

Abstract
Immunoprecipitation (IP) and coimmunoprecipitation (co-IP) are key techniques for studying protein-protein interactions. These methods utilize immobilized protein A or protein G to isolate antibody-bound target antigens. The main disadvantage of traditional immunoprecipitation and coimmunoprecipitation is that the conditions used to elute the precipitated antigen also release the antibody, contaminating the antigen and destroying the antibody support. To overcome these problems, we describe two methods to generate a reusable antibody support by cross-linking the antibody to immobilized protein A or protein G, or by coupling it directly to the resin. Our studies have demonstrated that the immobilization efficiency for the antibody coupling method was similar for several species of antibody. Furthermore, we illustrate that using both methods of antibody immobilization yields IP and co-IP results similar to traditional protocols but eliminates the antibody heavy and light chains contamination.
INTRODUCTION
Immunoprecipitation (IP) is a powerful immunochemical technique that has been used to study antigen characteristics such as antigen presence and quantity, relative molecular weight, rate of synthesis or degradation, posttranslational modifications, and interactions with proteins, nucleic acids, or ligands [1, 2, 3]. The IP procedure involves extracting antigens from cells in an appropriate lysis buffer, incubating the lysate with antibody to allow formation of immune complexes, and precipitating those complexes with immobilized protein A or protein G.
Coimmunoprecipitation (co-IP) is a key technique used to study protein-protein interactions [4]. Co-IP has been widely used to study receptor-ligand interactions [5], enzyme-substrate interactions [6], and interactions of subunits within a protein complex [7]. Co-IP of cell or tissue extract is also used to confirm yeast two-hybrid screening results [8, 9, 10]. Typically, an antibody specific for one protein is incubated with a cell lysate or a protein mixture to form an immune complex with the target protein (antigen). The target protein may be interacting with one or other more proteins to form a protein complex (co-complex). The entire co-complex is then precipitated using immobilized protein A or protein G.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by staining, autoradiography, or Western blot analysis is typically used to detect the interacting partners. If the antigen or its interaction partner(s) and the antibody heavy and light chains have similar relative molecular weights then, under reducing conditions, they will comigrate, making analysis of the IP results problematic. Several alternatives are currently used to circumvent this problem. One of these methods is to eliminate the reducing agent in the Laemmli buffer to cause the whole antibody molecule to migrate at the top of the gel, thus separating it from most proteins [11]. This technique, however, utilizes milder sample denaturing conditions which may not disrupt strong interactions within protein complexes and, therefore, may not be useful for co-IP experiments. A second alternative is to probe Western blots with biotinylated primary antibodies [12]. This method is generally less sensitive but must be exercised when the antibodies used for IP and immunoblotting have been generated in the same animal species. In this paper, we present two quick and easy IP and co-IP methods (seize technology) to eliminate antibody contamination in precipitated proteins: the antibody cross-linking method and the antibody coupling method (Seize Technology is a trademark of Pierce Biotechnology, Inc, Rockford, Ill—Scheme 1) that improve protein-protein interaction detection. The first approach uses a chemical cross-linker, disuccinimidyl suberate (DSS), to attach the Fc portion of an antibody to immobilized protein A or protein G. This novel procedure combines cross-linking and affinity chromatography to generate an oriented antibody-protein A or protein G support. The second method couples the antibody directly onto an activated support via lysine residues. This coupling procedure eliminates the need for protein A or protein G and offers universal coupling of all antibody species and subclasses; even chicken IgY and mouse IgG2a can be coupled equally well. Moreover, the antibody supports generated by both methods are reusable.
Scheme 1.
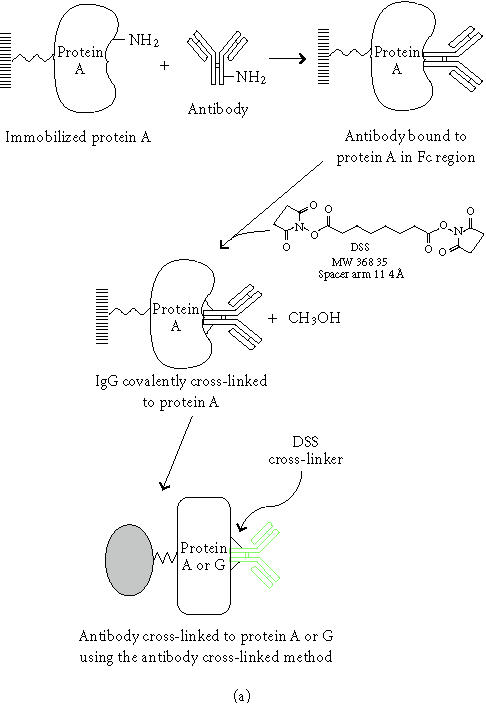
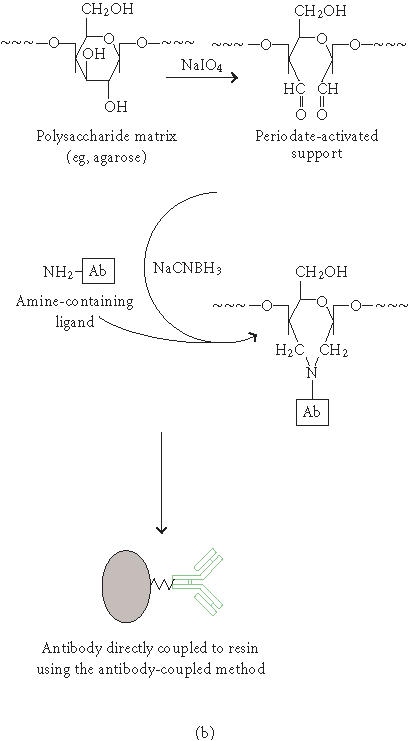
Diagram of the antibody bioconjugation chemistry. (a) Description of the antibody cross-linked method. (b) Explanation of the antibody-coupled method.
We have compared the traditional (non-cross-linked), the antibody cross-linked, and the antibody-coupled IP techniques. The traditional IP method gave higher recovery of target protein but contained strong contamination of antibody heavy and light chains while the improved methods showed no antibody contamination. In addition, several protein complexes were precipitated showing that the benefits of antibody-coupled resins extend to co-IP applications as well.
MATERIALS AND METHODS
Materials
Antibodies. Mouse monoclonal anti-T7-tag antibody was purchased from Novagen, Inc (Madison, Wis). The anti-MDM2 monoclonal antibody was bought from Oncogene Research Products (Boston, Mass). The mouse monoclonal antibody to 20S proteasome subunit α6 was purchased from Affinity Research Products Ltd (Exeter, UK). The polyclonal goat anti-GFP (green fluorescent protein) antibody was procured from Pierce Chemical Co (Pierce, Rockford, Ill). During the antibody immobilization processes, all centrifugation steps were performed at 80 × g for 1 minute.
Plasmid DNAs. Plasmid DNA, pGEM-Hsp53, and pGEM-HsMDM2 were kindly provided by Dr Arthur Haas (Medical College of Wisconsin, Milwaukee, Wis), and pET-T7-tag-Max and pET-c-Myc were a gift from Dr Kent Wilcox (Medical College of Wisconsin).
SDS-PAGE. All precast SDS-PAGE gels utilized in our experiments were the Novex brand (Invitrogen, Carlsbad, Calif). Standard electrophoresis conditions recommended by the gel manufacturer were employed. Prestained protein molecular weight marker (BlueRanger) was obtained from Pierce.
Reagents. For reagents and supplies which are not described herein the vendor was Pierce.
Cross-linking antibodies to protein G agarose
Protein G agarose (Pierce) was dispensed into a spin column and washed 2–3 times with modified Dulbecco's phosphate-buffered saline (PBS, 8 mM Na2PO3, 2 mM K2PO3, 140 mM NaCl, 10 mM KCl, pH 7.4). A 100–200-μg aliquot of monoclonal or polyclonal antibody was incubated with 400-μL protein G agarose (50% slurry) for one hour at room temperature (RT). After the unbound antibodies were washed away, antibody-bound protein G agarose was resuspended in 400-μL modified Dulbecco's PBS, and 0.1 mL of 13 mg/mL DSS cross-linker freshly prepared in dimethyl sulfoxide (DMSO) was added. The cross-linking reaction was performed at RT for one hour. The excess DSS was removed by washing the resin 4 times with 400-μL of Tris-buffered saline (TBS, 25 mM Tris, 150 mM NaCl, pH 7.2), 4 times with 0.1 M glycine (pH 2.8) to remove free antibody, and finally 3 times with TBS. The cross-linking efficiency was evaluated by A280. The antibody-protein G agarose was stored as 50% slurry at 4°C.
Coupling of antibodies to agarose resin
Coupling of antibodies to agarose resin was performed using AminoLink Plus Coupling Gel (Pierce) in a spin column. Briefly, coupling gel was washed twice with PBS (100 mM Na2PO3, 150 mM NaCl, pH 7.2). Affinity-purified antibody diluted in PBS containing 50 mM sodium cyanoborohydride was added to the resin and the mixture was inverted at RT for 4 hours at 1-hour intervals. The flow-through was spun out and the resin was washed once with PBS to remove any uncoupled antibody. A 30-minute incubation with 1 M Tris-HCl, pH 7.4, and 50 mM sodium cyanoborohydride blocked the remaining sites on the resin. The resin was washed 6 times with 1 M NaCl and equilibrated in PBS containing 0.02% sodium azide for storage at 4°C. The flow-through and the first wash were evaluated by A280 to determine the coupling efficiency.
In vitro transcription/translation
The TNT T7/Sp6-Coupled Reticulocyte Lysate System (Promega Corp, Madison, WI) was used for the in vitro synthesis of 35S-labeled proteins directly from DNA templates containing T7 or SP6 RNA polymerase promoters. The DNA template (typically 1 μg) was incubated with the transcription/translation mix in a total volume of 50 μL at 30°C for 90 minutes. The synthesized protein products were analyzed by SDS-PAGE and visualized by autoradiography.
Coimmunoprecipitation
Co-IP of 20S proteasome complex with mouse monoclonal antibody to 20S proteasome subunit α6
A 100-μL aliquot of mouse monoclonal anti-α6 (100–1000μg) was cross-linked onto protein G agarose (100-μL of settled resin) (see above procedure). Three flasks (75 cm2) of 80% confluent HeLa cells (∼ 5 × 107 cells) were lysed in 3 mL of M-PER mammalian lysis buffer (Pierce). The cell lysate was then diluted in an equal volume of modified Dulbecco's PBS and precleared with 100-μL protein G resin for 1 hour at 4°C with rotation. The immunoprecipitation was performed overnight at 4°C using 100-μL of the antibody-protein G resin (settled resin). The resin was washed three times with 400 μL of TBS, and the protein complexes were eluted three times with 100-μL 0.1 M glycine (pH 2.8). Elutions were pooled and concentrated using Ultrafree-0.5 centrifugal filtration devices (Millipore, Bedford, Mass). SDS-PAGE (12% gel) and silver staining [13] were carried out for protein detection. For comparison, traditional IP was performed using the same conditions without cross-linking the antibody to the protein G resin.
To demonstrate the antibody specificity, HeLa cell lysate (30 μL) was separated by 12% SDS-PAGE and transferred onto nitrocellulose membrane. The blot was probed with 1 μg/mL mouse monoclonal antibody to 20S proteasome subunit α6 and detected with SuperSignal West Pico chemiluminescent substrate (Pierce).
Co-IP of c-Myc and T7-tagged Max with mouse monoclonal antibody to T7-tag
Two hundred micrograms of mouse anti-T7 tag antibody were cross-linked to protein G agarose (200 μL settled resin). Plasmid DNA pET-T7-tag-Max and pET-c-Myc were used for the in vitro synthesis of 35S-labeled proteins (see the procedures above). Equal amounts of 35S-labeled T7-tagged Max and c-Myc (5 μL) were incubated together for 30 minutes at 30°C. This mixture was added to a spin column which contained antibody-protein G agarose (100-μL of settled resin) in 400-μL modified Dulbecco's PBS. The co-IP was carried out at 4°C for 2 hours with constant rotation. The resin was washed four times with 400-μL TBS and proteins were eluted three times with 0.1 M glycine (pH 2.8) and concentrated. The eluted protein complexes were resolved on 4–20% SDS-PAGE. The gel was washed in Milli-Q water for 5 minutes, soaked in Amplify (Amersham Biosciences, Piscataway, NJ) for 15–30 minutes, dried, and exposed to Kodak MS film with intensifying screens (Kodak, Rochester, NY) at -70°C overnight. Luciferase (35S-labeled) was incubated with T7-tagged Max as a negative control.
Co-IP of human p53 and MDM2 with mouse monoclonal antibody to human MDM2
Plasmid DNA pGEM-Hsp53 and pGEM-HsMDM2 were used for the in vitro synthesis of 35S-labeled proteins (see the procedure above). One hundred micrograms of anti-human MDM2 antibody were coupled to 200 μL agarose using the antibody coupling method (see the procedure above). Equal amounts of 35S-labeled human p53 and MDM2 (4 μL) were incubated together for 30 minutes at 30°C. This mixture was added to the antibody-coupled agarose (60 μL settled resin) in 200 μL modified Dulbecco's PBS with protease inhibitors (Roche Molecular Biochemicals, Indianapolis, Ind) and rotated for 2 hours at 4°C. The resin was then washed four times with TBS and proteins were eluted three times with 0.1 M glycine (pH 2.8) and concentrated. The eluted protein complexes were resolved by 4–12% SDS-PAGE and detected with autoradiography. Luciferase (35S-labeled) was incubated with MDM2 as a negative control.
Figure 1.
Comparison of co-IP using antibody cross-linked protein G agarose and non-cross-linked antibody with protein G agarose. Mouse monoclonal antiproteasome subunit α6 antibody (100-μL) and 100-μL protein G resin were used for both co-IPs. HeLa cell (∼ 5 × 107 cells) lysate was precleared with protein G agarose and co-IP was performed at 4°C overnight. (a) Co-IP of 20S proteasome complex using antisubunit α6 antibody cross-linked protein G agarose. The eluted proteasome complex was concentrated and separated on 12% SDS-PAGE and silver stained. Lane M, BlueRanger prestained protein molecular weight marker mix; lane 1, crude HeLa cell lysate; lane 2, elutions 1 and 2; lane 3, elutions 3 and 4. (b) Co-IP of 20S proteasome complex using traditional antisubunit α6 antibody with protein G agarose. One sixth of total eluted proteasome-antibody complex was separated on 12% SDS-PAGE and silver stained. (c) Immunoblot detection of 20S proteasome subunit α6 in crude HeLa cell lysate using anti-α6 antibody.
Immunoprecipitation
E coli BL21 cells (Novagen) containing 6×His-tagged GFP plasmid were induced and lysed with B-PER bacterial protein extraction reagent (Pierce). The 6×His-GFP protein was partially purified using nickel resin (Pierce). Anti-GFP antibody (130 μg) was coupled onto AminoLink Plus Coupling Gel, cross-linked to protein G agarose (see above procedures), or just mixed with protein G agarose (all used 100-μL of settled resin). The 6×His-GFP fusion protein (135 μg) purified with nickel resin was mixed separately with the three antibody resins in 100-μL modified Dulbecco's PBS in spin columns. The IP was carried out at 4°C for one hour with rotation. The resin-bound antigen was washed three times with 400-μL TBS, and the bound antigen was eluted three times with 100-μL of 0.1 M glycine (pH 2.8). One fifth of each elution fraction was resolved on 12% SDS-PAGE under reducing conditions and detected by Coomassie staining.
RESULTS AND DISCUSSION
Antibody conjugation chemistry
The two novel procedures for antibody immobilization combine linker and resin chromatography techniques to generate either a cross-linked or coupled antibody activated support. Chemical reactions in the antibody immobilization process are described in Scheme 1. For further details of the bioconjugate chemistry, see [14, 15]. The chemistry of conjugation is briefly discussed below. The scheme also depicts a representation of the antibody-bound resin for the two developed methods.
Cross-linked antibody method and co-IP applications
In the antibody cross-linked method, DSS is used to covalently link the Fc portion of the antibody to the protein G or protein A agarose, generating a reusable antibody support. DSS is a water-insoluble, noncleavable, homobifunctional N-hydroxysuccinimidyl (NHS) ester cross-linker [14, 15]. This cross-linker is widely used for conjugating radiolabeled ligands to cell surface receptors [16]. Accessible α-amine groups present on the N-termini of peptides and proteins react with NHS-esters. However, α-amines are seldom available on a protein, so the reaction with side chains of amino acids becomes important. While five amino acids contain amine groups in their side chains, only the ɛ-amine of lysine reacts significantly with NHS-ester. A phosphate-buffered system was chosen for our coupling buffer since any amine groups present in the buffer would quench the reaction. DSS was dissolved in DMSO at a concentration of 13 mg/mL and used at a final concentration of 2.6 mg/mL. Because it contains a primary amine, TBS was used as a blocking reagent and a washing buffer after the cross-linking reaction was completed. Glycine at pH 2.8 was used to remove any free antibody after the cross-linking step and to elute the antigen. The average cross-linking efficiency was over 80% (data is not shown).
Co-IP of proteins from cellular extracts is the most convincing evidence that two or more proteins physically interact with each other. The 26S proteasome is a key enzyme in the ubiquitin/ATP-dependent pathway of protein degradation [17, 18]. The catalytic core of this unusually large complex (Mr ∼ 700,000) is formed by the 20S proteasome, a barrel-shaped structure comprised of four rings each containing seven subunits, α 7 β 7 β 7 α 7 [19, 20]. The fourteen different subunits of mammalian 20S proteasome have molecular weights ranging from 18 to 33 kd [21]. The α6 subunit is located on the outer rings of the 20S proteasome [19]. The mouse monoclonal antibody to human 20S proteasome subunit α6 recognizes a 33- kd band on a Western blot of total HeLa cell lysate (Figure 1). The antisubunit α6 antibody was cross-linked to protein G agarose and used to coimmunoprecipitate the whole 20S proteasome complex from HeLa cell lysate. The eluted protein complex contains a series of proteins ranging from 18 to 33 kd, which is the typical pattern of 20S proteasome subunits (Figure 1a). The higher molecular weight proteins ranging from 45 to 100 kd (Figure 1a, lane 2) are the regulatory subunits of the 26S proteasome complex which were coimmunoprecipitated with the catalytic core complex [22]. As a comparison, a co-IP with the traditional method (non-cross-linked antibody) was performed using the same conditions. Although only one sixth of the total eluent was analyzed on SDS-PAGE, a strong contamination of antibody heavy and light chains was observed (Figure 1b). This result demonstrates that the cross-linked antibody can efficiently coimmunoprecipitate a large protein complex and eliminate the antibody contamination. Therefore, this method could be scaled up to affinity purify protein complexes for downstream assays and protein characterization studies.
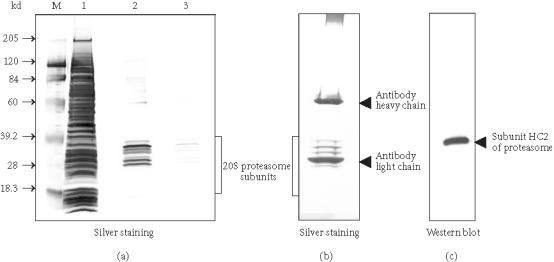
In vitro binding and co-IP assays are very useful when studying the interactions of proteins that become complex only at a certain point in the cell cycle or of a subset of proteins belonging to a larger protein complex [22, 23]. Co-IP can also be used to confirm protein-protein interaction results from an in vivo yeast two-hybrid screen [8]. Max and c-Myc are a pair of interacting proteins that form heterodimers to regulate the transcription of genes which have been shown to contribute to carcinogenesis [24]. In this experiment, 35S-labeled Max (T7-tagged) and c-Myc were translated in vitro in the presence of L-[35S] methionine using a rabbit reticulocyte lysate. The two separately synthesized 35S-labeled proteins were incubated at 30°C for 30 minutes. The anti-T7 tag antibody was cross-linked to protein G agarose and used to coimmunoprecipitate T7-tagged Max and c-Myc. Luciferase was used as a negative control for T7-tagged Max. Figure 2 shows that c-Myc can be coimmunoprecipitated with Max, whereas luciferase does not coimmunoprecipitate with Max. The entire co-IP experiment was performed in a single spin column, which limited contamination of radioactive material, and prevented the loss of resin between the washes, thereby enhancing the recovery [25]. As little as 25 μL of antibody-protein G agarose per sample was used up to five times without detectable loss of the activity. This advantage could be very useful for confirming in vivo yeast two-hybrid screening results because the protein G agarose cross-linked to an antibody against the “bait” protein can be used repeatedly to confirm the interaction between the “bait” protein and each “prey” protein. We also have used protein G agarose cross-linked with the c-Myc tag antibody to successfully coimmunoprecipitate SV40 large-T antigen with c-Myc-tagged p53 (data is not shown).
Figure 2.
Coimmunoprecipitation of T7-tagged Max and c-Myc using anti-T7 tag antibody cross-linked protein G agarose. Max (T7-tagged), c-Myc, and luciferase were in vitro translated and 35S-labeled (lanes 1, 2, and 3) using the TNT-Coupled Reticulocyte Lysate System. Before co-IP, Max and c-Myc, and Max and luciferase were mixed proportionally and incubated at 30°C for 1 hour. Co-IPs were carried out with anti-T7 tag antibody cross-linked protein G agarose at 4°C for 2 hours (lanes 4 and 5) and the eluted protein complexes were separated on 4–20% SDS-PAGE. The 35S-labeled proteins were detected by autoradiography. Star (∗): degradation product of c-Myc.
Although cross-linking antibody to protein G or protein A agarose is a good approach to immobilize the antibodies in the correct orientation, the cross-linking efficiency and specificity varied depending upon the concentrations of antibody, protein G agarose, and cross-linking reagent. Binding capacity after cross-linking also varied from antibody to antibody. The drop in binding capacity could be caused by a change in conformation when cross-linking occurs on the surface of the antibody molecule or when cross-linking occurs within the binding sites (see data below). Therefore, optimal DSS and antibody concentrations need to be determined empirically for each antibody. This led us to expand our investigation for a more universal method of antibody attachment with improved preservation of antibody binding activity.
Antibody-coupled method and co-IP applications
The antibody-coupled procedure utilizes reductive amination to directly link the antibody to the agarose bead. The coupling resin is provided in an activated state containing aldehyde groups formed by mild oxidation of adjacent diols using sodium meta-periodate [14]. Primary and secondary amine groups on the antibody react with the aldehydes to form Schiff bases that are then reduced by sodium cyanoborohydride to form secondary and tertiary amine linkages [14]. Since this procedure links the antibody to the resin in every direction, not all antibody molecules will present an active orientation. Typical coupling efficiencies for various species of antibodies are shown in Figure 3a. The coupling efficiencies were determined by spectrophotometric analysis of antibody solutions before and after coupling. On average, 88 percent of the antibody was coupled in 4 hours when using 200 μg of antibody and 200 μL of settled resin. Scalability was demonstrated when immobilization using 50 and 100-μL of settled coupling resin yielded comparable coupling efficiencies (data is not shown). All species of antibody exhibited the same relationship with respect to time and coupling. Therefore, this method is not limited to antibody species that only bind strongly to protein G or protein A. Figure 3b illustrates the relationship between protein concentration and the rate of the coupling reaction. As expected, the rate of coupling increases with increased protein concentration. These results show that this is a universal technique with no need to optimize for each antibody.
Figure 3.
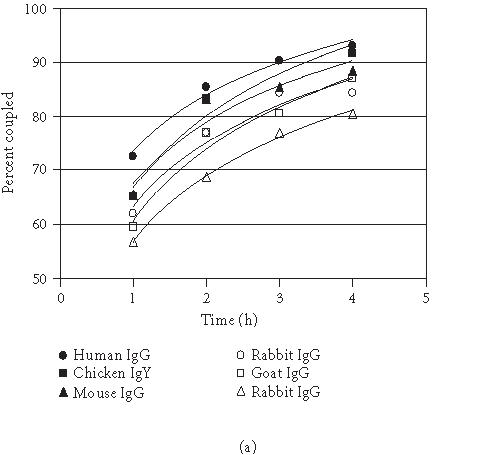
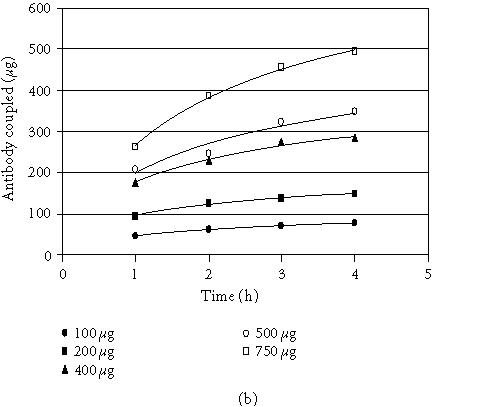
Coupling of mammalian and avian antibodies. (a) Antibody from various species (200 μg) was coupled to 200 μL of coupling gel (settled gel) at 1-hour intervals for 4 hours at RT. For the chicken antibody, 500 μL was used. (b) Normal chicken IgY antibody (100–750μg) was coupled to 200 μL of coupling gel (settled gel) at 1-hour intervals for 4 hours at RT.
MDM2 oncoprotein plays a central role in the regulation of p53 tumor suppressor protein [26, 27, 28]. MDM2 binds to p53 and blocks its activity as a tumor suppressor and promotes its degradation in many tumor cells [6, 29]. In our experiment, human p53 and MDM2 genes were transcribed/translated and 35S-labeled in a reticulocyte lysate. The co-IP result using coupled anti-MDM2 antibody shows that MDM2 interacts with p53 but not luciferase (Figure 4). We have used as little as 20 μL of the antibody-coupled resin and have reused the resin up to five times without obvious loss of activity.
Figure 4.
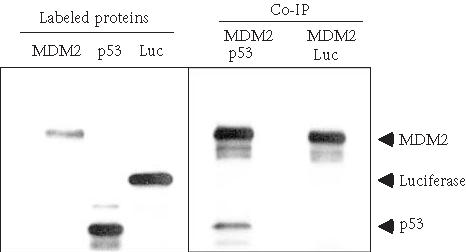
Coimmunoprecipitation of p53 and MDM2 using anti-MDM2 antibody-coupled agarose. MDM2, p53, and luciferase were in vitro translated and 35S-labeled using TNT-Coupled Reticulocyte Lysate System. p53 and MDM2 were combined and incubated at 30°C for 30 minutes. Co-IP was performed at 4°C for 2 hours with 60 μL anti-MDM2 antibody-coupled agarose. Luciferase was used as a negative control protein to incubate with MDM2. Eluted proteins were resolved on 4–12% SDS-PAGE and visualized by autoradiography.
Binding capacity of antibody-coupled resin or antibody cross-linked resin
The antibody cross-linked and antibody-coupled procedures eliminate the contamination problem by preventing the antibody from co-eluting with the antigen. Another benefit to both methods is that the antibody-resin is reusable, thereby conserving valuable antibody. We compared the traditional, antibody cross-linked and antibody-coupled procedures to evaluate the relative amount of antigen recovered using the same amount of goat anti-GFP antibody (Figure 5). Under these conditions, a significant increase in recovered antigen is seen when using the antibody-coupled procedure versus the antibody cross-linked method. Although the traditional method yielded a greater quantity of antigen, the presence of antibody light chains in the eluent distorted the recovered GFP band, since they have comparable molecular weights.
Figure 5.
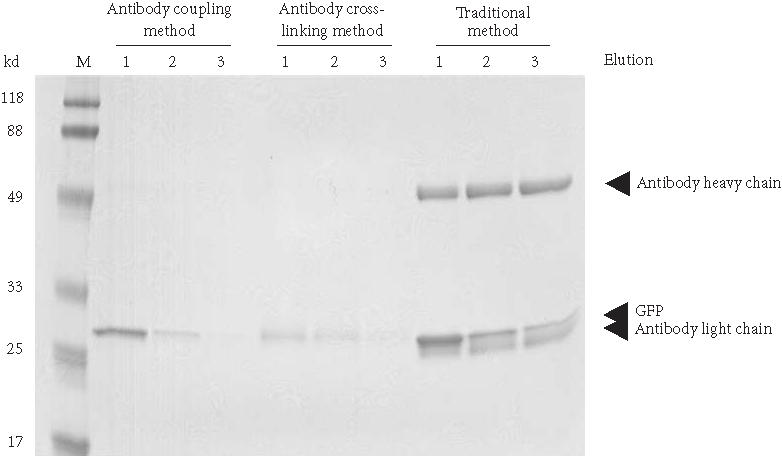
Comparison of IP using antibody cross-linked protein G agarose, antibody-coupled agarose, and non-cross-linked antibody with protein G agarose. In each case, 130 μg of affinity-purified goat anti-GFP antibody was used with 100-μL of settled protein G gel or coupling gel. IP was performed using 135 μg of partially purified GFP. Ten percent of the elution volume was electrophoresed on a 12% polyacrylamide reducing gel and stained with Coomassie. Lane M, BlueRanger molecular weight marker; lanes 1–3 elutions from different antibody immobilization methods.
CONCLUSIONS
We have developed two methods to immunoprecipitate and coimmunoprecipitate proteins that eliminate antibody contamination. The first method properly orients the antibody for antigen recognition by binding its Fc portion to protein A or protein G resin. Subsequently, the antibody is cross-linked to the resin to prevent leaching into the eluent. The second method achieves the same goal yet is universal for all antibody classes and species because it couples the antibody directly to the resin matrix. Both methods give comparable results to traditional IP although overall antigen-binding efficiency is not always as high as with traditional IP. The precipitated proteins from our IP and co-IP methods can be easily detected and characterized in downstream applications such as mass spectrometry or enzymatic assays. Furthermore, the stabilized antibody-linked resin from either technique can be regenerated and reused multiple times thereby conserving precious antibody samples.
References
- Williams N E. Immunoprecipitation procedures. Methods Cell Biol. 2000;62:449–453. [PubMed] [Google Scholar]
- Harlow E d, Lane D. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1999. Using Antibodies: A Laboratory Manual. [Google Scholar]
- Otto J J, Lee S W. Immunoprecipitation methods. Methods Cell Biol. 1993;37:119–127. [PubMed] [Google Scholar]
- Ransone L J. Detection of protein-protein interactions by coimmunoprecipitation and dimerization. Methods Enzymol. 1995;254:491–497. doi: 10.1016/0076-6879(95)54034-2. [DOI] [PubMed] [Google Scholar]
- Shi Y, Ullrich S J, Zhang J, et al. A novel cytokine receptor-ligand pair. Identification, molecular characterization, and in vivo immunomodulatory activity. J Biol Chem. 2000;275(25):19167–19176. doi: 10.1074/jbc.M910228199. [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420(1):25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Yamauchi J, Kaziro Y, Itoh H. C-terminal mutation of G protein beta subunit affects differentially extracellular signal-regulated kinase and c-Jun N-terminal kinase pathways in human embryonal kidney 293 cells. J Biol Chem. 1997;272(12):7602–7607 . doi: 10.1074/jbc.272.12.7602. [DOI] [PubMed] [Google Scholar]
- Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999;447(1):5–9. doi: 10.1016/s0014-5793(99)00254-9. [DOI] [PubMed] [Google Scholar]
- Wong C, Naumovski L. Method to screen for relevant yeast two-hybrid-derived clones by coimmunoprecipitation and colocalization of epitope-tagged fragments–application to Bcl-xL. Anal Biochem. 1997;252(1):33–39. doi: 10.1006/abio.1997.2284. [DOI] [PubMed] [Google Scholar]
- Estojak J, Brent R, Golemis E A. Correlation of two-hybrid affinity data with in vitro measurements. Mol Cell Biol. 1995;15(10):5820–5829. doi: 10.1128/mcb.15.10.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C, Galande S. Elimination of reducing agent facilitates quantitative detection of p53 antigen. Biotechniques. 2001;30(5):960–963. doi: 10.2144/01305bm07. [DOI] [PubMed] [Google Scholar]
- Berryman M, Bretscher A. Immunoblot detection of antigens in immunoprecipitates. Biotechniques. 2001;31(4):744–746. [PubMed] [Google Scholar]
- Scopes P K, Smith J A. Analysis of protein. In: Ausubal F M, Brent R, Kingston R E, et al., editors. John Wiley & Sons; New York, NY: 1994. p. 10.6.3. Current Protocols in Molecular Biology . [Google Scholar]
- Hermanson G T, Mallia A K, Smith P K. Academic Press; San Diego, Calif: 1992. Immobilized Affinity Ligand Techniques. [Google Scholar]
- Hermanson G T. Academic Press; San Diego, Calif: 1996. Bioconjugate Techniques. [Google Scholar]
- Cox G W, Mathieson B J, Giardina S L, Varesio L. Characterization of IL-2 receptor expression and function on murine macrophages. J Immunol. 1990;145(6):1719–1726. [PubMed] [Google Scholar]
- Rechsteiner M, Hoffman L, Dubiel W. The multicatalytic and 26S proteases. J Biol Chem. 1993;268(9):6065–6068. [PubMed] [Google Scholar]
- Eytan E, Armon T, Heller H, Beck S, Hershko A. Ubiquitin C-terminal hydrolase activity associated with the 26S proteasome complex. J Biol Chem. 1993;268(7):4668–4674. [PubMed] [Google Scholar]
- Groll M, Ditzel L, Lowe J, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386(6624):463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Tanahashi N, Tsurumi C, Tamura T, Tanaka K. Molecular structure of 20S and 26S proteasomes. Enzyme Protein. 1993;47(4–6):241–251. doi: 10.1159/000468683. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Tsurumi C. The 26S proteasome: subunits and functions. Mol Biol Rep. 1997;24(1-2):3–11. doi: 10.1023/a:1006876904158. [DOI] [PubMed] [Google Scholar]
- Chu-Ping M, Vu J H, Proske R J, Slaughter C A, DeMartino G N. Identification, purification, and characterization of a high molecular weight, ATP-dependent activator (PA700) of the 20 S proteasome. J Biol Chem. 1994;269(5):3539–3547. [PubMed] [Google Scholar]
- DaFonseca C J, Shu F, Zhang J J. Identification of two residues in MCM5 critical for the assembly of MCM complexes and Stat1-mediated transcription activation in response to IFN-gamma. Proc Natl Acad Sci USA. 2001;98(6):3034–3039. doi: 10.1073/pnas.061487598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood T D, Amati B, Land H, Evan G I. Max and c-Myc/Max DNA-binding activities in cell extracts. Oncogene. 1992;7(9):1783–1792. [PubMed] [Google Scholar]
- Brymora A, Cousin M A, Roufogalis B D, Robinson P J. Enhanced protein recovery and reproducibility from pull-down assays and immunoprecipitations using spin columns. Anal Biochem. 2001;295(1):119–122. doi: 10.1006/abio.2001.5215. [DOI] [PubMed] [Google Scholar]
- Juven-Gershon T, Oren M. Mdm2: the ups and downs. Mol Med. 1999;5(2):71–83. [PMC free article] [PubMed] [Google Scholar]
- Freeman D A, Wu L, Levine A J. Functions of the MDM2 oncoprotein. Cell Mol Life Sci. 1999;55(1):96–107. doi: 10.1007/s000180050273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines D S. The mdm2 proto-oncogene. Leuk Lymphoma. 1997;26(3-4):227–238. doi: 10.3109/10428199709051772. [DOI] [PubMed] [Google Scholar]
- Haines D S, Landers J E, Engle L J, George D L. Physical and functional interaction between wild-type p53 and mdm2 proteins. Mol Cell Biol. 1994;14(2):1171–1178. doi: 10.1128/mcb.14.2.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]