previously with minor modifications17,18. The bacterial strains Staphylococcus aureus ATCC 6538, Micrococcus luteus ATCC 15307, Bacillus subtilis ATCC 6633, Salmonella enterica ATCC 10708, Escherichia coli ATCC 12435, Mycobacterium aurum ATCC 23366, in addition to the fungal strain Saccharomyces cerevisiae ATCC 204508 were used as model strains for antimicrobial susceptibility assays. All strains were grown in appropriate liquid or on agar plates using tryptic soy broth (BD211825) for S. aureus and M. luteus, LB medium (BD244620) for B. subtilis and E. coli, nutrient broth (BD 234000) for S. enterica, Middlebrook 7H9 with OADC enrichment (Sigma-Aldrich M0178-500G) for M. aurum, and YAPD (ATCC medium number 1069) for S. cerevisiae. Individual strains were grown in 5 mL of medium for 16 h at 37 °C with shaking (250 rpm). An aliquot of a fully grown culture (100 μL) was diluted to using sterile liquid medium to OD600 ~ 0.1. Aliquots (160 μL) of each diluted culture were then transferred into the individual wells of a 96-well plate supplied with 2 μL of the tested compound. Various maximum final concentration of tested compounds (~10 μg/mL) with serial dilutions were maintained to assess the antimicrobial activities and compared to the negative control containing vehicle (DMSO) alone and positive controls (rifampicin and ciprofloxacin for M. aurum, amphotericin B for S. cerevisiae, and kanamycin for the other microorganisms. The culture plates were incubated at 28–37 °C for 16–48 h with shaking (160 rpm), and then the OD600 of each well measured using a FLUOstar Omega scanning microplate spectrofluorometer (BMG Labtech). The acquired OD600 values were normalized to the negative control wells (100% viability). The minimal concentration of the tested compound that caused growth inhibition was recorded as the MIC.
Cell Viability Assay
A resazurin-based cytotoxicity assay, also known as AlamarBlue assay, was used to assess the cytotoxicity of agents against the human lung non-small cell carcinoma cell line A549 and human prostate cancer cell lines PC3 where the degree of cytotoxicity was based upon residual metabolic activity as assessed via reduction of resazurin (7-hydroxy-10-oxido-phenoxazin-10-ium-3-one) to its fluorescent product resorufin. A549 and PC3 cells, purchased from ATCC (Manassas, VA, USA), were grown in DMEM/F-12 Kaighn’s modification and MEM/EBSS media, (Thermo scientific HyClone, Logan, UT, USA), respectively, with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine. Cells were seeded at a density 5 × 103 cells per well onto 96-well culture plates with a clear bottom (Corning, NY, USA), incubated 24 hrs at 37 °C in a humidified atmosphere containing 5% CO2 and were exposed to test agents for 2 days (positive controls: 1.5 mM hydrogen peroxide, 10 μg/ml actinomycin D). Resazurin (150 μM final concentration) was subsequently added to each well and the plates were shaken for 10 seconds and were incubated for another 3 h at 37 °C to allow viable cells to convert resazurin into resorufin. The fluorescence intensity for resorufin was detected on a scanning microplate spectrofluorometer FLUOstar Omega (BMG LABTECH GmbH, Ortenberg, Germany) using an excitation wavelength of 560 nm and an emission wavelength of 590 nm. The assay was repeated in three independent experimental replications. In each replication, the emission of fluorescence of resorufin values in treated cells were normalized to, and expressed as a percent of, the mean resorufin emission values of untreated control (metabolically active cells; 100%, all cells are viable).
Supplementary Material
Acknowledgments
We are thankful to Prof. Dr. Stephanie Grond, Eberhard-Karls-Universität Tübingen, for providing Streptomyces sp. strain Gö-GS12. This work was supported in part by the National Institutes of Health Grant AI087849 (S.V.L.), AI52188 (J.S.T.), and OD21479 (J.S.T.); the National Center for Advancing Translational Sciences Grant UL1TR000117 (S.V.L. and J.S.T.); the University of Kentucky College of Pharmacy (S.V.L. and J.S.T.); and the University of Kentucky Markey Cancer Center (J.S.T.)
Footnotes
The authors declare no competing financial interest.
Supporting Information Available: 1H NMR, 13C NMR, HSQC, HMBC, NOESY, HR-ESI-MS and CD spectra of compounds 1−5. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Brockmann H. Angew Chem. 1960;72:939–947. [Google Scholar]
- 2.Mauger AB, Lackner H. In: Anticancer agents from natural products. Cragg GM, Kingston DG, Newman D, editors. CRC press; Boca Raton Fl: 2011. pp. 363–382. Chapter 15. [Google Scholar]
- 3.Bitzer J, Streibel M, Langer H-J, Grond S. Org Biomol Chem. 2009;7:444–50. doi: 10.1039/b815689a. [DOI] [PubMed] [Google Scholar]
- 4.Lackner H, Hülsmann H, Heinze S, Simon H, Bär H, Zimmer C, Gräfe U. J Antibiot. 2000;53:84–87. doi: 10.7164/antibiotics.53.84. [DOI] [PubMed] [Google Scholar]
- 5.Lackner H, Bahner I, Shigematsu N, Pannell LK, Mauger AB. J Nat Prod. 2000;63:352–356. doi: 10.1021/np990416u. [DOI] [PubMed] [Google Scholar]
- 6.Bitzer J, Gesheva V, Zeeck A. J Nat Prod. 2006;69:1153–1157. doi: 10.1021/np060063g. [DOI] [PubMed] [Google Scholar]
- 7.Waring MJ. Sequence-specific DNA binding agents. Vol. 6 Royal Society of Chemistry; 2006. [Google Scholar]
- 8.Kamitori S, Takusagawa F. J Am Chem Soc. 1994;116:4154–4165. [Google Scholar]
- 9.Farber S, D’Angio GJ, Evans A, Mitus A. Annals of the New York Acad Sci. 1960;89:421–424. doi: 10.1111/j.1749-6632.1960.tb20165.x. [DOI] [PubMed] [Google Scholar]
- 10.D’Angio GJ, Evans A, Breslow N, Beckwith B, Bishop H, Farewll V, Goodwin W, Leape L, Palmer N, Sinks L, Sutow W, Tefft M, Wolff J. Cancer. 1981;47:2302–2311. doi: 10.1002/1097-0142(19810501)47:9<2302::aid-cncr2820470933>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Shaaban KA, Elshahawi S, Ponomareva LV, Sunkara M, Copley GC, Hower JC, Morris AJ, Kharel MK, Thorson JS. J Antibiot. 2014;67:571–575. doi: 10.1038/ja.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanada M, Sugawara K, Nishiyami Y, Kamei H, Hatori M, Konishi M. J Antibiot. 1992;45:20–28. doi: 10.7164/antibiotics.45.20. [DOI] [PubMed] [Google Scholar]
- 13.Kamitoro S, Takusagawa F. J Mol Biol. 1992;225:445–456. doi: 10.1016/0022-2836(92)90931-9. [DOI] [PubMed] [Google Scholar]
- 14.Shinomiya M, Chu W, Carlson RG, Weaver RF, Takusagawa F. Biochemistry. 1995;34:8481–8491. [PubMed] [Google Scholar]
- 15.Takasagawa F, Takusagawa KT, Calson RG, Weaver RF. Bioorg Med Chem. 1997;5:1997–1207. doi: 10.1016/s0968-0896(97)00062-x. [DOI] [PubMed] [Google Scholar]
- 16.Schaefer M, Sheldrick GM, Bahner I, Lackner H. Angew Chem Int Ed Engl. 1998;37:2381–2384. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2381::AID-ANIE2381>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 17.Wiegand I, Hilpert K, Hancock RE. Nat Protoc. 2008;3:163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 18.Wayne P. National committee for clinical laboratory standards. Performance standards for antimicrobial disc susceptibility testing. 2002;12 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.