

Introduction
Approximately 170 million people are chronically infected with HCV and 350 million are chronically infected with HBV worldwide. It is estimated that more than one million patients die from complications related to chronic viral hepatitis, mainly HCC which is one of the most frequent cancers in many countries, especially Africa, the Middle East and Asia. HCV drug development has been impressive, and this revolution led to several direct-acting antiviral agents achieving an HCV cure after only 6–12 weeks. This progress could theorically lead to HCV global elimination making HCV and its consequences a rarity. HBV research and development programs can learn from the HCV experience, to achieve an HBV functional or sterilizing cure. This review will summarize key steps which have been realized for an HCV cure, and discuss the next steps to achieve for an HCV elimination. And also, how this HCV revolution has inspired scientists and clinicians to achieve the same for HBV.
Keywords: HCV elimination, direct-acting antivirals, HBV cure, cccDNA, capsid inhibitors
Major steps toward HCV elimination
The replicon system has been an important tool to evaluate direct-acting antiviral agents (DAAs)
HCV, identified in 1989, is an enveloped virus with a 9.6 kb single-stranded RNA genome [1], a member of the Flaviviridae family, genus Hepacivirus. The development of a subgenomic HCV RNA replicon capable of replication in the human hepatoma cell line, Huh 7, has been a significant advance in this field [2, 3]. This replicon model, together with other replicon-based systems, improve our understanding of HCV replication as well as resistance and allow evaluation of potential antiviral molecules for efficacy and cytotoxicity using real-time PCR. The HCV replication cycle begins with virion attachment to its specific receptor. The HCV RNA genome serves as a template for viral replication and as viral messenger RNA for viral production. It is translated into a polyprotein that is cleaved by proteases. Viral assembly then occurs. Potentially, each step of the viral replication cycle including viral entry is a target for drug development. Knowledge of the structures of HCV protease and HCV polymerase has allowed structure-based drug design to develop inhibitors to these enzymes [4, 5].
Direct-acting antivirals (DAA)
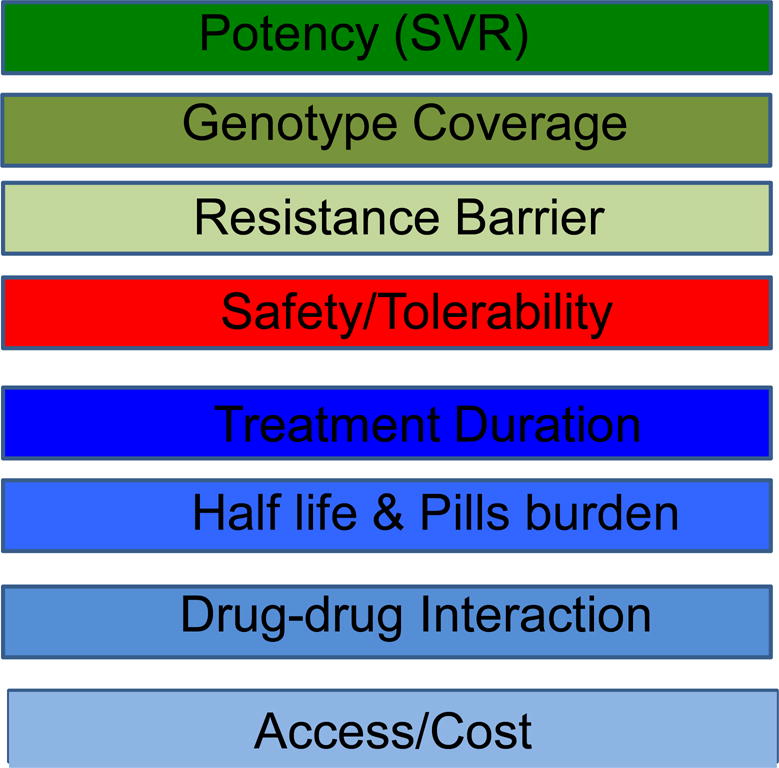
The proof-of-concept for the first DAA (a protease inhibitor) was developed in 2002, and was a revolution in HCV drug discovery [6, 7]. All the HCV enzymes – NS2-3 and NS3-4A proteases, NS3 helicase and NS5B RdRp – are essential for HCV replication, and are potential drug discovery targets. In addition, NS5A DAA have produced picomolar HCV inhibitors such as Daclastavir. Therefore, DAA with different viral targets, including NS3 protease inhibitors, nucleoside/nucleotide analogs and non-nucleoside inhibitors of the RNA-dependent RNA polymerase and NS5A inhibitors, were developed [8]. There is a strong rational to combine different DAA for HCV treatment. This allows high efficacy, reduced risk of resistance and reduced duration of treatment. High priorities achieved with DAA are listed in Box 1. (please insert Box 1)
BOX 1. Top Priorities for Direct-Acting Antivirals.
Protease inhibitors
The NS3 serine protease is located in the N-terminal region of NS3. The NS3 serine protease domain is associated with the NS4A cofactor to cleave four specific sites. This enzyme has been extensively characterized at the biochemical level and its structure is known [4, 5]. The serine protease activity of NS3 is an attractive target for new drugs that could effectively block viral replication. The NS3/4A protease inhibitors can be divided into two chemical classes: macrocyclic inhibitors and linear tetra-peptide a-ketoamid derivatives. In 2003, a macrocyclic protease inhibitor (BILN 2061; ciluprevir) that blocks HCV replication in the replicon model was shown to be effective in humans [6, 9].
Although first-generation protease inhibitors are potent, they have several potential limitations. Protease inhibitors are highly specific and as the amino acid sequence of the NS3 protease domain differs significantly between HCV genotypes, they exhibit varying activities across genotypes. The genetic barrier to resistance is defined as the number of amino acid substitutions required to confer full resistance to a drug. Usually, DAA with a low genetic barrier to resistance require only one or two amino acid substitutions for high resistance. DAA with a high barrier to resistance usually require three or more amino acid substitutions in the same region to confer loss of activity.
The main weaknesses of the first-generation PIs were their low genetic barrier to resistance and the fact that their effectiveness was limited to GT-1 patients. Second-wave PIs have a higher barrier to resistance, better potency against multiple genotypes, more convenient dosing schedules and improved safety and tolerance. Second-generation PIs are broadly active against all genotypes and against viral isolates that carry resistance mutations for first-generation PIs. In combination with NA, the new PIs appear to achieve greater SVR rates than the first-generation PIs. These new treatments allow for more convenient administration schedules (one or two administrations per day); this could result in improved pharmacokinetics and better patient compliance. Besides, the safety profile seems to be good. The pan-genotypic activity of these new treatments provides new therapeutic options for a greater number of patients, in particular for those infected with GT-4 [10].
Polymerase inhibitors
Nucleoside polymerase inhibitors in their 5′-triphosphate form interfere with viral replication by binding to the NS5B RNA-dependent RNA polymerase. NS5B RNA polymerase inhibitors can be divided into two different types – nucleoside inhibitors (NI) and non-nucleoside inhibitors (NNI). NI mimic the natural substrates of the polymerase and are incorporated into the RNA chain causing direct chain termination [11]. NI require bioconversion to an active triphosphate form. As the active site of NS5B is highly conserved, NI are generally pan-genotypic (effective against all the different genotypes). However, single amino acid substitutions in every position of the active site may result in loss of function of the NI, but resistance to nucleoside analog inhibitors is typically very low in humans as this virus has reduced fitness.
In contrast, NNI bind to several discrete sites outside of the HCV polymerase active centre, which results in conformational protein change before the elongation complex is formed [11]. NS5B is structurally organized in a characteristic ‘right-hand motif’ containing finger, palm and thumb domains, and offers at least four NNI-binding sites, namely, benzimidazole (thumb 1)-binding, thiophene (thumb 2)-binding, benzothiadiazine (palm1)-binding and benzofuran-(palm 2)-binding sites. Resistance is more frequent with NNI compared with NI. However, mutations at NNI-binding sites do not necessarily lead to impaired function of the enzyme.
NS5A inhibitors
The NS5A is a membrane-associated phosphoprotein present in basally phosphorylated (p56) and hyperphosphorylated (p58) forms [12–13]. It was previously reported that only p58-defective mutants could be complemented in trans, and NS5A is involved in HCV virion production, suggesting that different forms of NS5A exert multiple functions at various stages of the viral replication cycle [12–13]. The N terminus of NS5A (domain I) has been crystallized in alternative dimeric forms and contains both zinc- and RNA-binding domains, properties that have been demonstrated in vitro. NS5A has been shown to interact with a number of host proteins and plays a role in interferon resistance in vivo [12–13]. Daclastavir is active at picomolar concentrations in vitro in HCV replicons expressing a broad range of HCV genotypes and acts in an additive to synergistic fashion with interferon and other DAA [12–13]. The resistance profile of daclastavir reveals inhibitor sensitivity maps to the N terminus of domain 1 of NS5A [14]. It has been demonstrated that NS5A inhibitors could block hyperphosphorylation of NS5A, which is believed to play an essential role in the viral replication cycle.
What are the Goals obtained by achieving sustained virological response ?
A remarkable revolution has been recently achieved with the availability of DAA with high chance to cure and good tolerability. The primary goal of treatment is to achieve a sustained virologcal response (SVR) which is usually defined as undetectable serum HCV RNA 12 weeks after the end of treatment [15]. The SVR has been shown to be durable on long-term follow-up and associated with the eradication of HCV infection confirmed by undetectable HCV RNA in serum and the liver [16]. An SVR indicates that viral infection has been cured. In addition, viral eradication is associated with the regression of fibrosis, the possible reversibility of cirrhosis as well as significant improvement of the clinical outcome and survival with a decreased incidence of complications, especially HCC. Goals obtained by achieving an SVR are listed in Box 2. (please insert Box 2)
BOX 2. Goals obtained by achieving Sustained Virological Response (SVR) ≈ cure.
Eradicate the virus (HCV clearance)
Reduce Necroinflammation
Stop Fibrosis progression
Prevent Cirrhosis & complications
Prevent Hepatocellular carcinoma
Reduce extra-hepatic manifestations
Increase Survival
Patients with cirrhosis treated with DAAs should continue to be closely monitored for HCC (ultrasound each 6 months). Surveillance is also advocated in SVR patients with any histological stage of hepatitis C who carry comorbidities as alcohol cunsomption and diabetes, which are well-recognized independent risk factors for HCC. The benefit of treating HCV in patients with past or present HCC is under evaluation.
What are the Gaps to achieve HCV elimination?
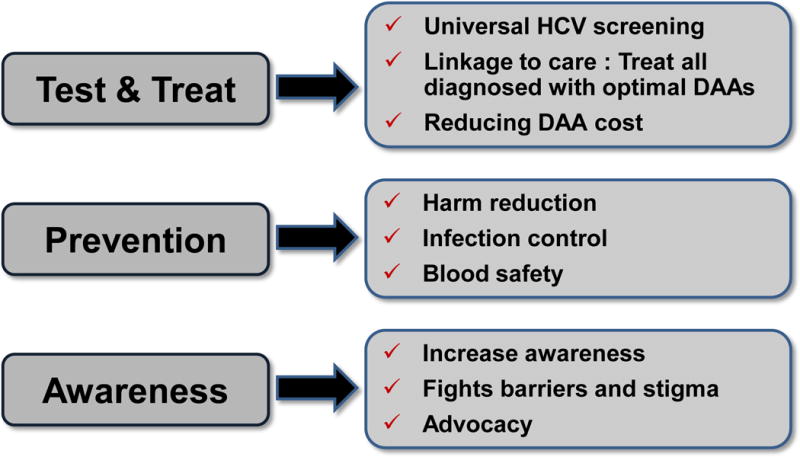
HCV elimination is an achievable goal, since several, all oral, IFN–free DAA combinations are now able to cure HCV in more than 95% of HCV infected individuals after 8 to 12 weeks of treatment. The programme to eradicate HCV must include increased screening (universal), linkage to care, as well as increased access to treatment worldwide [Box 3]. Reducing DAAs cost will also be important [17]. Real-life data on treatment efficacy, tolerability and adherence are mandatory. Compliance to drug regimens will become a major issue to avoid failure. How far we can go with shortened treatment duration remains to be studied (2, 3, 4, 6 or 8 weeks). However a pilot study conducted in China in genotype 1b patients with three potent DAA (NA, NS5A and PI) resulted in 100 % SVR [18]. Patients who failed previous treatment and developed resistance-associated variants (RAV) to NS5A, should be rescued with future combinations.
BOX 3. Strategies for HCV elimination.
HBV infection
Chronic hepatitis B (CHB) infection affects over 400 million people worldwide, with long-term morbidity such as cirrhosis and hepatocellular carcinoma (HCC) accounting for around 600,000 deaths annually [19]. Fibrosis is the most important prognosis predictor. Serum biomarkers to diagnose fibrosis are under development [20].
In the sixties, Blumberg identified the hepatitis B virus (Australia antigen (Ag)) [21–22]. Rapidly after the identification of hepatitis B, vaccines were a large success [23–26]. Blumberg was awarded the Nobel Prize in Medicine in 1976 for both the description of HBV and the notion of a life saving effective HBV vaccine.
HBV Current therapy
The goal of therapy for CHB is to improve quality of life, preventing viral transmission, and survival by preventing progression to severe disease and HCC [27]. Previously, HBV DNA suppression with long-term lamivudine (LAM) was associated with a reduction in the incidence of hepatic decompensation and HCC [28–29]. Treatment goals can be achieved by sustained suppression of HBV replication, thereby reducing the histological activity of CHB and reducing the risk of fibrosis progression. Suppression of HBV replication is critical, but must be maintained if optimal treatment outcomes are to be achieved. Currently, there are two main treatment strategies for both hepatitis B “e” (HBe) antigen-positive (HBeAg+ve) and antigen-negative (HBeAg-ve) patients: 2 finite treatment courses of interferon alpha (IFN)/pegylated IFN (PEG-IFN) or as long-term therapy with nucleoside analogs (NA). A one year treatment with PEG-IFN offers the potential for immune-mediated control of HBV infection, with higher rates of HBe seroconversion and the possibility of off-treatment viral suppression, with loss of hepatitis B surface antigen (HBsAg) in a proportion of patients who maintain undetectable HBV DNA. However, PEG-IFN needs to be administered by subcutaneous injection and is associated with frequent side effects such as depression; it is also contraindicated in patients with decompensated cirrhosis or relevant autoimmune disease, during pregnancy.
In contrast, NA suppress HBV by direct antiviral activity, and if compliance to treatment is good, more than 95% of patients treated with the newer, highly potent NA tenofovir disoproxil fumarate (TDF) and entecavir (ETV) achieve virological remission. NAs are administered orally, and tolerance is good, although the safety of these drugs over lifelong therapy is unknown. One potentially serious drawback of long-term NA therapy is potential renal toxicity (in the cse of TDF) and the development of drug resistance; however, although common with earlier less potent NA such as lamivudine (LAM) and adefovir (ADV), resistance has become considerably less of a problem with the highly potent NA TDF and ETV.
HBV current therapy: What have we learned recently?
Long-term efficacy and real-world data
Long-term clinical data up to six years and beyond are emerging for the newer NA that are providing reassuring data on their efficacy and safety. There is cumulative evidence that complete long-term suppression of HBV replication by the most potent drugs (ETV and TDF) results in an improved long-term outcome with a decreased risk of progression to cirrhosis and complications such as liver failure, HCC and improved survival. In addition, a recent study assessing liver histology in patients treated with TDF for 5 years demonstrated that fibrosis regressed in most patients [30–31]. Moreover, unlike what is generally believed, the reversal of cirrhosis was observed during treatment in 75% of patients with cirrhosis, probably associated with a decreased risk of HCC and improved survival. Further long-term data are emerging from studies using newer potent NA in routine clinical practice confirming safety and efficacy of these agents in the “real-world” setting.
Combination therapy (TDF plus PEG-IFN)
In a recent study, Marcellin et al. evaluated HBsAg loss in patients receiving the combination of TDF and PEG-IFN for a finite duration [32]. In an open-label, active-controlled study, 740 patients with CHB were randomly assigned to receive TDF plus PEG-IFN for 48 weeks (group A), TDF plus PEG-IFN for 16 weeks followed by TDF for 32 weeks (group B), TDF for 120 weeks (group C), or PEG-IFN for 48 weeks (group D). At week seventy-two, 9.1% of subjects in group A had HBsAg loss compared with 2.8% of subjects in group B, none of the subjects in group C, and 2.8% of subjects in group D. A significantly higher proportion of subjects in group A had HBsAg loss than in group C (p < 0.001) or group D (p = 0.003). However, the proportions of subjects with HBsAg loss did not differ significantly between group B and group C (p = 0.47) or group D (p = 0.88). HBsAg loss in group A occurred in hepatitis B e antigen-positive and hepatitis B e antigen-negative patients with all major viral genotypes.
Finally, a significantly greater proportion of patients receiving TDF plus PEG-IFN for 48 weeks had HBsAg loss than those receiving TDF or PEG-IFN alone.
Can we stop therapy of nucleosides?
The European Association for the Study of the Liver (EASL) [27] and the American Association for the Study of Liver Disease (AASLD) guidelines [33] suggest HBsAg loss as the ideal end point for oral therapy. However, this rarely occurs and is mainly seen in HBeAg-negative patients. Therefore, patients should probably be treated for life. On the other hand, the Asian Pacific Association for the Study of the Liver (APASL) recommendations [34] for stopping NA are sustained suppression of HBV replication, (three measurements showing HBeAg loss or undetectable HBV DNA within 1 year), which is frequent during NAs therapy. Thus, one of the key issues when monitoring patients who are receiving NA therapy is when can patients stop therapy with no risk of relapse ?
The retrospective study by Chen et al. [35] in HBeAg-positive and negative patients reports that end of treatment HBsAg levels < 300 IU/ml, 300–1000 IU/ml and >1000 UI/ml in HBeAg-positive patients were associated with sustained HBeAg loss in 55.6%, 7.7% and 3.3%, respectively. EOT HBsAg cut-offs <120 IU/mL, 120–1000 IU/mL and >1000 IU/ml in HBeAg-negative patients were associated with HBsAg loss in 79.2%, 14.3% and 0%, respectively, and HBsAg cut-offs < 200 IU/mL, 200–1000 IU/mL and >1000 IU/mL were associated with an SVR in 93%, 11.1% and 15.4%, respectively. Similar results were reported in patients whose treatment was discontinued according to APASL guidelines. An end of treatment HBsAg level <100 IU/mL was associated with high SVR, while >1000 IU/mL was associated with a 1 year post-treatment relapse in 70% [36–38].
HBsAg quantification: a new tool for HBV monitoring
Quantifying HBsAg is certainly an important new tool for predicting the severity of disease, to distinguish inactive carriers from patients with HBeAg-negative chronic active hepatitis; and also help tailor follow-up and treatment management [39–40]. In addition, a decline in quantitative HBsAg during therapy is a strong predictor of SVR after PEG-IFN therapy and of the probability of HBsAg loss, which is the ultimate goal of therapy [41]. In patients treated with analogs, a decline in HBsAg levels is also a predictor of HBsAg loss, allowing therapy to be discontinued.
Future strategies
There is considerable interest in the potential for finite therapy in patients following successful HBsAg seroconversion. Studies are underway to determine if it is possible to successfully combine the potent effects of NA with an immunomodulatory therapy to allow more patients to stop therapy. Furthermore, there are three drugs, which are all prodrug of tenofovir, in development. Among these, tenofovir alafenamide, or TAF, is already approved in the USA for HIV treatment.
Knowledge of HBV replication cycle is important for drug development to identify targets. Goals of therapy is to obtain a virological cure, or at least a functional cure
HBsAg synthesis during the HBV viral replication cycle is complex and usually occurs in the endoplasmic reticulum [42, 43]. Upon entry into the hepatocyte the virion sheds its protein coat and its genome is transported into the nucleus where it resides as stable fully double stranded cccDNA and acts as a template for the transcription of the viral gene [44]. HBsAg is translated from mRNA with the transcriptional template-active cccDNA, which is the reflection of the number of infected hepatocytes. The clinical relevance of HBsAg levels is inferred from the relationship of this marker to the intrahepatic amount of cccDNA. There is a correlation between serum HBsAg concentrations and the intrahepatic levels of cccDNA, with the highest levels occurring in HBeAg positive hepatitis B and the lowest in patients with resolved hepatitis [45–47]. Through this association, the amount of circulating HBsAg is thought to indirectly measure the control of infection by the immunological response independent from the antiviral response, which can be assessed by measuring HBV DNA levels in serum. Serum HBsAg can be considered to be a surrogate marker of the number of infected cells.
Since NA cannot eliminate cccDNA, persisting in the nucleus of hepatocyte, HBV infection cannot be cured. Currently, there are many compounds in development for chronic hepatitis B, and they can be categorized into two groups.
The DAA and the host-targeting antiviral agents (HTA). Goals to achieve for future treatment are listed in Box 4 [48]. (please insert Box 4)
BOX 4. Definition of a cure.
Functional cure
Sustained off-drug suppression of serum HBsAg, HBeAg, viral DNA, and cccDNA.
Normalization of liver function (normal levels of serum ALT and AST).
Comparable with individuals with naturally resolved infection.
Absolute cure – virologic cure
Sustained off-drug suppression of serum HBsAg, HBeAg, and viral DNA.
Normalization of liver function (normal levels of serum ALT and AST).
Elimination of cccDNA.
Presence of HBsAb.
Comparable with uninfected individuals.
Entry inhibitors
Yan et al. reported in 2012 that sodium taurocholate cotransporting polypeptide (NTCP) is a major functional receptor for HBV (and HDV) to enter the hepatocyte [49]. Yan et al. started by isolating primary liver cells from treeshrews, and then used a combination of advanced purification and mass spectrometry analysis to show that the NTCP on the surface of the cells interacts with the pre-S1 domain in HBV. The authors then performed a series of gene knockdown experiments on liver cells of both human and treeshrew origin: when the gene that codes for NTCP was silenced, HBV infection was greatly reduced. Moreover, they were able to transfect HepG2 cells—which are widely used in research into liver disease, but are not susceptible to HBV and HCV infection—with NTCP from humans and treeshrews to make them susceptible.
Myrcludex-B is a synthetic lipopeptide that is derived from pre-S1 domain of HBV envelope protein [50–51]. Since it contains NTCP-binding pre-S1 domain of HBV envelope protein, it competitively binds to NTCP, thereby inhibit attachment of HBV to NTCP. Currently Myrcludex-B is in Phase II clinical trial, and there are other drugs under investigation, such as cyclosporine A and ezetimibe.
Capsid effectors: nucleocapsid assembly inhibitors affect cccDNA
These drugs inhibit formation and assembly of core particles leading to the failure of mature viron production as well as blocking cccDNA replenishment (Table 1). (please insert Table 1). cccDNA is an excellent target, since to achieve HBV cure, elimination, suppression or control of cccDNA is warranted (Box 5) [52–53]. (please insert Box 5)
Table 1.
DAAs: Inhibitors of nucleocapsid assembly
| Drug name | Target | Compound | Stage of Development |
|---|---|---|---|
| GLS4 | Interfere with capsid formation/stability | Heteroaryldihydropyrimidine (HAPs) | Phase II |
| Bay 41-4109 | Viral nucleocapsid inhibitor | HAPs | Phase I |
| AT-130 | Inhibition of HBV capsid assembly | Phenylpropenamide derivatives | Preclinical and early clinical phase |
| NVR-3-778 (NVR1221) | Inhibition of HBV capsid assembly | Small molecule | Phase Ib |
BOX 5. Relevance of cccDNA as a targuet.
cccDNA persistence is the cause of chronic HBV disease
cccDNA exists as a minichromosome in the nucleus
cccDNA persists in the absence of active viral replication
cccDNA levels reduced, but not eliminated with treatment/liver regeneration
HBV Cure: elimination, suppression or control of cccDNA
Capsid effectors disrupt the capsid and indirectly affect the stabiity of cccDNA, by inhibiting the formation of cccDNA, and/or destroying the cccDNA per se. However, only three of these drugs (from Novira now J&J, and a Chinese company) have progressed to clinical trials, but many others are in early preclinical phases [54–56]. GLS4 is currently in Phase II study, and the results are awaited [54]. NVR 3-778 is a small molecule that inhibits the HBV capsid protein by oral administration [56]. Oral HBV core inhibitor NVR 3-778 appears to be effective against HBV with synergistic activity when combined with NA.
DAA: RNA interference
There are several RNA interference drugs under clinical trial. ARC-520, which is currently in Phase II/III trial, is a compound that contains HBV mRNA-targeting siRNA [57]. Upon administration, the siRNAs in ARC-520 associates with the viral mRNA and forms a double-stranded RNA, leading to degradation. Therefore, it can reduce the production of viral protein and viral replication per se.
The product targets highly conserved regions of all five of the mRNAs produced by HBV. This approach potentially interferes with the production of all of the viral proteins, as well as the stability of the pre-genomic RNA that the virus uses as a template for DNA synthesis. ARC-520 is claimed to have a dual mechanism of action, both reducing viral replication and restoring the immune system’s ability to clear the infection from hepatocytes.
A recent report of the phase IIa trial has shown that a single injection of ARC-520 resulted in significant reduction in HBsAg for up to 43 days. When given as a single, IV administration, ARC-520 was well tolerated up to and including a dose of 3 mg/kg in CHB subjects, who were also receiving entecavir, and up to 4 mg/kg in normal volunteers [57]. A single injection of ARC-520 resulted in significant reduction in HBsAg for up to 43 days.
Gene editing approaches: CRISPR/Cas 9
Using lentiviral transduction of a bacterial Cas9 gene and single guide RNA (sgRNA) specific for HBV, effective inhibition of HBV DNA production in in vitro models of both chronic and de novo HBV infection can be observed. Cas9/sgRNA combinations specific for HBV can reduce total viral DNA levels by up to 1,000-fold and HBV cccDNA levels by up to 10-fold and also mutationally inactivates the majority of the residual viral DNA [58, 59].
Conclusions
There has been a revolution in the treatment of HCV infection. Several oral regimens combining direct-acting antiviral (DAA) agents result in an increase in SVR rates to above 95% and reduce the duration of treatment from 12 to 8 weeks. Remaining issues include increasing screening and access to care. Resolution of these issues will result in access to care so that HCV may become the first chronic viral infection eradicated worldwide.
There is increasing research in the field of HBV infection. The new goal of HBV therapy is to achieve “functional cure” or even “absolute cure” with HBsAg loss/seroconversion and clearance of cccDNA. New agents (DAA and HTA) for CHB are starting to emerge: HBV entry inhibitors, small interfering RNA, capsid inhibitors, CRISPR/Cas9 are promising, but early in development. New drugs aimed to decrease or eliminate cccDNA and/or HbsAg are promising.
KEY POINTS.
HCV cure
Combining DAAs results in high SVR (>95%) and short duration (6–12 weeks).
Shortened duration treatments (3 to 8 weeks) are under evaluation.
HCV elimination require improvement in screening, prevention and access to treatment.
HBV cure
The new goal of HBV therapy is to achieve “virological cure” with HBsAg loss/seroconversion and cccDNA clearance.
cccDNA is an excellent target, since elimination/control of cccDNA lead to HBV cure.
Inhibitors of nucleocapsid inhibit formation and assembly of core particles leading to the failure of mature viron production as well as blocking cccDNA replenishment.
Acknowledgments
This work was supported in part by NIH CFAR grant P30-AI050409 (to RFS).
Abbreviations
- AE
adverse event
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- cccDNA
covalently closed circular DNA
- CHB
chronic hepatitis B
- CHC
chronic hepatitis C
- DAA
direct-acting antivirals
- EOT
end of treatment
- ETV
entecavir
- GT
genotype
- HBeAg
hepatitis B e antigen
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- HCC
hepatocellular carinoma
- HCV
hepatitis C virus
- HTA
host-targeting antiviral agents
- IFN
interferon
- LAM
lamivudine
- NA
nucleoside analogs
- NI
nucleoside inhibitors
- NNI
non-nucleoside inhibitors
- NTCP
sodium taurocholate cotransporting polypeptide
- PEG-IFN
pegylated-interferon
- PI
protease inhibitors
- QD
once daily
- RAV
resistance-associated variants
- RBV
ribavirin
- RdRp
RNA-dependent RNA polymerase
- SVR
sustained virological response
- TAF
tenofovir alafenamide
- TDF
tenofovir disoproxil fumerate
Footnotes
Conflicts of interest:
Raymond Schinazi is the founder and major shareholder of Cocrystal Pharma, Inc. Tarik Asselah is a speaker and investigator for Abbvie, BMS, Boehringer-Ingelheim, Tibotec, Janssen, Gilead, Roche and MSD
Contributor Information
Raymond F Schinazi, Frances Winship Walters Professor of Pediatrics, Center for AIDS Research, Emory University School of Medicine, Atlanta, Georgia 30033, USA., Tel: +1 404 727 1414, Fax: +1 404 727 1330.
Tarik Asselah, Professor of Medicine, UMR1149, Team Physiopathology and Treatment of Viral Hepatitis, Centre de Recherche sur l’Inflammation, and Université Denis Diderot Paris 7, Hepatology Department, AP-HP, Beaujon Hospital, 100 Bd du Général Leclerc, Clichy 92110, France., Tel: +33(0) 140875579, Fax: +33(0) 147309440.
References
- 1.Choo QL, Kuo G, Weiner AJ, et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–62. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 2.Lohmann V, Korner F, Koch J, et al. Replication of sub- genomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–3. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 3.Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–4. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 4.Kim JL, Morgenstern KA, Lin C, et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell. 1996;87:343–55. doi: 10.1016/s0092-8674(00)81351-3. [DOI] [PubMed] [Google Scholar]
- 5.Yao N, Hesson T, Cable M, et al. Structure of the hepatitis C virus RNA helicase domain. Nat Struct Biol. 1997;4:463–7. doi: 10.1038/nsb0697-463. [DOI] [PubMed] [Google Scholar]
- 6.Lamarre D, Anderson PC, Bailey M, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–9. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- 7.Zeuzem S, Soriano V, Asselah T, et al. Faldaprevir and deleobuvir for HCV genotype 1 infection. N Engl J Med. 2013;369:630–9. doi: 10.1056/NEJMoa1213557. [DOI] [PubMed] [Google Scholar]
- 8.Schinazi R, Halfon P, Marcellin P, Asselah T. HCV direct-acting antiviral agents: the best interferon-free combinations. Liver Int. 2014;34(Suppl. 1):69–78. doi: 10.1111/liv.12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinrichsen H, Benhamou Y, Wedemeyer H, et al. Shortterm antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology. 2004;127:1347–55. doi: 10.1053/j.gastro.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Asselah T, Hezode C, Qaquish RB, et al. Ombitasvir, paritaprevir, and ritonavir plus ribavirin in adults with hepatitis C virus genotype 4 infection and cirrhosis (AGATE-I): a multicentre, phase 3, randomised open-label trial. Lancet Gastroenterology & Hepatology. 2016;1:25–35. doi: 10.1016/S2468-1253(16)30001-2. [DOI] [PubMed] [Google Scholar]
- 11.Koch U, Narjes F. Recent progress in the development of inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. Curr Top Med Chem. 2007;7:1302–29. doi: 10.2174/156802607781212211. [DOI] [PubMed] [Google Scholar]
- 12.Gao M, Nettles RE, Belema M, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gish RG, Meanwell NA. The NS5A replication complex inhibitors: difference makers? Clin Liver Dis. 2011;15:627–39. doi: 10.1016/j.cld.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 14.Halfon P, Locarnini S. Hepatitis C virus resistance to protéase inhibitors. J Hepatol. 2011;55:192–206. doi: 10.1016/j.jhep.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 15.Martinot-Peignoux M, Stern C, Maylin S, et al. Twelve weeks posttreatment follow-up is as relevant as 24 weeks to determine the sustained virologic response in patients with hepatitis C virus receiving pegylated interferon and ribavirin. Hepatology. 2010;51:1122–6. doi: 10.1002/hep.23444. [DOI] [PubMed] [Google Scholar]
- 16.Maylin S, Martinot-Peignoux M, Moucari R, et al. Eradication of hepatitis C virus in patients successfully treated for chronic hepatitis C. Gastroenterology. 2008;135(3):821–9. doi: 10.1053/j.gastro.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 17.Hagan LM, Sulkowski MS, Schinazi RF. Cost analysis of sofosbuvir/ribavirin versus sofosbuvir/simeprevir for genotype 1 hepatitis C virus in interferon-ineligible/intolerant individuals. Hepatology. 2014;60(1):37–45. doi: 10.1002/hep.27151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lau G, Benhamou Y, Chen G, et al. Efficacy and safety of three-week response-guided triple direct-acting antiviral therapy: a phase 2, proof-of-concept study. Lancet Gastroenterology & Hepatology. 2016;1(2):97–104. doi: 10.1016/S2468-1253(16)30015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.World Health Organization. Hepatitis B Fact sheet No. 204. 2012 Cited 24 Oct 2012. Available from URL: http://www.who.int/mediacentre/factsheets/fs204/en/
- 20.Appourchaux K, Dokmak S, Resche-Rigon M, et al. MicroRNA-based diagnostic tools for advanced fibrosis and cirrhosis in patients with chronic hepatitis B and C. Sci Rep. 2016 Oct 12;6:34935. doi: 10.1038/srep34935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blumberg BS, Alter HJ, Visnich A. A ‘new’ antigen in leukemia sera. J Amer Med Assoc. 1965;191:541–6. doi: 10.1001/jama.1965.03080070025007. [DOI] [PubMed] [Google Scholar]
- 22.Blumberg BS, Hepatitis B. The hunt for a killer virus. Princeton, New Jersey: Princeton University Press; 2003. [Google Scholar]
- 23.Krugmann S, Giles JP, Hammond J. Viral hepatitis, type B (MS-2 strain) prevention with specific hepatitis B immune serum globulin. J Amer Med Assoc. 1971;218:1665–70. [PubMed] [Google Scholar]
- 24.Soulier JP, Blatix C, Courouce AM, et al. Prevention of virus B hepatitis (SH hepatitis) Am J Dis Child. 1972;123:429. doi: 10.1001/archpedi.1972.02110100161061. [DOI] [PubMed] [Google Scholar]
- 25.Szmuness W, Stevens C, Harley E, et al. Hepatitis B vaccine: demonstration of efficacy in a controlled clinical trial in a high-risk population in the United-States. N Engl J Med. 1980;303:833–41. doi: 10.1056/NEJM198010093031501. [DOI] [PubMed] [Google Scholar]
- 26.Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22,707 men in Taiwan. Lancet. 1981;2:1129–33. doi: 10.1016/s0140-6736(81)90585-7. [DOI] [PubMed] [Google Scholar]
- 27.European Association for the Study of the Liver. EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J Hepatol. 2012;57:167–85. doi: 10.1016/j.jhep.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Lai CL, Chien RN, Leung NW, et al. A one-year trial of lamivudine for chronic hepatitis B. Asia Hepatitis Lamivudine Study Group. N Engl J Med. 1998;339:61–8. doi: 10.1056/NEJM199807093390201. [DOI] [PubMed] [Google Scholar]
- 29.Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521–31. doi: 10.1056/NEJMoa033364. [DOI] [PubMed] [Google Scholar]
- 30.Marcellin P, Gane E, Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. 2013;318:468–75. doi: 10.1016/S0140-6736(12)61425-1. [DOI] [PubMed] [Google Scholar]
- 31.Marcellin P, Asselah T. Long-term therapy for chronic hepatitis B: hepatitis B virus DNA suppression leading to cirrhosis reversal. J Gastroenterol Hepatol. 2013;28:912–23. doi: 10.1111/jgh.12213. [DOI] [PubMed] [Google Scholar]
- 32.Marcellin P, Ahn SH, Ma X, et al. Combination of tenofovir disoproxil fumarate and peginterferon alpha-2a increases loss of hepatitis B surface antigen in patients with chronic hepatitis B. Gastroenterology. 2016;150(1):134–44. doi: 10.1053/j.gastro.2015.09.043. [DOI] [PubMed] [Google Scholar]
- 33.Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH, American Association for the Study of Liver Diseases AASLD guidelines for treatment of chronic hepatitis B. Hepatology. 2016 Jan;63(1):261–83. doi: 10.1002/hep.28156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liaw YF, Kao JH, Piratvisuth T, et al. Asian-Pacific consensus statement on the management of chronic hepatitis B: a 2012 update. Hepatol Int. 2012;6:531–561. doi: 10.1007/s12072-012-9365-4. [DOI] [PubMed] [Google Scholar]
- 35.Chen CH, Lu SN, Hung CH, et al. The role of hepatitis B surface antigen quantification in predicting HBSAg loss and HBV relapse after discontinuation of lamuvidine treatment. J Hepatol. 2014;61:515–22. doi: 10.1016/j.jhep.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 36.Liang Y, Jiang J, Su M, et al. Predictors of relapse in chronic hepatitis B after discontinuation of antiviral therapy. Aliment Pharmacol Ther. 2011;34:344–52. doi: 10.1111/j.1365-2036.2011.04738.x. [DOI] [PubMed] [Google Scholar]
- 37.Chan HLY, Wong GLH, Chim AML, et al. Prediction of off-treatment response to lamivudine by serum hepatitis B surface antigen quantification in hepatitis B e antigen-negative chronic hepatitis B patients? Antivir Ther. 2011;16:1249–57. doi: 10.3851/IMP1921. [DOI] [PubMed] [Google Scholar]
- 38.Cai W, Xie Q, An B, et al. On-treatment serum HBsAg level is predictive of sustained off-treatment virologic response to telbivudine in HBeAg-positive chronic hepatitis B patients. J Clin Virol. 2010;48:22–6. doi: 10.1016/j.jcv.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 39.Martinot-Peignoux M, Lapalus M, Asselah T, Marcellin P. HBsAg quantification: useful for monitoring natural history and treatment outcome. Liver Int. 2014;34(Suppl. 1):97–107. doi: 10.1111/liv.12403. [DOI] [PubMed] [Google Scholar]
- 40.Martinot-Peignoux M, Carvalho-Filho R, Lapalus M, et al. Hepatitis B surface antigen serum level is associated with fibrosis severity in treatment-naıve, e antigen-positive patients. J Hepatol. 2013;58:1089–95. doi: 10.1016/j.jhep.2013.01.028. [DOI] [PubMed] [Google Scholar]
- 41.Moucari R, Mackiewicz V, Lada O, et al. Early serum HBsAg drop: a strong predictor of sustained virological response to pegylated interferon alfa-2a in HBeAg negative patients. Hepatology. 2009;49:1151–7. doi: 10.1002/hep.22744. [DOI] [PubMed] [Google Scholar]
- 42.Dienes HP, Gerlich WH, Worsdorfer M, et al. Hepatic expression patterns of the large and middle hepatitis B surface proteins in viremic and non viremic chronic hepatitis B. Gastroenterology. 1990;98:1017–23. doi: 10.1016/0016-5085(90)90028-y. [DOI] [PubMed] [Google Scholar]
- 43.Lau JYN, Bain VG, Davies SE, et al. Export of intracellular HBsAg in chronic hepatitis B virus infection is related to viral replication. Hepatology. 1991;14:416–21. [PubMed] [Google Scholar]
- 44.Ganem D, Prince AM. Hepatitis B virus infection-natural history and clinical consequences. N Engl J Med. 2004;350:118–29. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 45.Werle-Lapostolle B, Bowden S, Locarnini S, et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–8. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 46.Chan HL, Wong VW, Tse AM, et al. Serum hepatitis B surface antigen quantitation can reflect hepatitis B virus in the liver and predict treatment response. Clin Gastroenterol Hepatol. 2007;5:1462–8. doi: 10.1016/j.cgh.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 47.Jaroszewicz J, Calle Serrano B, Wursthorn K, et al. Hepatitis B surface antigen (HBsAg) levels in the natural history of hepatitis B virus (HBV)-infection: a European perspective. J Hepatol. 2010;52:514–22. doi: 10.1016/j.jhep.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 48.Boucle S, Bassit L, Ehteshami M, Schinazi RF. Toward elimination of hepatitis B virus using novel drugs, aapproaches, and combined modalities. Clin Liver Dis. 2016;20(4):737–749. doi: 10.1016/j.cld.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan H, Zhong G, Xu G, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Urban S, Schulze A, Schieck A, et al. 10 preclinical studies on Myrcludex B, a novel entry inhibitor for hepatitis B and hepatitis delta virus (HDV) infections. J Hepatol. 2010;52(Supplement 1):S5. [Google Scholar]
- 51.Volz T, Allweiss L, Ben MM, et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J Hepatol. 2013;58(5):861–7. doi: 10.1016/j.jhep.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 52.Guo H, Jiang D, Zhou T, et al. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol. 2007;81(22):12472–84. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai D, Mills C, Yu W, et al. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob Agents Chemother. 2012;56(8):4277–88. doi: 10.1128/AAC.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu G, Liu B, Zhang Y, et al. Preclinical characterization of GLS4, an inhibitor of hepatitis B virus core particle assembly. Antimicrob Agents Chemother. 2013;57(11):5344–54. doi: 10.1128/AAC.01091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang XY, Wei ZM, Wu GY, et al. In vitro inhibition of HBV replication by a novel compound, GLS4, and its efficacy against adefovir-dipivoxil-resistant HBV mutations. Antivir Ther. 2012;17(5):793–803. doi: 10.3851/IMP2152. [DOI] [PubMed] [Google Scholar]
- 56.Yuen MF, Kim DL, Weilert F, et al. Phase 1b efficacy and safety of NVR 3-778, a first-in-class HBV core inhibitor, in HBeAg-positive patients with chronic HBV infection. Hepatology. 2015;62:1385A. [Google Scholar]
- 57.Yuen MF, Chan HLY, Liu SHK, et al. ARC-520 produces deep and durable knockdown of viral antigens and DNA in a phase II study in patients with chronic hepatitis B. Hepatology. 2015;62:LB9. [Google Scholar]
- 58.Lin SR, Yang HC, Kuo YT, et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol Ther Nucleic Acids. 2014;3:e186. doi: 10.1038/mtna.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kennedy EM, Bassit LC, Mueller H, et al. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology. 2015;476:196–205. doi: 10.1016/j.virol.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]