

Abstract
Background
Lipopolysaccharide (endotoxin, LPS) is a strong inducer of the innate immune response. It is widespread in our environment, e.g. in house dust and contributes to asthma. Compared to humans, horses are even more sensitive to LPS. However, data on LPS effects on the equine transcriptome are very limited. Using RNA-seq we analysed LPS-induced differences in the gene expression in equine peripheral blood mononuclear cells at the gene and gene-network level in two half-sib families and one group of unrelated horses.
Results
24 h-LPS challenge of equine immune cells resulted in substantial changes in the transcriptomic profile (1,265 differentially expressed genes) showing partial overlap with human data. One of the half-sib families showed a specific response different from the other two groups of horses. We also identified co-expressed gene modules that clearly differentiated 24 h-LPS- from non-stimulated samples. These modules consisted of 934 highly interconnected genes and included genes involved in the immune response (e.g. IL6, CCL22, CXCL6, CXCL2), however, none of the top ten hub genes of the modules have been annotated as responsive to LPS in gene ontology.
Conclusions
Using weighted gene co-expression network analysis we identified ten co-expressed gene modules significantly regulated by in vitro stimulation with LPS. Apart from 47 genes (5%) all other genes highly interconnected within the most up- and down-regulated modules were also significantly differentially expressed (FDR < 0.05). The LPS-regulated module hub genes have not yet been described as having a role in the immune response to LPS (e.g. VAT1 and TTC25).
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3390-y) contains supplementary material, which is available to authorized users.
Keywords: Lipopolysaccharides, LPS, Differential expression, Gene modules, WGCNA, Horse, RNA-seq
Background
Innate immune response is the front line of the immune defence and therefore plays a crucial role for the organism’s survival starting at the time of birth. The innate immunity is non-specifically induced by invariant molecular structures present in pathogens, so called pathogen-associated molecular patterns (PAMPs) [1]. The best studied PAMP, lipopolysaccharide (LPS, also known as endotoxin due to its content of a toxic lipid A), is a component of the outer membrane of most gram-negative bacteria [2, 3].
When LPS enters the blood stream, most commonly through an intestinal lesion, it is opsonized by serum LPS-binding protein [4], which is recognized by Toll-like receptor 4 (TLR4) with the help of its co-receptor CD14 and another cell surface molecule, MD-2 [5, 6]. This recognition leads to a signalling cascade through MyD88- and TRIF-dependent pathways [7], activating the NF-κB transcription factor and finally inducing expression and release of numerous cytokines, including TNF-α, IL-1, IL-6, and IL-10 [8, 9]. This response and cascade are evolutionarily well conserved and thus very similar across species [10].
Sepsis is defined as a disrupted regulation of inflammation in the face of bacterial or other microbial infection, which can ultimately lead to tissue damage, organ failure, and death. This response may consist of the systemic inflammatory response (SIRS) or/and the compensatory anti-inflammatory response (CARS), and the balance between SIRS and CARS is crucial for the host survival [11, 12]. SIRS often results from infections caused by gram-negative bacteria [13, 14]. When sepsis and SIRS lead to clinically evident cardiovascular compromise, the result is termed septic shock [15], which is an uncontrolled life-threatening condition responsible for a large proportion of deaths of hospitalized patients worldwide [16]. Circulating LPS is believed to be the principal trigger of the septic shock [6].
In contrast to the above-described systemic effects of endotoxaemia (the presence of endotoxin in the blood), inhaled LPS can induce airflow obstruction and neutrophilic inflammation in healthy individuals. In asthmatic patients the effect of LPS may be either beneficial or harmful [17, 18], depending on the timing and dosage of the LPS exposure, as well as other environmental and genetic factors [17, 19–21]. In rats, early exposure to LPS, prior to sensitization with an allergen, can attenuate inflammatory processes in lungs, eosinophilia, and bronchial hypersensitivity, whereas endotoxin doses that are inhaled later on (6 days after allergen exposure) increase airway inflammation and edema [17, 22]. Low doses of LPS that induce a ‘normal’ Th1 type response may be beneficial as they are directing the Th1/Th2 balance towards Th1, thereby reducing the effects of the allergy related Th2 type response. However, it has also been shown that higher doses of LPS can contribute to occupational asthma [23]. The positive modulatory and negative exacerbating effects of LPS may be dependent on further microbial antigens and genetic effects [19, 24–26]. For instance, sequence variants in the TLR4 gene can affect the responsiveness to LPS, and thereby influence the prevalence of asthma in a population [17, 19].
The degree of LPS sensitivity seems to be species-specific and horses are one of the most sensitive animals in their response to LPS exposure, while rodents appear to be much more resistant [11, 27, 28]. Endotoxaemia plays a major role in many equine diseases, particularly in intestinal disorders like acute colitis and ischemic bowel diseases, which often present with colic as the principal clinical manifestation [29]. Consequently, endotoxaemia is one of the main causes of mortality and morbidity in horses [30]. Horses suffering from colic have been shown to have increased LPS plasma concentrations and fatal colic cases had significantly higher LPS levels than non-fatal colic cases [31].
In equine recurrent airway obstruction (RAO, ‘equine asthma’), LPS does not cause the disease per se, which, similar to the pathophysiology of asthma, is due to a hypersensitivity to allergens in hay with a strong genetic basis [32–37]. However, hay dust can also contain high concentrations of LPS and inhaled endotoxin contributes to airway inflammation in RAO [38–40]. Furthermore, it is recognized that environmental exposure to microbial compounds, that do not result in clinical disease, but act through innate immune response mechanisms, influences the development of adaptive immunity and consequently allergy. Dendritic cells (DCs) are essential for priming T helper-2 differentiation of naïve T cells towards aeroallergens. However, contamination of antigens with PAMPs, such as LPS, is required to activate DCs to mount an immune response. Hammad et al. demonstrated in a mouse model of asthma that TLR4 triggering of epithelial cells, resulting in the release of innate pro-allergic cytokines, is necessary to drive allergic inflammation via activation of mucosal DCs [41]. To our knowledge equine global gene expression changes after LPS stimulation have not yet been analysed and reported. Genes function within networks that are typically redundant and regulatory mechanisms frequently assure that up- or down-regulation of a specific gene will be compensated by other genes [42]. It has been shown that studying groups of co-expressed genes, called gene modules, may better represent pathways of genes, which are co-regulated and/or interact with each other [43–47]. Weighted gene co-expression network analysis (WGCNA) is a tool that distinguishes modules of co-expressed genes by correlation and clustering analysis. The most highly connected genes within a module, ‘hub genes’, can then serve as good biomarkers that are characteristic for a phenotype studied, e.g. a disease.
Peripheral blood mononuclear cells (PBMCs) are a mix of several immune cell types circulating in the blood. They include cells that are involved in both the innate (monocytes, dendritic cells) and the acquired (lymphocytes) immune response systems. Therefore, PBMCs to a certain extent reflect the immune system status [48] and are widely used in systemic immune response studies [49–55].
Gene expression changes upon LPS challenge of immune cells have been intensively studied and well characterized in many mammalian species, but data in horses are limited despite the important impact of LPS both in equine gastrointestinal and respiratory diseases [29–31, 38–40]. Using a large RNA-seq dataset generated in the context of previous studies [50, 56], we report here the effect of 24 h in vitro LPS-stimulation on the transcriptome of equine PBMCs and the effect of genetic background on these LPS-induced transcriptomic changes.
Methods
Ethics statement
All animal experiments were performed according to the local regulations and with the consent of the horse owners. This study was approved by the Animal Experimentation Committee of the Canton of Bern, Switzerland (BE33/07, BE58/10 and BE10/13). The sample collection was previously described in detail in an earlier publication [49].
Samples, RNA extraction and RNA-seq
This study is based on the results generated in a previously reported experiment and therefore all methods in detail are described elsewhere [49, 50, 56]. Briefly, blood samples were collected from 41 adult Warmblood horses (free of RAO) from three cohorts: two half-sibling families (Fam1: 7 horses, Fam2: 9 horses) and a group of unrelated horses (Un: 25 horses). About 8 million PBMCs from each horse were stimulated with LPS or left unstimulated (mock) for 24 h and subsequently frozen at -80 °C until RNA was extracted. High quality RNA (RIN > 8) was used for paired-end library preparation and sequenced on an Illumina HiSeq 2500 with 2 x 50 sequencing cycles. After quality control the sequencing reads were mapped to the horse reference genome (EquCab2). Raw data in binary-sequence alignment format (BAM) are available from European Nucleotide Archive (ENA) (http://www.ebi.ac.uk/ena/data/view/PRJEB7497) and Short Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?study=ERP007230).
Differential expression analysis
Reads with mapping quality ≥ 20 were counted gene-wise with HTSeq [57] using the horse genome annotation from Ensembl (version 72) and default parameters. The differential expression analysis was performed with edgeR R package (Additional file 1) [58]. From the whole dataset used in our previous study [50, 56] we used 41 control PBMC samples that were cultured without any stimulating factor and 41 PBMC samples from the same horses stimulated with LPS and applied a filtering step in which we discarded all the genes with less than 10 counts in more than 90% of samples. As an initial quality control step we performed principal component analysis (PCA) using variance stabilized counts with individual horse effect removed using the removeBatchEffect function of limma R package (Additional file 1) [59, 60]. We next excluded outliers based on their Euclidean distances and visual inspection of the sample dendrogram (Fig. 1). Only horses with both mock and LPS stimulation were kept for further analysis (39 horses, 78 samples, Additional file 2).
Fig. 1.
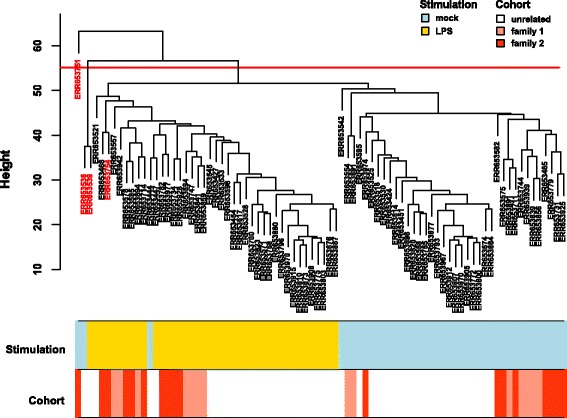
Sample hierarchical clustering. The samples clustered based on their Euclidean distance calculated with variance stabilized expression levels. The red line indicates the threshold line that excludes outliers from further analysis and excluded samples are marked in red in the dendrogram
For the model fitting, we applied a design that takes into account the effects of stimulation and cohort with interactions’ effects on the expression level:
The unrelated group of horses was set as the reference group. The model was then fitted using the generalized linear model implemented in edgeR [58]. The effect of each factor on gene expression was tested and genes that were differentially expressed with a significance level of false discovery rate [61], FDR < 0.001 were assumed to be differentially expressed genes (DEGs). The stringent FDR threshold was set based on the large number of DEGs identified, please see Results for details.
The DEGs identified were compared with two other (microarray) studies available in public databases, where human PBMCs were stimulated with LPS for 24 h: [62, 63]. Using the GEO2R tool [64] we retrieved the DEGs between the control and 24 h-LPS stimulated human PBMCs. While many probes in a microarray can represent a single gene, we used only those genes, for which all probes were consistently significantly regulated by LPS with the same direction of regulation (up or down). From our list of DEGs we used genes with a gene symbol (genes without associated gene names were filtered out for this comparison as no human homologous gene could be identified). A stringent significance threshold of FDR < 0.001 was applied for enlisting the DEGs.
To get insight into the biological function of the DEGs, they were subjected to enrichment analyses with PANTHER overrepresentation test (release 20160715) [65–67] using the gene ontology (GO) biological process terms [68] and all 20,374 Equus caballus genes available from the PANTHER database as the reference set. Only the most specific GO subclasses from a group of all related parent classes in ontology were considered. The PANTHER default Bonferroni corrected p-value (P-value) significance threshold of 0.05 was used.
Co-expressed network analysis
Signed co-expression networks were built using the WGCNA package in R [69] using variance stabilized counts with individual horse effect removed (Additional file 1) [59, 60]. BlockwiseModules function of the WGCNA package, which allows for network construction from the entire dataset, was used. For each set of genes a pair-wise correlation matrix was computed across the samples, and an adjacency matrix was calculated by raising the correlation matrix to the power of 12 using the scale-free topology criterion as suggested in [69].
Network interconnectedness (topological overlap measure) for each pair of the genes is calculated based on the adjacency matrix. The resulting topological overlap matrix is then converted to a dissimilarity measure and submitted to hierarchical clustering. The clustering produces a dendrogram, the branches of which represent similarly expressed genes, with the most highly connected nodal points or "hubs" located at the branch tips [69]. To cut the branches (cluster individual branches in separate "modules”), we used the hybrid dynamic tree-cutting because it leads to robustly defined modules [69]. We set the minimum module size to 40 genes and the minimum height for merging modules to 0.25 (Additional file 1).
Each module was summarized by the first principal component (i.e. eigengene) of the scaled (standardized) module expression profiles. The module eigengene is a single number that corresponds to the weighted average expression of all module genes in a sample [69]. For each module, the correlation between each gene expression values and module eigengene defines module membership (kME). The closer the absolute value of kME is to 1, the stronger the evidence that the gene belongs to the module represented by the module eigengene.
Significance of the LPS stimulation or cohort effect on the gene module was calculated using expression values of the module’s eigengene using limma package [60] with the same linear model as the one used for gene-wise differential expression analysis, without the horse effect removed before (Additional file 1). The genes initially assigned to the top two significant LPS-related modules and with high module membership (kME ≥ 0.7) were used for the enrichment analyses with PANTHER overrepresentation test [65] as described for DEGs.
Results
Differential expression analysis
We used a subset of a previously published RNA-seq dataset [50, 56]. Briefly, the 82 samples were derived from three different cohorts of healthy Warmblood horses: one group of horses that were unrelated at the parent level and two half-sibling families. The PCA showed a clear clustering of the samples according to the stimulation factor (Additional file 3). However, we removed three outliers based on the sample dendrogram (Fig. 1) plus one additional sample that matched the same horse as an outlier removed in order to keep only the samples with both mock and LPS stimulations required for the differential expression analysis. We identified 1,265 out of 12,855 analysed genes to be differentially expressed genes (DEGs) between LPS-stimulated and unstimulated samples in the reference group of 24 unrelated horses (FDR < 0.001, Additional file 4). The list of DEGs with associated gene names available (n = 1,167, 92% of all DEGs) partially overlapped DEGs from the other two studies on human PBMCs stimulated with LPS [62, 63]: 287 genes from the DEGs identified in this study were shared with at least one of the two other studies and 44 were shared by all three studies (Fig. 2). Log 2 fold changes (log2FC) of the DEGs identified in equine PBMCs ranged from -6.77 (insulin-like growth factor I, IGF1) to 4.81 (C-X-C motif chemokine 6 precursor, CXCL6).
Fig. 2.
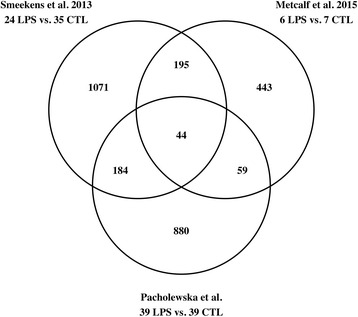
Number of common and study-specific DEGs. The venn diagram shows the overlap between the differentially expressed genes (DEGs) identified in equine (Pacholewska et al.) and human (Smeekens at al, Metcalf et al.) PBMCs stimulated with LPS for 24 h. From the equine gene list only genes with orthologous human gene symbols were examined. The number of replicates is denoted in the graph: LPS – samples stimulated with LPS, CTL – unstimulated control samples
The PANTHER overrepresentation test [65] showed that the DEGs identified are involved in many immune response related processes (Fig. 3a). Ten most specific GO subclasses with the highest fold enrichments (FEs) are shown in Fig. 3a. The FE among the ten GO terms ranged from 4.69 to 2.81 (P-value range: 2.80e-6 – 4.67e-2). The only significantly enriched PANTHER pathway was the inflammation mediated by chemokine and cytokine signalling pathway (P-value = 6.10e-3; FE = 2.22). Of the 1,265 DEGs 37 genes (3%) were not matched to the PANTHER horse genome reference set of genes.
Fig. 3.
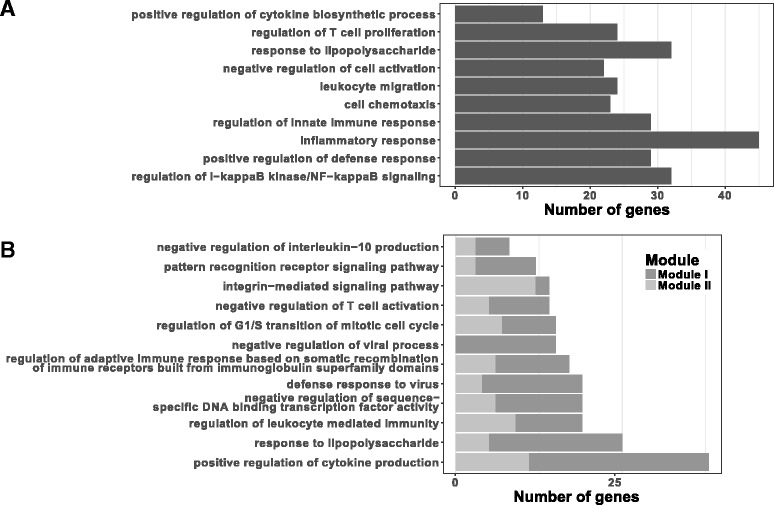
Number of DEGs annotated in the gene ontology (GO) biological processes. Most enriched and significant (Bonferroni corrected p-value < 0.05) biological processes annotated in the GO database and enriched by LPS-regulated genes identified in a) gene-wise and b) gene module-wise expression analysis (based on the GO database, release 2016-10-27 [65–67]). Each bar represents the number of genes enriched in the GO biological process. Bars are sorted by the fold enrichment with the most enriched processes on top
Influence of the genetic background
We identified 16 genes (Additional file 4) that responded differently to stimulation with LPS in family 1 and 155 in family 2 compared to the unrelated cohort (Additional file 4, LPS:Fam1 and LPS:Fam2 effects). The LPS:Fam2 genes significantly enriched two GO biological processes: the nucleic acid metabolic process (32 genes, P-value = 1.86e-2), and cellular macromolecule metabolic processes (65 genes, P-value = 3.54e-6).
Co-expression analysis
Normalized read counts per gene for the 78 samples grouped into two clusters accordingly to non- or LPS-stimulated group (Fig. 1). WGCNA identified 30 modules of co-expressed genes that consisted of 53 to 1,542 genes (Additional file 5). Only 636 genes (5%) were not included in any of the modules (by default WGCNA groups such genes into a dummy “grey” module). Every module was represented by its first principal component called the module eigengene (see methods for details). The association of eigengene expression with LPS stimulation and cohort was investigated using the linear model as explained in the Methods section. Of the 30 modules, ten were significantly associated with LPS stimulation in the reference group of 24 unrelated horses (FDR < 0.001), five modules were significant for the LPS:Fam2 interaction effect (module eigengenes responded differently to LPS stimulation in family 2 compared to unrelated cohort), and the most significant LPS:Fam2 module was also significant for the LPS:Fam1 effect (Additional file 6).
The top significantly LPS-related modules (I and II) clearly divided the samples into two separated clusters of LPS-stimulated or unstimulated cells as shown in Fig. 4. The barplots represent the expression of the module eigengene and heatmaps the expression of the module genes (kME ≥ 0.7) across the samples. The genes with the strongest correlation (membership) with the top two LPS-related modules’ eigengenes are listed in Table 1.
Fig. 4.
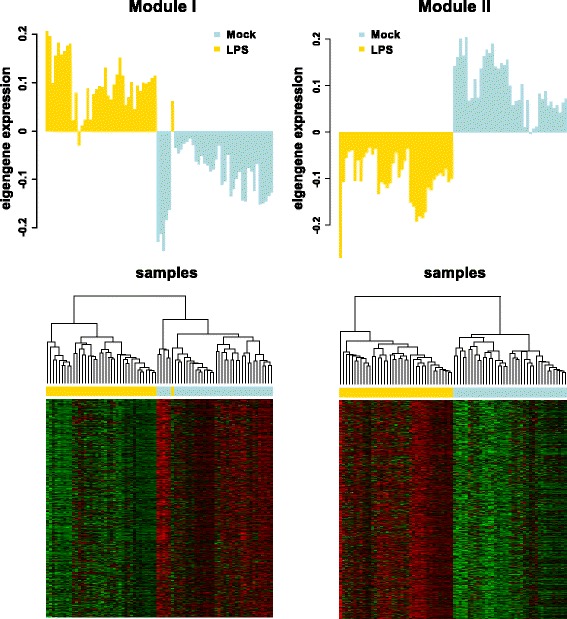
The two most significantly LPS-associated gene modules across samples. Bar plots represent the expression level of the module eigengenes across the samples. The light blue colour represents control mock-stimulated samples; the yellow colour represents samples stimulated with LPS. Heatmaps show the expression levels of all the module gene members with kME ≥ 0.7 (rows) across samples (columns). The expression values were scaled by rows and denoted by colour from red (low) to green (high). Dendrograms of samples were produced using hierarchical cluster analysis
Table 1.
The genes with the strongest membership with the top two LPS-related modules
| Module | Module hub genes | Module membership | LPS log2FC | DE FDR |
|---|---|---|---|---|
| module I | BIRC3 | 0.97 | 1.26 | 1.89e-12 |
| module I | VPS37A | 0.95 | 0.53 | 3.97e-9 |
| module I | STARD13 | 0.94 | 0.80 | 1.01e-4 |
| module I | SOD2 | 0.94 | 1.75 | 8.83e-16 |
| module I | SLC1A2 | 0.94 | 2.83 | 3.62e-18 |
| module I | ARHGEF3 | 0.94 | 0.53 | 3.39e-8 |
| module I | TTC25 | 0.94 | 0.84 | 5.30e-8 |
| module I | SLC39A14 | 0.93 | 1.80 | 2.80e-12 |
| module I | ENSECAG00000002023, superoxide dismutase | 0.93 | 1.79 | 5.69e-16 |
| module I | TREX1 | 0.93 | 0.85 | 2.25e-7 |
| module II | VAT1 | 0.95 | −1.13 | 1.68e-16 |
| module II | SDC3 | 0.94 | −2.27 | 1.16e-9 |
| module II | FCGRT | 0.94 | −1.39 | 1.42e-10 |
| module II | SLC46A1 | 0.94 | −1.80 | 8.21e-18 |
| module II | NAAA | 0.94 | −0.94 | 2.53e-20 |
| module II | CSF1R | 0.93 | −1.54 | 1.43e-11 |
| module II | MAN2B1 | 0.93 | −0.44 | 5.77e-10 |
| module II | ARRB1 | 0.92 | −1.03 | 3.19e-9 |
| module II | GAMT | 0.92 | −2.40 | 3.77e-19 |
| module II | S100A4 | 0.92 | −1.10 | 4.29e-12 |
For every module the ten most correlated genes are listed with the following features: module membership, log2 fold change of the expression regulated by LPS, false discovery rate for the gene-wise differential expression (DE)
The I and II module genes (with kME ≥ 0.7) were further subjected to the enrichment analysis. We retrieved all 12 significant GO biological processes with FE ≥ 2.81 (FE ≥ of the tenth most enriched biological process by the LPS-induced DEGs). FE among the 12 GO terms ranged from 12.74 to 3.11 (P-value range: 1.27e-5 – 3.1e-2). The processes were mostly related to the immune response and included the response to LPS, as well as a cell cycle related process (Fig. 3b). The most enriched PANTHER GO-Slim biological process was the negative regulation of apoptotic process (P-value = 4.72e-2), and none of the PANTHER pathway was significantly enriched. Of the 934 module I and II genes 20 (2%) were not matched to the PANTHER horse genome reference set of genes.
Discussion
LPS is known to be a strong inducer of the innate immune response and changes in the gene expression can be observed within one hour post LPS challenge [3, 70]. Actually, the majority of the studies on the LPS-induced immune response focus on short-term effects. In contrast, this study investigated the consequences of a 24 h LPS-challenge, which may better reflect continued exposure to LPS, which is the cause of increased morbidity and mortality in equine gastrointestinal [29–31] and respiratory diseases [38–40]. As expected, in the equine PBMCs studied here, 24 h LPS stimulation affected the regulation of a substantial number of genes, almost 10% of all genes studied at a FDR < 0.001. This fraction increased to more than 24% of the studied genes when a less stringent FDR threshold of < 0.05 was applied. The magnitude of this effect was in accordance with previous reports using genome-wide expression analyses demonstrating that LPS strongly affects the transcriptomic profile of blood cells revealed by in vitro and in vivo studies in humans [62, 63, 71–74].
While the horse is more sensitive to LPS than other mammals that have been studied (including humans) [27], the innate immune response to LPS is very similar across species. This includes specific cell types and mediators involved in the inflammatory cascade following LPS challenge and is likely due to the fact that innate immunity is a highly conserved, evolutionarily old immune response [10].
Interestingly, our results from the differential expression analysis agreed in part with similar studies on human LPS-stimulated PBMCs [62, 63] (Fig. 2). Almost a quarter of DEGs identified in our study overlapped DEGs from both the studies on human PBMCs. It has to be also taken into account that the overlap between the two human studies was also surprisingly small: only 32% of DEGs identified by Metcalf et al. was identified in the study of Smeekens et al.; and only 15% vice versa. From a total of 239 common DEGs between the two studies on human PBMCs, 18% were also identified in our study. While these two studies focused on the defence against Candida albicans and the age effect on innate response, they employed the same duration of 24 h-LPS-stimulation as in our study [62, 63]. Moreover, many of the equine LPS-regulated genes (8%) did not have human homologs and therefore had to be excluded from this comparison.
Although the immunological reaction to LPS has already been described in several species, the response to LPS is still poorly investigated in horses with only 41 unique equine proteins listed under the GO term ‘response to LPS’ (GO: 0032496, state of 23rd September 2016) with evidence based on computational analysis. Of these 41 protein-coding genes 18 were also supported by our study at FDR < 0.05 (CD40, CXCL2, CXCL6, CXCL8, F2R, IL18, MIP-2BETA, PPBP, TLR4, TNFRSF11B, TNFRSF18, TNFRSF1B, TNFRSF21, TNFRSF4, TNFRSF8, ENSECAG00000012397, ENSECAG00000012560, ENSECAG00000015037) and 11 when the stringent threshold was applied (FDR < 0.001). The most up-regulated gene in our dataset, the chemokine CXCL6, attracts neutrophils that are known to be involved in the first line of the immune response [75, 76]. Among the up-regulated DEGs, there were also genes previously identified as LPS-induced genes in humans at the mRNA (using reverse transcriptase quantitative PCR) or protein level (e.g. IL1B, IL6, IL8 (also known as CXCL8), CCL22, CXCL6) [75].
Previous genome-wide transcriptome profiling of whole blood samples derived from paediatric patients with SIRS, sepsis, or septic shock revealed putative use of circulating IL27 in the blood as a biomarker for sepsis in human patients [77, 78]. Interestingly, in our study both genes encoding for the two subunits of IL27 (IL27 and EBI3) were up-regulated in LPS stimulated cells.
It is worth mentioning that sepsis is characterized by a ‘cytokine storm’ in the first phase, followed by a second phase, after approximately 24 h, where the immune response is ‘paralysed’ due to the apoptosis of activated lymphocytes [79–81]. The suppression of the immune response increases the risk of secondary infections in patients with sepsis, which in turn represent a major cause of death in these patients [82]. The apoptosis during immune paralysis in sepsis is believed to be induced by caspase signals, namely death receptor induced caspase 8 (CASP8) or mitochondrial induced caspase 9 (CASP9) [83, 84]. Although we did not identify significant changes in the expression levels of CASP8 or CASP9, the most markedly down-regulated gene by the LPS stimulation was the insulin-like growth factor 1 gene (IGF1). IGF1 has been shown to have an anti-apoptotic effect [85–88]. This decrease was in accordance with another study in which LPS was shown to suppress the expression of IGF1 in post mortem derived human microglia on both mRNA and protein levels [89].
Because many genes’ functions are still unknown and because genes often work within complex networks, we hypothesized that the analyses of co-expressed groups of genes could facilitate a deeper insight into LPS-induced immune responses. Therefore, we assigned genes to modules based on their co-expression. Ten out of 30 modules identified possessed significant relationships with LPS stimulation.
Interestingly, our analysis revealed that horse family 2 is more distant from the unrelated group in their response to LPS than family 1, based on both differential expression and co-expression analysis (Additional files 4 and 6). We noticed earlier that occurrence of equine asthma (RAO) is correlated with an increased resistance against parasites in unrelated Warmblood horses and in family 1, but not in family 2 [49, 50, 90, 91]. Hub genes of the three LPS:Fam2 specific modules that were not significant for LPS:Fam1 may thus play a role in the response to parasites.
As expected, some of the LPS-related modules identified in this study confirmed previous findings in regards to our knowledge of LPS-induced immune responses, but our analysis also revealed interesting novel aspects. Genes of the two most strongly LPS-related modules showed clear differences in the expression in stimulated versus unstimulated PBMCs, as shown in Fig. 4. Among the top significant LPS-related module genes there were genes involved in immune response, including the response to LPS (Fig. 3b). Indeed, the WGCNA analysis revealed 272 genes from the two modules not identified as LPS-regulated based on gene-wise differential expression analysis at FDR < 0.001. Only 47 of these were not DEGs at the more permissive FDR threshold of 0.05.
Although module I included one of the hallmark-genes of LPS-stimulation, IL6, the hub genes in this module were BIRC3, encoding the baculoviral IAP repeat containing 3, and VPS37A, encoding vacuolar protein sorting 37 homolog A. BIRC3 was already implicated in the pro-inflammatory cytokine induction [92] and VPS37A in the inhibition of viral infection [93]. The third hub gene was StAR related lipid transfer domain containing 13 (STARD13), which is believed to supress cell proliferation and motility [94] and therefore may be important for the regulation of the immune response upon stimulation with LPS.
Even more interesting are hub genes from the LPS-downregulated module: albeit the function of the vesicle amine transport 1 protein encoded by VAT1 gene has not been fully described, it has been shown that this gene is expressed in human neutrophils and VAT1 protein is located peripherally in unstimulated neutrophils with a more granular pattern upon stimulation with a macrophage activator [95, 96]. Since our collection of PBMCs did not contain neutrophils, the function of VAT1 in PBMCs requires further investigation.
Surprisingly, we identified up- and down-regulated genes involved in both positive and negative regulation of apoptosis, e.g. the anti-apoptotic IGF1 gene, which was the most down-regulated DEG in LPS-stimulated samples. Apart from IGF1 module I hub genes included guanidinoacetate N-gethyltransferase gene (GAMT) that was also decreased in LPS-samples. This gene may have also an anti-apoptotic role as it has been reported that fibroblast cell lines from two GAMT-deficient patients showed increased production of mitochondrial reactive oxygen species and apoptotic rate [97]. As apoptosis induces immunosuppression [98], the reduced expression of the anti-apoptotic genes (IGF1, GAMT) may have a role in the immune suppression phase of sepsis and support the concept of LPS induced immune regulation/paralysis.
Another indication of the immune regulation by long-term stimulation with LPS in our study was the down-regulation of another hub gene, syndecan 3 (SDC3, module I). SDC3 serves as a synovial binding site for IL-8 on endothelial cells [99] thus down-regulation of SDC3 results in a decrease in binding sites for the inflammatory IL8. The immune regulation/paralysis could be further supported by the down-regulation of the N-acylethanolamine acid amidase (NAAA) and Fc fragment of IgG receptor and transporter gene (FCGRT), module II hub genes. NAAA degrades N-acylethanolamines in macrophages and is involved in inflammatory processes [100] and has previously been reported as potential anti-inflammatory agent [101–104]. FCGRT protects immunoglobulin G from degradation, thus its expression reduced may be the result of the immune response regulatory mechanisms [105].
What is noteworthy is that none of the 20 hub genes of the I and II modules listed in Table 1 has been annotated in the GO term response to lipopolysaccharides (GO:0032496). Among these hub genes was also the tetratricopeptide repeat domain 25 gene (TTC25) that has been poorly characterized so far. Nonetheless, this gene seems to play an important biological role in the respiratory tract and other organ systems that depend on intact ciliary function, as it has been recently reported that mutations in the TTC25 lead to primary ciliary dyskinesia [106]. However, we also need to caution that this study was performed on RNA-seq data only and individual gene specific function and their role in LPS-related pathways would still require further functional analysis.
Conclusions
The WGCNA revealed novel aspects in an already much studied field, such as the response to LPS. We identified two co-expressed gene modules that clearly separated LPS- from non-stimulated cells. Apart from known LPS-response genes (e.g. IL6, IL8, CXCL6) the two modules identified comprised novel genes with potential roles in LPS-response pathways (e.g. VAT1 and TTC25). Moreover, the regulation of genes involved in the apoptotic process in our data suggests that 24 h stimulation with LPS may reflect the ‘immune paralysis’ phase observed in patients with bacterial sepsis. In addition, we further confirmed that the family 2 horses differ in their immune response from family 1 and unrelated horses.
Acknowledgements
The authors would like to thank all participating horse owners and veterinarians for their support of this study. We would like to thank also our colleagues involved in the design and the experimental part of the project: Emmanouil T. Dermitzakis, Michaela Drögemüller, Jolanta Klukowska-Rötzler, Simone Lanz, Eman Hamza, and Muriel Fragnière. We also appreciate the support of the Next Generation Sequencing Platform of the University of Bern, the Genomics Platform at the University of Geneva Medical Center, and the Vital-IT high-performance computing centre of the Swiss Institute of Bioinformatics (http://www.vital-it.ch). Moreover, we thank the reviewer for his/her thorough review and valuable comments.
Funding
The study was funded by Swiss National Science Foundation: grant No. 310030-138295 and 310000-116803/1; and Swiss Institute of Equine Medicine Research.
Availability of data and materials
Raw data in binary-sequence alignment format (BAM) are available from European Nucleotide Archive (http://www.ebi.ac.uk/ena/data/view/PRJEB7497).
Authors' contributions
VG, TL, and EM conceived and designed the experiments. AP and VJ analyzed the data. AP, VJ, TL, VG, and EM wrote the paper. All authors read and approved the final manuscript.
Authors’ information
Current address for AP: Center for non-coding RNA in Technology and Health, Department of Veterinary Clinical and Animal Sciences, University of Copenhagen , Grønnegårdsvej 3, 1870 Frederiksberg C, Denmark.
Competing interests
The authors have declared that no competing interests exist.
Abbreviations
- CARS
Compensatory anti-inflammatory response
- DC
Dendritic cell
- DEG
Differentially expressed gene
- FDR
False discovery rate
- FE
Fold enrichment
- GO
Gene ontology
- kME
Module membership
- Log2FC
Log 2 fold change
- LPS
Lipopolysaccharides
- PAMP
Pathogen associated molecular pattern
- PBMC
Peripheral blood mononuclear cell
- PCA
Principal component analysis
- RAO
Recurrent airway obstruction (horse asthma)
- SIRS
Systemic inflammatory response
- WGCNA
Weighted gene co-expression network analysis.
Additional files
Script used for the data analysis. An R script used for the gene-wise differential expression analysis with edgeR [58] and weighted gene co-expression network analysis with WGCNA R package [69]. (PDF 38 kb)
Data used for the analysis in R. An RData file that contains compressed objects required for the differential expression and co-expression analyses with the script from Additional file 1. It contains the count and design tables. (RDATA 2007 kb)
Principal component analysis. Principal components were calculated on variance stabilized counts before and after implementing the removeBatchEffect function of limma R package [59, 60]. All 82 samples were plotted across two first principal components and coloured according to the stimulation (blue – unstimulated; yellow – stimulated with LPS). (PDF 42 kb)
Lists of DEGs between LPS stimulated and unstimulated PBMCs. For every effect studied (LPS, and interaction effects: LPS:Fam1, LPS:Fam2) the list of genes differentially expressed at false discovery rate < 0.001 with log 2 fold changes is given. (XLSX 198 kb)
Dendrogram of genes assigned to the modules. The colours in the bar below the dendrogram represent the modules. The grey colour represents genes not assigned to any of the modules. The dendrogram was obtained by average linkage hierarchical clustering [69]. (PDF 255 kb)
Co-expressed gene modules. For every module the list of genes with module membership, and FDR values of the module association with LPS stimulation and cohort is given. (XLSX 6286 kb)
Contributor Information
Alicja Pacholewska, Email: alicja@rth.dk.
Eliane Marti, Email: eliane.marti@vetsuisse.unibe.ch.
Tosso Leeb, Email: tosso.leeb@vetsuisse.unibe.ch.
Vidhya Jagannathan, Email: vidhya.jagannathan@vetsuisse.unibe.ch.
Vincent Gerber, Email: vinzenz.gerber@vetsuisse.unibe.ch.
References
- 1.Janeway CA. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54 Pt 1:1–13. doi: 10.1101/SQB.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Osborn MJ, Rosen SM, Rothfield L, Zeleznick LD, Horecker BL. Lipopolysaccharide of the Gram-negative cell wall. Science. 1964;145:783–9. doi: 10.1126/science.145.3634.783. [DOI] [PubMed] [Google Scholar]
- 3.Schütt C. Fighting infection: the role of lipopolysaccharide binding proteins CD14 and LBP. Pathobiology. 1999;67:227–9. doi: 10.1159/000028097. [DOI] [PubMed] [Google Scholar]
- 4.Tobias PS, Soldau K, Ulevitch RJ. Isolation of a lipopolysaccharide-binding acute phase reactant from rabbit serum. J Exp Med. 1986;164:777–93. doi: 10.1084/jem.164.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 6.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–7. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 7.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 8.Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85(2):85–95. doi: 10.1016/S0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- 9.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 10.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–73. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;4:854–65. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 12.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–8. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 13.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 14.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest. 1992;101(6):1644–55. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein B, Giroir B, Randolph A. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005;6(1):2–8. doi: 10.1097/01.PCC.0000149131.72248.E6. [DOI] [PubMed] [Google Scholar]
- 16.Ulloa L, Tracey KJ. The “cytokine profile”: A code for sepsis. Trends Mol Med. 2005;11(2):56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Liu AH. Endotoxin exposure in allergy and asthma: Reconciling a paradox. J Allergy Clin Immunol. 2002;109(3):379–92. doi: 10.1067/mai.2002.122157. [DOI] [PubMed] [Google Scholar]
- 18.Michel O. Systemic and local airways inflammatory response to endotoxin. Toxicology. 2000;152:25–30. doi: 10.1016/S0300-483X(00)00288-2. [DOI] [PubMed] [Google Scholar]
- 19.Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–91. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 20.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196:1645–51. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braun-Fahrländer C, Riedler J, Herz U, Eder W, Waser M, Grize L, et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N Engl J Med. 2002;347:869–77. doi: 10.1056/NEJMoa020057. [DOI] [PubMed] [Google Scholar]
- 22.Tulić MK, Wale JL, Holt PG, Sly PD. Modification of the inflammatory response to allergen challenge after exposure to bacterial lipopolysaccharide. Am J Respir Cell Mol Biol. 2000;22:604–12. doi: 10.1165/ajrcmb.22.5.3710. [DOI] [PubMed] [Google Scholar]
- 23.Reed CE, Milton DK. Endotoxin-stimulated innate immunity: A contributing factor for asthma. J Allergy Clin Immunol. 2001;108(2):157–66. doi: 10.1067/mai.2001.116862. [DOI] [PubMed] [Google Scholar]
- 24.Yazdani N, Amoli MM, Naraghi M, Mersaghian A, Firouzi F, Sayyahpour F, et al. Association between the functional polymorphism C-159 T in the CD14 promoter gene and nasal polyposis: Potential role in asthma. J Investig Allergol Clin Immunol. 2012;22:406–11. [PubMed] [Google Scholar]
- 25.Baldini M, Lohman IC, Halonen M, Erickson RP, Holt PG, Martinez FD. A Polymorphism* in the 5’ flanking region of the CD14 gene is associated with circulating soluble CD14 levels and with total serum immunoglobulin E. Am J Respir Cell Mol Biol. 1999;20:976–83. doi: 10.1165/ajrcmb.20.5.3494. [DOI] [PubMed] [Google Scholar]
- 26.Sackesen C, Birben E, Soyer OU, Sahiner UM, Yavuz TS, Civelek E, et al. The effect of CD14 C159T polymorphism on in vitro IgE synthesis and cytokine production by PBMC from children with asthma. Allergy Eur J Allergy Clin Immunol. 2011;66:48–57. doi: 10.1111/j.1398-9995.2010.02428.x. [DOI] [PubMed] [Google Scholar]
- 27.Singh Suri S, Janardhan KS, Parbhakar O, Caldwell S, Appleyard G, Singh B, et al. Expression of toll-like receptor 4 and 2 in horse lungs. Vet Res. 2006;37:541–51. doi: 10.1051/vetres:2006017. [DOI] [PubMed] [Google Scholar]
- 28.Warren HS. Editorial: Mouse models to study sepsis syndrome in humans. J Leukoc Biol. 2009;86:199–201. doi: 10.1189/jlb.0309210. [DOI] [PubMed] [Google Scholar]
- 29.Werners AH, Bull S, Fink-Gremmels J. Endotoxaemia: a review with implications for the horse. Equine Vet J. 2005;37:371–83. doi: 10.2746/0425164054529418. [DOI] [PubMed] [Google Scholar]
- 30.Sykes BW, Furr MO. Equine endotoxaemia - A state-of-the-art review of therapy. Aust Vet J. 2005;83:45–50. doi: 10.1111/j.1751-0813.2005.tb12191.x. [DOI] [PubMed] [Google Scholar]
- 31.Senior JM, Proudman CJ, Leuwer M, Carter SD. Plasma endotoxin in horses presented to an equine referral hospital: Correlation to selected clinical parameters and outcomes. Equine Vet J. 2011;43:585–91. doi: 10.1111/j.2042-3306.2010.00328.x. [DOI] [PubMed] [Google Scholar]
- 32.Gerber V, Baleri D, Klukowska-Rötzler J, Swinburne JE, Dolf G. Mixed inheritance of equine recurrent airway obstruction. J Vet Intern Med. 2009;23:626–30. doi: 10.1111/j.1939-1676.2009.0292.x. [DOI] [PubMed] [Google Scholar]
- 33.Jost U, Klukowska-Rötzler J, Dolf G, Swinburne JE, Ramseyer A, Bugno M, et al. A region on equine chromosome 13 is linked to recurrent airway obstruction in horses. Equine Vet J. 2007;39:236–41. doi: 10.2746/042516407X171110. [DOI] [PubMed] [Google Scholar]
- 34.Swinburne JE, Bogle H, Klukowska-Rötzler J, Drögemüller M, Leeb T, Temperton E, et al. A whole-genome scan for recurrent airway obstruction in Warmblood sport horses indicates two positional candidate regions. Mamm Genome. 2009;20:504–15. doi: 10.1007/s00335-009-9214-5. [DOI] [PubMed] [Google Scholar]
- 35.Ramseyer A, Gaillard C, Burger D, Straub R, Jost U, Boog C, et al. Effects of genetic and environmental factors on chronic lower airway disease in horses. J Vet Intern Med. 2007;21:149–56. doi: 10.1111/j.1939-1676.2007.tb02941.x. [DOI] [PubMed] [Google Scholar]
- 36.Gerber V, Tessier C, Marti E. Genetics of upper and lower airway diseases in the horse. Equine Vet J. 2014;1–8. [DOI] [PubMed]
- 37.Marti E, Gerber H, Essich G, Oulehla J, Lazary S. The genetic basis of equine allergic diseases. 1. Chronic hypersensitivity bronchitis. Equine Vet J. 1991;23:457–60. doi: 10.1111/j.2042-3306.1991.tb03761.x. [DOI] [PubMed] [Google Scholar]
- 38.Pirie RS, Dixon PM, McGorum BC. Endotoxin contamination contributes to the pulmonary inflammatory and functional response to Aspergillus fumigatus extract inhalation in heaves horses. Clin Exp Allergy. 2003;33:1289–96. doi: 10.1046/j.1365-2745.2003.01679.x. [DOI] [PubMed] [Google Scholar]
- 39.Pirie RS, Dixon PM, Collie DD, McGorum BC. Pulmonary and systemic effects of inhaled endotoxin in control and heaves horses. Equine Vet J. 2001;33:311–8. doi: 10.2746/042516401776249732. [DOI] [PubMed] [Google Scholar]
- 40.Pirie RS, Collie DDS, Dixon PM, McGorum BC. Inhaled endotoxin and organic dust particulates have synergistic proinflammatory effects in equine heaves (organic dust-induced asthma) Clin Exp Allergy. 2003;33:676–83. doi: 10.1046/j.1365-2222.2003.01640.x. [DOI] [PubMed] [Google Scholar]
- 41.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15:410–6. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nowak MA, Boerlijst MC, Cooke J, Smith JM. Evolution of genetic redundancy. Nature. 1997;388:167–71. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- 43.Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–55. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 44.Aggarwal A, Guo DL, Hoshida Y, Yuen ST, Chu KM, So S, et al. Topological and functional discovery in a gene coexpression meta-network of gastric cancer. Cancer Res. 2006;66:232–41. doi: 10.1158/0008-5472.CAN-05-2232. [DOI] [PubMed] [Google Scholar]
- 45.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–94. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo F, Yang Y, Zhong J, Gao H, Khan L, Thompson D, et al. Constructing gene co-expression networks and predicting functions of unknown genes by random matrix theory. BMC Bioinformatics [Internet] BioMed Central. 2007;8:1–17. doi: 10.1186/1471-2105-8-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crowther D, Fairley GH, Sewell RL. Lymphoid cellular responses in the blood after immunization in man. J Exp Med. 1969;129:849–69. doi: 10.1084/jem.129.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lanz S, Gerber V, Marti E, Rettmer H, Klukowska-Rötzler J, Gottstein B, et al. Effect of hay dust extract and cyathostomin antigen stimulation on cytokine expression by PBMC in horses with recurrent airway obstruction. Vet Immunol Immunopathol. 2013;155:229–37. doi: 10.1016/j.vetimm.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 50.Pacholewska A, Jagannathan V, Drögemüller M, Klukowska-Rötzler J, Lanz S, Hamza E, et al. Impaired Cell Cycle Regulation in a Natural Equine Model of Asthma. PLoS One. 2015;10:e0136103. doi: 10.1371/journal.pone.0136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ernst M, Kern P, Flad H-D, Ulmer AJ. Effects of systemicin vivo interleukin-2 (IL-2) reconstitution in patients with acquired immune deficiency syndrome (AIDS) and AIDS-related complex (ARC) on phenotypes and functions of peripheral blood mononuclear cells (PBMC). J. Clin. Immunol. [Internet]. 6:170–81. Available from: http://dx.doi.org/10.1007/BF00918750 [DOI] [PubMed]
- 52.Moreno J, Nieto J, Chamizo C, González F, Blanco F, Barker DC, et al. The immune response and PBMC subsets in canine visceral leishmaniasis before, and after, chemotherapy. Vet Immunol Immunopathol. 1999;71:181–95. doi: 10.1016/S0165-2427(99)00096-3. [DOI] [PubMed] [Google Scholar]
- 53.Kierstead LS, Dubey S, Meyer B, Tobery TW, Mogg R, Fernandez VR, et al. Enhanced rates and magnitude of immune responses detected against an HIV vaccine: effect of using an optimized process for isolating PBMC. AIDS Res Hum Retroviruses. 2007;23:86–92. doi: 10.1089/aid.2006.0129. [DOI] [PubMed] [Google Scholar]
- 54.Hutchins WA, Kieber-Emmons T, Carlone GM, Westerink MA. Human immune response to a peptide mimic of Neisseria meningitidis serogroup C in hu-PBMC-SCID mice. Hybridoma. 1999;18:121–9. doi: 10.1089/hyb.1999.18.121. [DOI] [PubMed] [Google Scholar]
- 55.Meade KG, Gormley E, Park SDE, Fitzsimons T, Rosa GJM, Costello E, et al. Gene expression profiling of peripheral blood mononuclear cells (PBMC) from Mycobacterium bovis infected cattle after in vitro antigenic stimulation with purified protein derivative of tuberculin (PPD) Vet Immunol Immunopathol. 2006;113:73–89. doi: 10.1016/j.vetimm.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 56.Pacholewska A, Drögemüller M, Klukowska-Rötzler J, Lanz S, Hamza E, Dermitzakis ET, et al. The transcriptome of equine peripheral blood mononuclear cells. PLoS One. 2015;10:e0122011. doi: 10.1371/journal.pone.0122011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2014;31:166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huber W, von Heydebreck A, Sültmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data alibration and to the quantification of differential expression. Bioinformatics. 2002;18:S96–104. doi: 10.1093/bioinformatics/18.suppl_1.S96. [DOI] [PubMed] [Google Scholar]
- 60.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- 62.Smeekens SP, Ng A, Kumar V, Johnson MD, Plantinga TS, van Diemen C, et al. Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. Nat Commun. 2013;4:1342. doi: 10.1038/ncomms2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Metcalf TU, Cubas RA, Ghneim K, Cartwright MJ, Van GJ, Richner JM, et al. Global analyses revealed age-related alterations in innate immune responses after stimulation of pathogen recognition receptors. Aging Cell. 2015;14(3):421–32. doi: 10.1111/acel.12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: Archive for functional genomics data sets - Update. Nucleic Acids Res. 2013;41. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed]
- 65.Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: Expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016;44:D336–42. doi: 10.1093/nar/gkv1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–66. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaudet P, Chisholm R, Berardini T, Dimmer E, Engel SR, Fey P, et al. The gene ontology’s reference genome project: A unified framework for functional annotation across species. PLoS Comput. Biol. 2009;5. doi: 10.1371/journal.pcbi.1000431. [DOI] [PMC free article] [PubMed]
- 68.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suzuki T, Hashimoto S, Toyoda N, Nagai S, Yamazaki N, Dong HY, et al. Comprehensive gene expression profile of LPS-stimulated human monocytes by SAGE. Blood. 2000;96:2584–91. [PubMed] [Google Scholar]
- 71.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–7. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 72.Cobb JP, Moore EE, Hayden DL, Minei JP, Cuschieri J, Yang J, et al. Validation of the riboleukogram to detect ventilator-associated pneumonia after severe injury. Ann Surg. 2009;250:531–9. doi: 10.1097/SLA.0b013e3181b8fbd5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wong HR, Shanley TP, Sakthivel B, Cvijanovich N, Lin R, Allen GL, et al. Genome-level expression profiles in pediatric septic shock indicate a role for altered zinc homeostasis in poor outcome. Physiol Genomics. 2007;30:146–55. doi: 10.1152/physiolgenomics.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scicluna BP, van’t Veer C, Nieuwdorp M, Felsmann K, Wlotzka B, Stroes ESG, et al. Role of tumor necrosis factor-α in the human systemic endotoxin-induced transcriptome. PLoS One. 2013;8 doi: 10.1371/journal.pone.0079051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pu D, Wang W. Toll-like receptor 4 agonist, lipopolysaccharide, increases the expression levels of cytokines and chemokines in human peripheral blood mononuclear cells. Exp Ther Med. 2014;8:1914–8. doi: 10.3892/etm.2014.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wuyts A, Struyf S, Gijsbers K, Schutyser E, Put W, Conings R, et al. The CXC chemokine GCP-2/CXCL6 is predominantly induced in mesenchymal cells by interleukin-1beta and is down-regulated by interferon-gamma: comparison with interleukin-8/CXCL8. Lab Invest. 2003;83:23–34. doi: 10.1097/01.LAB.0000048719.53282.00. [DOI] [PubMed] [Google Scholar]
- 77.Scicluna BP, van der Poll T. Interleukin-27: a potential new sepsis biomarker exposed through genome-wide transcriptional profiling. Crit Care. 2012;16:188. doi: 10.1186/cc11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wong HR, Cvijanovich NZ, Hall M, Allen GL, Thomas NJ, Freishtat RJ, et al. Interleukin-27 is a novel candidate diagnostic biomarker for bacterial infection in critically ill children. Crit. Care. 2012. p. R213. doi: 10.1186/cc11847. [DOI] [PMC free article] [PubMed]
- 79.Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM, et al. Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun. 2010;78:1582–92. doi: 10.1128/IAI.01213-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–51. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 81.Hotchkiss RS, Opal SM. Immunotherapy for Sepsis: a new approach against an ancient foe. N Engl J Med. 2010;363:87–9. doi: 10.1056/NEJMcibr1004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Otto GP, Sossdorf M, Claus RA, Rödel J, Menge K, Reinhart K, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit. Care. BioMed Central; 2011;15:R183–R183. [DOI] [PMC free article] [PubMed]
- 83.Hotchkiss RS, Osmon SB, Chang KC, Wagner TH, Coopersmith CM, Karl IE. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J Immunol. 2005;174:5110–8. doi: 10.4049/jimmunol.174.8.5110. [DOI] [PubMed] [Google Scholar]
- 84.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813–22. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 85.Fernández M, Sánchez-Franco F, Palacios N, Sánchez I, Fernández C, Cacicedo L. IGF-I inhibits apoptosis through the activation of the phosphatidylinositol 3-kinase/Akt pathway in pituitary cells. J Mol Endocrinol. 2004;33:155–63. doi: 10.1677/jme.0.0330155. [DOI] [PubMed] [Google Scholar]
- 86.Castrillo A, Bodelón OG, Boscá L. Inhibitory effect of IGF-I on type 2 nitric oxide synthase expression in Ins-1 cells and protection against activation-dependent apoptosis: Involvement of phosphatidylinositol 3-kinase. Diabetes. 2000;49:209–17. doi: 10.2337/diabetes.49.2.209. [DOI] [PubMed] [Google Scholar]
- 87.Willaime-Morawek S, Arbez N, Mariani J, Brugg B. IGF-I protects cortical neurons against ceramide-induced apoptosis via activation of the PI-3 K/Akt and ERK pathways; is this protection independent of CREB and Bcl-2? Mol Brain Res. 2005;142:97–106. doi: 10.1016/j.molbrainres.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 88.Kooijman R. Regulation of apoptosis by insulin-like growth factor (IGF)-I. Cytokine Growth Factor Rev. 2006;17:305–23. doi: 10.1016/j.cytogfr.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 89.Suh H-S, Zhao M-L, Derico L, Choi N, Lee SC. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: differential regulation by inflammatory mediators. J Neuroinflammation. 2013;10:37. doi: 10.1186/1742-2094-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Neuhaus S, Bruendler P, Frey CF, Gottstein B, Doherr MG, Gerber V. Increased parasite resistance and recurrent airway obstruction in horses of a high-prevalence family. J Vet Intern Med. 2010;24:407–13. doi: 10.1111/j.1939-1676.2009.0465.x. [DOI] [PubMed] [Google Scholar]
- 91.Nussbaumer Schleuniger P, Frey CF, Gottstein B, Swinburne JE, Dolf G, Gerber V. Resistance against strongylid nematodes in two high prevalence Equine Recurrent Airway Obstruction families has a genetic basis. Pferdeheilkunde. 2011;27:664–9. doi: 10.21836/PEM20110613. [DOI] [Google Scholar]
- 92.Jin J, Xiao Y, Hu H, Zou Q, Li Y, Gao Y, et al. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat Commun. 2015;6:5930. doi: 10.1038/ncomms6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karjalainen M, Rintanen N, Lehkonen M, Kallio K, Mäki A, Hellström K, et al. Echovirus 1 infection depends on biogenesis of novel multivesicular bodies. Cell Microbiol. 2011;13:1975–95. doi: 10.1111/j.1462-5822.2011.01685.x. [DOI] [PubMed] [Google Scholar]
- 94.Leung TH-Y, Ching Y-P, Yam JWP, Wong C-M, Yau T-O, Jin D-Y, et al. Deleted in liver cancer 2 (DLC2) suppresses cell transformation by means of inhibition of RhoA activity. Proc Natl Acad Sci U S A. 2005;102:15207–12. doi: 10.1073/pnas.0504501102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Faugaret D, Chouinard FC, Harbour D, El azreq M-A, Bourgoin SG. An essential role for phospholipase D in the recruitment of vesicle amine transport protein-1 to membranes in human neutrophils. Biochem Pharmacol. 2011;81:144–56. doi: 10.1016/j.bcp.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 96.Panaro MA, Mitolo V. Cellular responses to FMLP challenging: a mini-review. Immunopharmacol Immunotoxicol. 1999;21:397–419. doi: 10.3109/08923979909007117. [DOI] [PubMed] [Google Scholar]
- 97.Alcaide P, Merinero B, Ruiz-Sala P, Richard E, Navarrete R, Arias Á, et al. Defining the pathogenicity of creatine deficiency syndrome. Hum Mutat. 2011;32:282–91. doi: 10.1002/humu.21421. [DOI] [PubMed] [Google Scholar]
- 98.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–1. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 99.Patterson AM, Gardner L, Shaw J, David G, Loreau E, Aguilar L, et al. Induction of a CXCL8 binding site on endothelial syndecan-3 in rheumatoid synovium. Arthritis Rheum. 2005;52:2331–42. doi: 10.1002/art.21222. [DOI] [PubMed] [Google Scholar]
- 100.Sun YX, Tsuboi K, Zhao LY, Okamoto Y, Lambert DM, Ueda N. Involvement of N-acylethanolamine-hydrolyzing acid amidase in the degradation of anandamide and other N-acylethanolamines in macrophages. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. 2005;1736:211–20 [DOI] [PubMed]
- 101.Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S, et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci U S A. 2009;106:20966–71. doi: 10.1073/pnas.0907417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li Y, Yang L, Chen L, Zhu C, Huang R, Zheng X, et al. Design and synthesis of potent N-acylethanolamine-hydrolyzing acid amidase (NAAA) inhibitor as anti-inflammatory compounds. PLoS One. 2012;7(8):e43023. doi: 10.1371/journal.pone.0043023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bandiera T, Ponzano S, Piomelli D. Advances in the discovery of N-acylethanolamine acid amidase inhibitors. Pharmacol Res. 2014;86:11–7. doi: 10.1016/j.phrs.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fiasella A, Nuzzi A, Summa M, Armirotti A, Tarozzo G, Tarzia G, et al. 3-Aminoazetidin-2-one derivatives as N-acylethanolamine acid amidase (NAAA) inhibitors suitable for systemic administration. Chem Med Chem. 2014;9:1602–14. doi: 10.1002/cmdc.201300546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kuo TT, Baker K, Yoshida M, Qiao SW, Aveson VG, Lencer WI, et al. Neonatal Fc receptor: From immunity to therapeutics. J Clin Immunol. 2010;30:777–89. doi: 10.1007/s10875-010-9468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wallmeier J, Shiratori H, Dougherty GW, Edelbusch C, Hjeij R, Loges NT, et al. TTC25 Deficiency Results in Defects of the Outer Dynein Arm Docking Machinery and Primary Ciliary Dyskinesia with Left-Right Body Asymmetry Randomization. Am J Hum Genet. 2016;99:460–9. doi: 10.1016/j.ajhg.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data in binary-sequence alignment format (BAM) are available from European Nucleotide Archive (http://www.ebi.ac.uk/ena/data/view/PRJEB7497).