

Abstract
Nakamurella lactea DLS-10T , isolated from rock in Korea, is one of the four type strains of the genus Nakamurella. In this study, we describe the high quality draft genome of N. lactea DLS-10T and its annotation. A summary of phenotypic data collected from previously published studies was also included. The genome of strain DLS-10T presents a size of 5.82 Mpb, 5100 protein coding genes, and a C + G content of 68.9%. Based on the genome analysis, emended description of N. lactea in terms of G + C content was also proposed.
Keywords: Frankineae, Rare actinobacteria, Nakamurellaceae, Bioactive natural product, Next generation sequencing
Introduction
The genus Nakamurella, belong to the order Nakamurellales [1] and is one of the rare genera in the class Actinobacteria [2]. The genus Nakamurella is the sole and type genus of the family Nakamurellaceae, which replaced the family Microsphaeraceae [2] in 2004 [3]. The genus and family names were assigned in honour of the microbiologist Kazonuri Nakamura [4].
Only four species with validly published names, Nakamurella multipartita [3, 5], Nakamurella panacisegetis [6, 7], Nakamurella flavida [6–8], and Nakamurella lactea [6, 7, 9], have been described, and only the genome of Nakamurella multipartita has been published [10].
N. lactea was originally described as Saxeibacter lacteus [9], which was the type species of one of the three genera comprising in the family Nakamurellaceae. Then, in the light of the 16S rRNA gene and rpoB gene sequences similarities and chemotaxonomic features [6], the species was reclassified into the genus Nakamurella. Nakamurella lactea is represented by the type strain DLS-10T (= DSM 19367 T = JCM 16024T = KCTC 19285T).
The availability of the genome of one more species in the genus will provide vital baseline information for better understanding of the ecology of these rare actinobacteria and their potential as source of bioactive natural products. In the present study, we summarise the phenotypic, physiological and chemotaxonomic, features of N. lactea DLS-10T together with the genomic data.
Organism information
Classification and features
N. lactea DLS-10T was isolated from a rock collected on the parasitic volcano Darangshi Oreum at 300 m above sea level in Jeju island, Republic of Korea (latitude 33.51, longitude 126.52) [9]. It has been shown by Lee et al. [9] and Kim et al. [4, 6] that its cells are aerobic, non-motile, non-spore and non-mycelium forming short rods with 0.4–0.7 μm and 0.9–1.0 μm of cell diameter and length, respectively (Fig. 1), producing cream-coloured colonies on TSA medium. A summary of the classification and general features of N. lactea strain DLS-10T is presented in the Table 1. Additional phenotypic features can be found in Lee et al. and Kim et al. [6, 9].
Fig. 1.
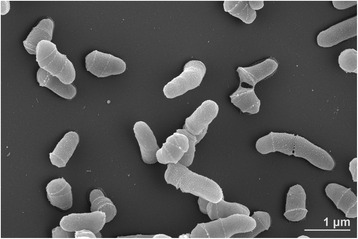
Scanning electron micrograph of N. lactea DLS-10T. The bacterium was grown on DSM medium 65 for 3 days at 28∘C
Table 1.
Classification and general features of Nakamurella lactea strain DLS-10T, according to the MIGS recommendations [36] as developed by [22]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [39] | |
| Phylum Actinobacteria | TAS [40] | ||
| Class Actinobacteria | TAS [2] | ||
| Order Nakamurellales | TAS [1] | ||
| Family Nakamurellaceae | TAS [41] | ||
| Genus Nakamurella | TAS [3, 41] | ||
| Species Nakamurella lactea
Type strain DLS-10 |
TAS [6, 9] | ||
| Gram stain | Positive | TAS [6, 9] | |
| Cell shape | Rod | TAS [6, 9] | |
| Motility | non-motile | TAS [6, 9] | |
| Sporulation | Non-sporulating | NAS [6, 9] | |
| Temperature range | 4–37 °C | TAS [6, 9] | |
| Optimum temperature | 25 °C | TAS [6, 9] | |
| pH range | 5.1–9.1 | TAS [6, 9] | |
| pH Optimum | 6.0–7.0 | ||
| Carbon source | L-Arabinose, myo-inositol and methyl α-D-mannoside, D-cellobiose, D-fructose, D-glucose, D-galactose, lactose, D-maltose, D-mannitol, D-mannose, L-rhamnose, salicin, sucrose and D-trehalose, D- turanose | TAS [6, 9] | |
| MIGS-6 | Habitat | Rock | TAS [9] |
| MIGS-6.3 | Salinity | Up to 3% NaCl | TAS [6, 9] |
| MIGS-22 | Oxygen requirement | Aerobic | TAS [9] |
| MIGS-15 | Biotic relationship | free-living | TAS [9] |
| MIGS-14 | Pathogenicity | non-pathogen | NAS |
| MIGS-4 | Geographic location | Korea | TAS [9] |
| MIGS-5 | Sample collection | Not reported | TAS [] |
| MIGS-4.1 | Latitude | 33.51 | TAS [9] |
| MIGS-4.2 | Longitude | 126.52 | TAS [9] |
| MIGS-4.4 | Altitude | 300 m | TAS [9] |
aEvidence codes are from of the Gene Ontology project [42]. TAS traceable author statement (i.e., a direct report exists in the literature)
Only four species isolated from soil (N. panacisegetis and N. flavida), rock (N. lactea) and sludge (N. mutipartita), respectively, are currently classified in the genus. Due to this limited number of the characterised species, the ecological diversity as well as the biotechnological potential of the members of the genus Nakamurella remain to be studied in depth.
Phylogenies based on 16S rRNA gene sequences included in this manuscript were performed using the GGDC web server [11] implementation of the DSMZ phylogenomics pipeline [12]. The multiple alignment was created with MUSCLE [13] and maximum likelihood (ML) and maximum parsimony (MP) trees were inferred from it with RAxML [14] and TNT [15], respectively. For ML, rapid bootstrapping in conjunction with the autoMRE bootstopping criterion [16] and subsequent search for the best tree was used; for MP, 1000 bootstrapping replicates were used in conjunction with tree-bisection-and-reconnection branch swapping and ten random sequence addition replicates. This analysis shows the family Nakamurellaceae [4] as the sister group of the families Cryptosporangiaceae, Sporichthyaceae, and Geodermatophilaceae. The monophyly of the genus Nakamurella was supported by (close to) maximum bootstrap values under ML and MP (Fig. 2).
Fig. 2.
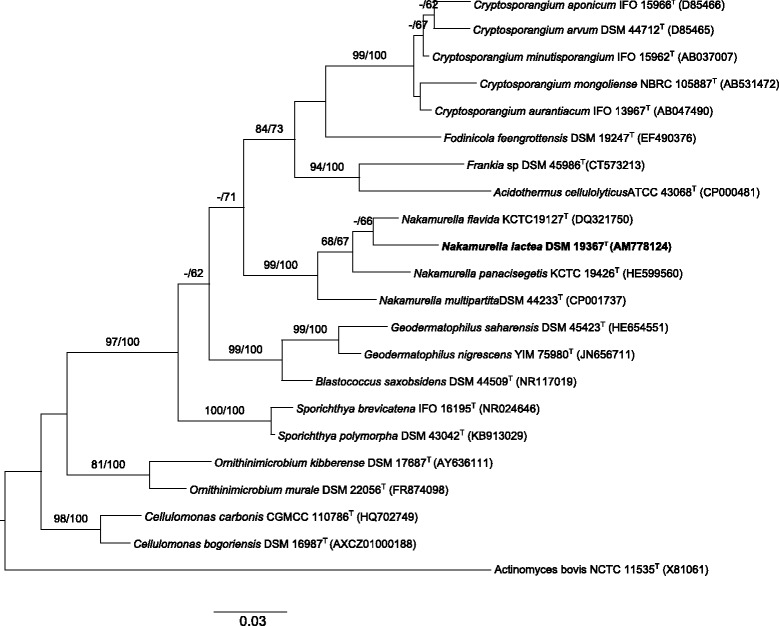
Maximum likelihood phylogenetic tree of N. lactea DLS-10T and related type strains within the related families constructed under the GTR + GAMMA model and rooted using Actinomyces bovis NCTC 11535T as outgroup. The branches are scaled in terms of the expected number of substitutions per site (see size bar). Support values from maximum-likelihood (left) and maximum-parsimony (right) bootstrapping are shown above the branches if equal to or larger than 60%
Chemotaxonomic data (optional, Heading 3)
Glucose, mannose, ribose and rhamnose were detected as the whole-cell sugars [5]. The pattern of polar lipid contains diphosphatidylglycerol, phosphatidylethanolamine, phosphatidylinositol, aminophospholipid, five unidentified phosphoglycolipids, and one unidentified glycolipid [6].
The diagnostic peptidoglican is the meso-diaminopimelic acid. The major fatty acids are anteiso-C15:0, C16:0, iso-C16:0, and anteiso-C17:0 [9]. MK-8(H4) and MK-9(H4) are the predominant menaquinones but MK-7(H4) was also revealed in a low amount [6].
Genome sequencing information
Genome project history
N. lactea DLS-10T (DSM 19367 T) was selected for sequencing on the basis of its phylogenetic position [17, 18], and is part of Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes project [19], a follow-up of the Genomic Encyclopedia of Bacteria and Archaea pilot project [20], which aims at increasing the sequencing coverage of key reference microbial genomes and to generate a large genomic basis for the discovery of genes encoding novel enzymes [21]. KMG-I is the first of the production phases of the “Genomic Encyclopedia of Bacteria and Archaea: sequencing a myriad of type strains” initiative [22] and a Genomic Standards Consortium project [23]. The project and the genome sequence are deposited in the Genome OnLine Database [24] and Genbank under the accession number AUFT00000000.1. In Table 2, we summarize genome sequence project.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Level 1: Standard Draft |
| MIGS-28 | Libraries used | NOHX |
| MIGS 29 | Sequencing platforms | Illumina, Illumina HiSeq 2000 |
| MIGS 31.2 | Fold coverage | NA |
| MIGS 30 | Assemblers | Allpaths/Velvet |
| MIGS 32 | Gene calling method | Prodigal 2.5 |
| Locus Tag | K340 | |
| Genbank ID | AUFT00000000.1 | |
| GenBank Date of Release | 2013-06-03 | |
| GOLD ID | Gi11889 | |
| BIOPROJECT | PRJNA195807 | |
| MIGS 13 | Source Material Identifier | DSM 19367T |
| Project relevance | GEBA-KMG, Tree of Life |
Growth conditions and genomic DNA preparation
A N. lactea DLS-10T culture was prepared in DSM medium 65 [25] at 28 °C. Genomic DNA was extracted using MasterPure™ Gram Positive DNA Purification Kit (Epicentre MGP04100) following the standard protocol provided by the manufacturer but modified by the incubation on ice overnight on a shaker, the use of additional 1 μl proteinase K, and the addition of 7.5 units achromopeptidase, 7.5 μg/μl lysostaphine, 1050.0 units lysozyme, and 7.5 units mutanolysine. DNA is available from DSMZ through the DNA Bank Network [26].
Genome sequencing and assembly
The draft genome of N. lactea DLS-10T was generated at the DOE Joint genome Institute (JGI) using the Illumina technology [27]. An Illumina standard shotgun library was constructed and sequenced using the Illumina HiSeq 2000 platform, which generated 13,910,936 reads totalling 2,086.6 Mb. All general aspects of library construction and sequencing performed at the JGI can be found at http://www.jgi.doe.gov. All raw Illumina sequence data was passed through DUK, a filtering program developed at JGI, which removes known Illumina sequencing and library preparation artefacts (unpublished results). Following steps were then performed for assembly: (1) filtered Illumina reads were assembled using Velvet (version 1.1.04) [28], (2) 1–3 kb simulated paired end reads were created from Velvet contigs using wgsim (https://github.com/lh3/wgsim), (3) Illumina reads were assembled with simulated read pairs using Allpaths–LG (version r42328) [29]. Parameters for assembly steps were: 1) Velvet (velveth:63 –shortPaired and velvetg: −very clean yes –exportFiltered yes –min contig lgth 500 –scaffolding no–cov cutoff 10) 2) wgsim (−e 0 –1 100 –2 100 –r 0 –R 0 –X 0) 3) Allpaths–LG (PrepareAllpathsInputs:PHRED 64 = 1 PLOIDY = 1 FRAG COVERAGE = 125 JUMP COVERAGE = 25 LONG JUMP COV = 50, RunAllpathsLG: THREADS = 8 RUN = std shredpairs TARGETS = standard VAPI WARN ONLY = True OVERWRITE = True). The final draft assembly contained 31 contigs in 27 scaffolds. The total size of the genome is 5.8 Mb and the final assembly is based on 712.8 Mb of Illumina data, which provides an average 122.5X coverage of the genome.
Genome annotation
The complete genome sequence was annotated using the JGI Prokaryotic Automatic Annotation Pipeline [30] with additional manual review using the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [31]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non redundant database, UniProt, TIGRFam, Pfam, KEGG, COG, and InterPro databases. The tRNAScanSE tool [32] was used to find tRNA genes, whereas ribosomal RNA genes were found by searches against models of the ribosomal RNA genes built from SILVA [33]. Other non–coding RNAs such as the RNA components of the protein secretion complex and the RNase P were identified by searching the genome for the corresponding Rfam profiles using INFERNAL [34]. Additional gene prediction analysis and manual functional annotation was performed within the Integrated Microbial Genomes (IMG) platform [35, 36] developed by the Joint Genome Institute, Walnut Creek, CA, USA [37].
Genome properties
The 5820860 bp of genome size of N. lactea DLS-10T presents 5100 protein-coding genes, 3 rRNA genes (5S, 16S, 23S RNA) and 59 tRNA genes. A G + C content of 68.9% was calculated. More genome details are listed in Tables 3 and 4.
Table 3.
Genome statistics
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 5820860 | 100.00 |
| DNA coding (bp) | 5332245 | 91.61 |
| DNA G + C (bp) | 4011790 | 68.92 |
| DNA scaffolds | 27 | 100.00 |
| Total genes | 5169 | 100.00 |
| Protein coding genes | 5100 | 98.67 |
| RNA genes | 69 | 1.33 |
| Pseudo genes | 231 | |
| Genes in internal clusters | 588 | 11.38 |
| Genes with function prediction | 4048 | 78.31 |
| Genes assigned to COGs | 3321 | 64.25 |
| Genes with Pfam domains | 4211 | 81.47 |
| Genes with signal peptides | 432 | 8.36 |
| Genes with transmembrane helices | 1206 | 23.33 |
| CRISPR repeats | 1 |
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | % | Description |
|---|---|---|---|
| J | 198 | 5.07 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.03 | RNA processing and modification |
| K | 392 | 10.04 | Transcription |
| L | 122 | 3.12 | Replication, recombination and repair |
| B | 1 | 0.03 | Chromatin structure and dynamics |
| D | 25 | 0.64 | Cell cycle control, Cell division, chromosome partitioning |
| V | 94 | 2.41 | Defence mechanisms |
| T | 137 | 3.51 | Signal transduction mechanisms |
| M | 144 | 3.69 | Cell wall/membrane biogenesis |
| N | 10 | 0.26 | Cell motility |
| U | 23 | 0.59 | Intracellular trafficking and secretion |
| O | 121 | 3.1 | Posttranslational modification, protein turnover, chaperones |
| C | 210 | 5.38 | Energy production and conversion |
| G | 648 | 12.31 | Carbohydrate transport and metabolism |
| E | 459 | 11.75 | Amino acid transport and metabolism |
| F | 91 | 2.33 | Nucleotide transport and metabolism |
| H | 219 | 5.61 | Coenzyme transport and metabolism |
| I | 255 | 6.53 | Lipid transport and metabolism |
| P | 244 | 6.25 | Inorganic ion transport and metabolism |
| Q | 154 | 3.94 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 443 | 11.34 | General function prediction only |
| S | 158 | 44.05 | Function unknown |
| - | 1848 | 35.75 | Not in COGs |
Conclusion
The genome of N. lactea will be used to study, for the first time, its potential as bioactive natural products source and the correlation between the rare soil bacteria and their habitat. According to [38], the within-species deviation in genomic G + C content is at most 1%. The range of 70.4–74.3% given in by Kim et al. [6] is thus too broad and too deviating from the 68.9% calculated in the genome sequence, much like the value 74.3% provided by Lee et al. [9]. This calls for an emendation of the species description [38].
Emended description of Nakamurella lactea (Lee et al. [9]) Kim et al. [6]
The properties are as given in the species description by Kim et al. [6] with the following emendation. Based on the genomic data the G + C content is 68.9%.
Acknowledgements
We thank Katja Steenblock (DSMZ) for her help in preparing the culture of N. lactea DSM 19367 T and Evelyne Brambilla (DSMZ) for her contribution in the DNA extraction. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported under Contract No. DE-AC02-05CH11231.
Authors’ contributions
IN and HPK conceived of the study and participated in its design and coordination. IN, LC and MCMC collaborated in acquisition of data, analysis of them and drafted the manuscript. MG and RM performed the phylogenetic analysis and SEM images, respectively. TW and NCK participated in genome sequencing, annotation and analysis. All authors contributed in improving the quality of the manuscript and approved the final version.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Sen A, Daubin V, Abrouk D, Gifford I, Berry AM, Normand P. Phylogeny of the class Actinobacteria revisited in the light of complete genomes. The orders 'Frankiales' and Micrococcales should be split into coherent entities: proposal of Frankiales ord. nov., Geodermatophilales ord. nov., Acidothermales ord. nov. and Nakamurellales ord. nov. Int J Syst Evol Microbiol. 2014;64:3821–3832. doi: 10.1099/ijs.0.063966-0. [DOI] [PubMed] [Google Scholar]
- 2.Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol. 1997;47:479–91. doi: 10.1099/00207713-47-2-479. [DOI] [Google Scholar]
- 3.Tao TS, Yue YY, Chen WX, Chen WF. Proposal of Nakamurella gen. nov. as a substitute for the bacterial genus Microsphaera Yoshimi et al. 1996 and Nakamurellaceae fam. nov. as a substitute for the illegitimate bacterial family Microsphaeraceae. Rainey et al. 1997. Int J Syst Evol Microbiol. 2004;54:999–1000. doi: 10.1099/ijs.0.02933-0. [DOI] [PubMed] [Google Scholar]
- 4.Kim KK, Lee JS. The Family Nakamurellaceae. In: Rosenberg E, Delong EF, Lory S, Stackebrandt E, Thompson F, editors. The Prokaryotes-Actinobacteria. Heidelberg: Springer-Verlag Berlin; 2014. [Google Scholar]
- 5.Yoshimi Y, Hiraishi A, Nakamura L. Isolation and characterization of Microsphaera multipartita gen. nov., sp. nov., a polysaccharide-accumulating Gram-positive bacterium from activated sludge. Int J Syst Bacteriol. 1996;46:519–25. doi: 10.1099/00207713-46-2-519. [DOI] [Google Scholar]
- 6.Kim KK, Lee KC, Lee JS. Nakamurella panacisegetis sp. nov. and proposal for reclassification of Humicoccus flavidus Yoon et al., 2007 and Saxeibacter lacteus Lee et al., 2008 as Nakamurella flavida comb. nov. and Nakamurella lactea comb. nov. Syst App Micro. 2012;35:291–6. doi: 10.1016/j.syapm.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 7.List Editor List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2012;62:2549–2554. doi: 10.1099/ijs.0.048033-0. [DOI] [Google Scholar]
- 8.Yoon JH, Kang SJ, Jung SY, Oh TK. Humicoccus flavidus gen. nov., sp. nov., isolated from soil. Int J Syst Evol Microbiol. 2007;57:56–9. doi: 10.1099/ijs.0.64246-0. [DOI] [PubMed] [Google Scholar]
- 9.Lee SD, Park SK, Yun YW, Lee DW. Saxeibacter lacteus gen. nov., sp. nov., an actinobacterium isolated from rock. Int J Syst Evol Micro. 2008;58:906–9. doi: 10.1099/ijs.0.65558-0. [DOI] [PubMed] [Google Scholar]
- 10.Tice H, Mayilra S, Sims D, Lapidus A, Nolan M, Lucas S, et al. Complete genome sequence of Nakamurella multipartita type strain (Y-104T) Stand Genomic Sci. 2010;2:168–75. doi: 10.4056/sigs.721316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meier-Kolthoff JP, Hahnke RL, Petersen J, Scheuner C, Michael V, Fiebig A, et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand Genomic Sci. 2014;10:2. doi: 10.1186/1944-3277-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goloboff PA, Farris JS, Nixon KC. TNT, a free program for phylogenetic analysis. Cladistics. 2008;24:774–86. doi: 10.1111/j.1096-0031.2008.00217.x. [DOI] [Google Scholar]
- 16.Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? J Comput Biol. 2010;17:337–54. doi: 10.1089/cmb.2009.0179. [DOI] [PubMed] [Google Scholar]
- 17.Göker M, Klenk HP. Phylogeny-driven target selection for large-scale genome-sequencing (and other) projects. Stand Genomic Sci. 2013;8:360–74. doi: 10.4056/sigs.3446951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol. 2010;33:175–82. doi: 10.1016/j.syapm.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Kyrpides NC, Woyke T, Eisen JA, Garrity G, Lilburn TG, Beck BJ, Whitman WB, Hugenholtz P, Klenk HP. Genomic Encyclopedia of Type Strains, Phase I: The one thousand microbial genomes (KMG-I) project. Stand Genomic Sci. 2013;17:9(3):1278–84. [DOI] [PMC free article] [PubMed]
- 20.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–60. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piao H, Froula J, Du C, Kim TW, Hawley ER, Bauer S, et al. Identification of novel biomass-degrading enzymes from genomic dark matter: Populating genomic sequence space with functional annotation. Biotechnol Bioeng. 2014;111:1550–65. doi: 10.1002/bit.25250. [DOI] [PubMed] [Google Scholar]
- 22.Kyrpides NC, Hugenholtz P, Eisen JA, Woyke T, Göker M, Parker CT, et al. Genomic Encyclopaedia of Bacteria and Archaea: sequencing a myriad of type strains. PLoS Biol. 2014;12. doi:10.1371/journal.pbio.1001920. [DOI] [PMC free article] [PubMed]
- 23.Field D, Amaral-Zettler L, Cochrane G, Cole JR, Dawyndt P, Garrity GM, et al. The Genomic Standards Consortium. PLoS Biol. 2011;9:8–10. doi: 10.1371/journal.pbio.1001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reddy TBK, Thomas AD, Stamatis D, Bertsch J, Isbandi M, Jansson J, et al. The Genomes OnLine Database (GOLD) v.5: a metadata management system based on a four level (meta) genome project classification. Nucleic Acids Res. 2015;43(Database issue):D1099–106. [DOI] [PMC free article] [PubMed]
- 25.List of growth media used at DSMZ. http://www.dsmz.de/microorganisms/media_list.php.
- 26.Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk H-P, Güntsch A, et al. Bank Network: The start from a German initiative. Biopreserv Biobank. 2011;9:51–5. doi: 10.1089/bio.2010.0029. [DOI] [PubMed] [Google Scholar]
- 27.Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5:433–8. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- 28.Zerbino D, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gnerre S, MacCallum I. High–quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108:1513–8. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huntemann M, Ivanova NN, Mavromatis K, Tripp HJ, Paez-Espino D, Palaniappan K, et al. The Standard Operating Procedure of the DOE-JGI Microbial Genome Annotation Pipeline (MGAP v.4) Stand Genomic Sci. 2015;10:86. doi: 10.1186/s40793-015-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG-ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–8. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 32.Lowe TM, Eddy SR. tRNAscan–SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pruesse E, Quast C, Knittel, Fuchs B, Ludwig W, Peplies J, Glckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nuc Acids Res. 2007;35:2188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.INFERNAL. Inference of RNA alignments. http://infernal.janelia.org.
- 35.Chen IM, Markowitz VM, Palaniappan K, Szeto E, Chu K, Huang J, Ratner A, Pillay M, Hadjithomas M, Huntemann M, Mikhailova N, Ovchinnikova G, Ivanova NN, Kyrpides NC. BMC Genomics. 2016;17:307. doi: 10.1186/s12864-016-2629-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Pillay M, et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42:D560–7. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joint Genome Institute. http://jgi.doe.gov/
- 38.Meier-Kolthoff JP, Klenk HP, Göker M. Taxonomic use of the G + C content and DNA:DNA hybridization in the genomic age. Int J Syst Evol Microbiol. 2014;64:352–356. doi: 10.1099/ijs.0.056994-0. [DOI] [PubMed] [Google Scholar]
- 39.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodfellow M. Phylum XXVI Actinobacteria phyl. nov. In: Goodfellow M, Kämpfer P, Busse HJ, Trujillo ME, Suzuki K, Ludwig W, Whitman WB, editors. Bergey's Manual of Systematic Bacteriology. New York: Springer; 2012. [Google Scholar]
- 41.Zhi XY, LiW J, Stackebrandt E. An update of the structure and 16S rRNAa gene sequence-based definition of higher ranks of the class Actinobacteria, with the proposal of two new suborders and four new families and emended descriptions of the existing higher taxa. Int J Syst Evol Microbiol. 2009;59:589–608. doi: 10.1099/ijs.0.65780-0. [DOI] [PubMed] [Google Scholar]
- 42.Ashburner M, Ball CA, Blake JA, Botstein D, But-ler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]