

Abstract
Epiregulin (EREG) belongs to the ErbB family of ligands. EREG binds to EGFR and ErbB4 receptor and stimulates homodimers of EGFR and ErbB4 in addition to all possible heterodimeric ErbB complexes, resulting in the activation of downstream signaling pathways. EREG is overexpressed in various human cancers and has been implicated in tumor progression and metastasis. Oncogenic activation of the MEK/ERK pathway plays a central role in the regulation of EREG expression. Non-small-cell lung cancers (NSCLCs) harboring KRAS, BRAF, or EGFR mutations overexpress EREG, and abrogation of such mutations or inhibition of MEK or ERK downregulates the expression of EREG. Elevated EREG expression in NSCLC is associated with aggressive tumor phenotypes and unfavorable prognosis, especially in oncogenic KRAS-driven lung adenocarcinomas. The finding that attenuation of EREG inhibits cell growth and induces apoptosis in KRAS-mutant and EREG-overexpressing NSCLC cell lines suggests that targeting EREG might be a treatment option for KRAS-mutant NSCLC, although further studies are necessary to elucidate its therapeutic value. These observations suggest that oncogenic mutations in the EGFR, KRAS, or BRAF genes induce EREG upregulation through the activation of MEK/ERK pathway in NSCLC cells, whereas overproduced EREG stimulates the EGFR/ErbB receptors and activates multiple downstream signaling pathways, leading to tumor progression and metastasis of these oncogene-driven NSCLCs. This paper reviews the current understanding of the oncogenic role of EREG and highlights its potential as a therapeutic target for NSCLC.
Keywords: epiregulin, NSCLC, KRAS mutation, therapeutic target
Introduction
Lung cancer is the leading cause of cancer mortality worldwide.1 Lung cancer is categorized into two main subtypes: small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC), the latter accounts for 80%–85% of all lung cancers.2 Lung adenocarcinoma is a major histological subtype of NSCLC, and its incidence is increasing in both men and women.3 The majority of patients with NSCLC have locally advanced or metastatic disease at initial diagnosis, and systemic cytotoxic chemotherapy such as platinum doublets has limited efficacy, with a median overall survival (OS) of 8–11 months.4 Therefore, there is an urgent need for the development of effective treatment modalities to improve the survival of patients with NSCLC.
The development of NSCLC involves a number of genetic and epigenetic alterations that accumulate over time.2 One of the functions of these molecular alterations is the activation of driver oncogenes that are essential for maintaining the malignant phenotype. Inactivation of a single oncogene is sufficient to kill cancer cells due to the phenomenon of “oncogene addiction”.5 Recent studies have identified several driver oncogenes that are potential therapeutic targets for NSCLC.6–10 KRAS and EGFR mutations are the common driver mutations in lung adenocarcinomas, and several fusion genes, including ones formed by rearrangements of ALK, RET, and ROS1, have also been identified as key driver oncogenes in terms of their therapeutic implications.11–16 To overcome such oncogene-driven tumors, molecularly targeted drugs, including tyrosine kinase inhibitors of EGFR and ALK, have been approved and are currently being used in the clinic.17 Mutations in the tyrosine kinase domain of EGFR have been widely studied;18 sensitive EGFR mutations such as in-flame deletions in exon 19 and L858R substitutions in exon 21 are well-known predictive biomarkers of the efficacy of EGFR-tyrosine kinase inhibitors (EGFR-TKIs).19–23 Soda et al identified ALK rearrangements11 that have been found as predictive biomarkers of the therapeutic efficacy of ALK-tyrosine kinase inhibitors in NSCLC.24,25 Currently, molecular testing for sensitizing EGFR mutations and EML4–ALK fusion oncogenes is performed in tumor samples.26
Although “personalized medicine” such as the use of EGFR-TKIs against EGFR-mutated NSCLC and ALK-tyrosine kinase inhibitors against ALK fusion-positive NSCLC is being applied into clinical practice, therapeutic modalities for KRAS-mutant NSCLC have not yet been established. KRAS encodes a small GTP-binding protein that is involved in many cellular processes, including cell growth, differentiation, and apoptosis.27,28 Wild-type KRAS has intrinsic GTP hydrolysis activity that catalyzes the conversion of KRAS into its GDP-bound (inactive) form, and KRAS mutations lock KRAS into its GTP-bound (active) form, resulting in oncogenic activation of downstream signaling pathways. KRAS mutations are attractive therapeutic targets because they are present in many human cancers, including cancers of the pancreas, colon, and lung.27,28 To establish therapeutic strategies for KRAS-mutant NSCLC, we performed a microarray analysis to compare the gene expression profiles of mutant KRAS-disrupted NSCLC clones with those of the mutant KRAS-expressing clones.29 Consequently, we identified epiregulin (EREG) as one of several putative transcriptional targets of oncogenic KRAS signaling.30 In this review, we describe the current understanding of the oncogenic role of EREG and its relationship with oncogenic KRAS, and we highlight the potential of EREG as a therapeutic target for NSCLC.
Epiregulin
EREG belongs to the ErbB family of ligands and was originally purified from conditioned medium of NIH 3T3 mouse tumorigenic fibroblasts.31 The human EREG gene is located on chromosome 4q13.3, and the AREG and BTC genes are also clustered at that location.32 EREG has 46 amino acid residues, and 24%–50% of its sequence is shared with those of other EGF family members.31 EREG is capable of binding to EGFR and ErbB4 receptor and stimulates homodimers of EGFR and ErbB4 in addition to heterodimers of ErbB2 and ErbB3, leading to the activation of their intrinsic kinase domain and the phosphorylation of specific tyrosine residues in the cytoplasmic tail of their receptors (Figure 1).33,34 Those phosphorylated residues serve as docking sites for intracellular signaling molecules, and therefore activate downstream signaling pathways, including the MEK/ERK pathway.33
Figure 1.

Binding specificity of EGF, transforming growth factor-α (TGF-α), amphiregulin (AREG), betacellulin (BTC), heparin-binding EGF (HB-EGF), EREG, and neuregulins (NRGs).
Notes: EGFR, TGF-α, and AREG bind specifically to EGFR. BTC, HB-EGF, and EREG bind both EGFR and ErbB4. NRGs are further categorized according to their capacity to bind ErbB3 and ErbB4 (NRG1 and NRG2) or only ErbB4 (NRG3 and NRG4). ErbB2 has no binding EGF family ligands, whereas it serves as a heterodimerization partner of the other ligands. ErbB3 lacks intrinsic kinase activity, but it can activate EGFR signaling pathways through heterodimerizing with another ErbB receptor.
Abbreviation: EREG, epiregulin.
Previous studies have reported the physiological role of EREG in the control of cell proliferation and differentiation of human airway epithelial cells. Coculturing human airway epithelial cells with lung fibroblasts, which express EREG, induces human airway epithelial differentiation accompanied by ErbB2 phosphorylation.35 Exposure of compressive stress increases EREG expression, and this phenomenon was shown to be suppressed by an EGFR inhibitor in human bronchial epithelial cells.36 These findings suggest that EREG activates ErbB receptors and their downstream signaling pathways in bronchial epithelial cells.
Role of EREG in cancer
EREG/EGFR pathways regulate diverse cellular processes, including cell proliferation, invasion, metastasis, angiogenesis, and resistance to apoptosis, conferring aggressive tumor behavior.37 EREG is overexpressed in many human cancers, such as pancreatic cancer, colon cancer, NSCLC, breast cancer, bladder cancer, prostate cancer, kidney cancer, liver cancer, ovarian cancer, oral cancer, thymic cancer, salivary adenoid cystic carcinoma, and malignant glioma, whereas EREG expression levels in normal adult tissues are extremely low.30,38–54 For instance, thymic carcinomas had a high percentage (91.7%) of immunohistochemical expression of EREG.50 EREG has been identified as one of the highly expressed genes in the hTERT-immortalized fibroblasts, and blockage of EREG inhibits the in vitro growth of the hTERT-immortalized cells, suggesting the critical role of EREG in hTERT-mediated immortalization and transformation.55 Zhu et al reported that pancreatic ductal adenocarcinomas (PDAs) exhibit higher levels of EREG mRNA than normal pancreatic and chronic pancreatitis tissues.39 They also found that in vitro cell growth of pancreatic cancer is significantly increased by human recombinant EREG in a dose-dependent manner. Given that whole-exome sequencing analysis of PDAs detected KRAS mutations in >90% of PDAs,56 it is possible that EREG is involved in the development of oncogenic KRAS-driven PDAs.
An oncogenic role of EREG has also been suggested in other human cancer types. In a COX2-overexpression mouse model of bladder carcinoma, EREG is the most significantly upregulated gene, and the expression of a recombinant EREG increases cell proliferation in bladder cancer cell lines.57 In a mouse model of hepatocellular carcinomas, EREG-knockout mice have fewer tumors that are smaller in size than EREG-wild-type mice.58 In addition, siRNA-mediated EREG knockdown suppresses in vitro hepatoma growth.47 These findings suggest that EREG is involved in bladder and hepatocellular carcinogenesis.
Several lines of evidence have implicated the role of EREG in tumor metastasis. A previous study of breast cancer cells with potential for lung metastasis identified the lung metastasis signature (LMS) genes, which include EREG, COX2, MMP1, and MMP2.59 shRNA-mediated simultaneous knockdown of these four genes in a xenograft model suppressed in vivo tumor growth, angiogenesis, and metastasis,60 indicating that EREG confers metastatic potential in breast cancer. Similarly, in a bladder cancer mouse model with lung metastasis, microarray analysis identified EREG as one of the upregulated genes in lung metastatic tumors.42 Gene expression profiling comparing colon cancers with and without liver metastasis also identified EREG as a metastasis-associated gene.61 Moreover, salivary adenoid cystic carcinoma cells with lung metastatic potential overexpress EREG, resulting in the promotion of migration and invasion through the activation of ERK and Akt.46 Recently, EREG was found to be upregulated by KAP1, a transcriptional regulator that promotes proliferation and metastasis in breast cancer.62 Collectively, these findings imply that EREG plays a key role in tumor progression and metastasis and confers high malignant potential in human cancers.
Interestingly, a possible link between EREG and cancer cell stemness has been suggested. A previous study that included microarray and immunohistochemistry analyses showed that EREG is expressed in LGR5-positive colon cancer cells, which possess cancer stem cell properties.63 Furthermore, in a metastatic xenograft model, an anti-EREG antibody exhibited antitumor activity against tumors derived from LGR5-positive colon cancer cells. Notably, LGR5 expression was reported to be related to larger tumor size, more advanced stage, and poor prognosis in lung adenocarcinoma.64 Therefore, EREG may be a therapeutic target for lung adenocarcinoma stem cells.
In contrast to the oncogenic roles of EREG, the negative aspect was also reported. EREG expression levels are undetectable in most SCLC cell lines,30 and EREG does not seem to be necessary for SCLC carcinogenesis. EREG expression was shown to be epigenetically silenced in gastric cancer cell lines by aberrant DNA methylation and histone demethylation.65 In addition, EREG promoter was hypermethylated in 30% of primary gastric tumor tissues. Thus, it is possible that EREG is inactivated in some human cancers, including gastric cancer and SCLC, and aberrant promoter methylation might be one of the mechanisms for EREG inactivation.
EREG as oncogenic KRAS-regulated gene
The Ras gene family includes three genes, KRAS, HRAS, and NRAS, all of which share very similar molecular structures and a common GTPase domain that binds to and hydrolyze guanine nucleotides.66 Oncogenic mutations of the Ras genes mainly occur at codons 12, 13, and 61, and these mutations lock Ras proteins into their GTP-bound (active) form, resulting in the constitutive activation of Ras downstream pathways and the promotion of oncogenesis.28 KRAS is the most commonly mutated isoform of the Ras genes, and KRAS mutations have been found in a variety of human cancers, including cancers of the pancreas and colon, and NSCLC.28 Several studies have found a relationship between EREG and KRAS. Baba et al conducted a polymerase chain reaction-based cyclic DNA subtraction library to compare genes that were differentially expressed between HCT116 colon cancer cells and KRAS-disrupted clones derived from HCT116 cells, and EREG was found to be upregulated by activation of Ras signaling pathways.67 In the KRAS-disrupted HCT116 clones, forced expression of exogenous EREG partially recovered in vivo tumorigenicity, indicating that EREG is involved in the tumorigenesis of KRAS-mutant colon cancer. Similarly, a previous microarray analysis demonstrated that EREG expression is upregulated in KRAS-transformed human prostate cancer cell; furthermore, MEK inhibition downregulated EREG expression, accompanied by downregulation of the ETS1 transcription factor, which binds to the EREG promoter.68 EREG expression is also upregulated in lung tumors from mice carrying mutant KRAS alleles.69
In addition to these findings, we identified EREG as one of the transcriptional targets of oncogenic KRAS signaling in KRAS-mutant NSCLC cells and immortalized bronchial epithelial cells, expressing ectopic mutant KRAS.29 In KRAS-mutant and EREG-overexpressing NSCLC cells, EREG expression is reduced by siRNAs targeting mutant KRAS (but not by siRNAs targeting wild-type KRAS) and by inhibitors of MEK and ERK.30 EREG is preferentially expressed in KRAS-mutant NSCLC cell lines and tumor specimens. Importantly, EREG expression is significantly correlated with KRAS copy number in a subgroup of KRAS-mutated NSCLC cell lines. Given that KRAS copy number gains are associated with increased mutant allele transcription and gene activities in NSCLC cells,70 KRAS copy number gains appear to enhance oncogenic KRAS-induced activation of the MEK/ERK pathway, resulting in the upregulation of EREG in NSCLC cells. Collectively, these findings suggest that EREG overexpression is likely induced by oncogenic KRAS via activation of the MEK/ERK pathway and plays an essential role in oncogenic KRAS-mediated tumorigenesis.
Clinicopathological and prognostic significance of EREG expression in cancer
Clinicopathological studies have suggested that EREG is associated with aggressive tumor phenotypes and unfavorable prognoses in several human cancers. The previous studies that had showed the prognostic significance of EREG expression were summarized in Table 1. EREG mRNA is highly expressed in bladder tumors with advanced T stages, and elevated EREG expression is significantly correlated with shorter survival.41 Elevated EREG expression is also associated with shorter survival in patients with oral squamous cell carcinoma.45 In colon cancer, EREG expression is significantly correlated with the depth of tumor invasion and distant metastasis.71 Furthermore, EREG expression was found to be associated with higher tumor grade and worse survival in glioblastoma.54
Table 1.
The studies evaluating the association between EREG expression and survival in human cancers
| Type of cancer | Method | Cutoff value | Number of points | Survival for EREG positive | Reference |
|---|---|---|---|---|---|
| NSCLC | IHC | ≥ score 100a | 356 | Poor* | 43 |
| Lung adenocarcinoma | qRT-PCR | ≥ median | 119 | Poorb | 49 |
| Bladder cancer | qRT-PCR | First cutoff: 2.4 and second cutoff: 4.8c | 73 | Poor | 41 |
| Oral squamous cell carcinoma | qRT-PCR | ≥0.1 | 30 | Poor | 45 |
| Glioblastoma | IHC | ≥ score 3d | 73 | Poor | 54 |
Notes: In all studies, overall survival was analyzed by the Kaplan–Meier method, and comparison between subgroups was examined by the log-rank test.
The staining score was quantified on the basis of staining intensity and extension (intensity × extension);
cox regression analysis was also performed, and the hazard ratio, adjusted by tumor stage, was 8.71 (95% CI: 1.90–39.80);
the first-cutoff point was median, and the second-cutoff point was arbitrarily chosen;
IHC positivity was determined according to the total score (intensity score + proportional score);
P=0.054 for the difference of overall survival, whereas the difference was more evident (P=0.014) after correction for differences in covariates (age, pathological nodal, tumor stage, and histological subtype).
Abbreviations: EREG, epiregulin; NSCLC, non-small-cell lung cancer; IHC, immunohistochemistry; CI, confidence interval; qRT-PCR, quantitative reverse transcription-polymerase chain reaction.
With regards to the prognostic significance of EREG in NSCLC, an immunohistochemical analysis of NSCLC biopsy samples revealed that 64.7% of the tumors stained positively for EREG, and that patients with EREG-positive tumors had poorer clinical outcomes than those with EREG-negative tumors.43 We also conducted a gene expression analysis of surgical tumor specimens to determine the clinicopathological and prognostic features of EREG expression in NSCLC.30 EREG mRNA expression levels are significantly higher in lung adenocarcinomas than in lung squamous cell carcinomas. EREG is predominantly expressed in NSCLCs with pleural involvement, lymphatic permeation, and vascular invasion, all of which confer aggressive tumor phenotypes. When we extended this clinicopathological study to include 136 surgical specimens from patients with lung adenocarcinoma, EREG expression was found to be significantly higher in tumors from elderly patients (≥70), male patients, and smokers.49 In this series, elevated EREG expression was also associated with pleural involvement positivity, lymphatic permeation positivity, and vascular invasion positivity. Patients with lung adenocarcinoma with EREG-high tumors have significantly shorter disease-free survival (DFS) and OS compared to those with EREG-low tumors. When the patients were divided into four groups according to the EREG expression and the KRAS mutation status, the patients with EREG-high/KRAS-mutant tumors had significantly shorter DFS and OS compared to those with EREG-low/KRAS-wild-type tumors. Multivariate analyses showed that EREG expression is a significant prognostic factor for DFS and OS. Taken together, EREG appears to contribute to the acquisition of aggressive tumor phenotypes and serves as a prognostic marker in NSCLC, especially KRAS-mutant lung adenocarcinoma.
Therapeutic potential for targeting EREG in NSCLC
Several lines of evidence suggest that targeting EREG in NSCLC has therapeutic potential. KRAS-mutant NSCLC cells preferentially express EREG, and siRNA-mediated EREG knockdown inhibits anchorage-dependent and anchorage-independent growth and induces apoptosis in KRAS-mutant NSCLC cells,30,49 suggesting that EREG is a therapeutic target for oncogenic KRAS-driven NSCLC. EREG overexpression was also found in the EGFR-mutant NSCLC cells,29,43,69 in the BRAF-mutant NSCLC cells, and in a subset of NSCLC cells with wild-type EGFR/KRAS/BRAF.29 In EREG-overexpressing NSCLC cells, inhibition of MEK or ERK reduces EREG expression, irrespective of mutation status.30 Thus, activation of the MEK/ERK pathway seems to be a common mechanism of EREG upregulation in NSCLC.
Previous studies have suggested that EREG has therapeutic potential for EGFR-mutant NSCLC. EREG is downregulated by siRNA-mediated EGFR knockdown and EGFR inhibitors in EGFR-mutant NSCLC cells.29,43 Lung tumors of mutant EGFR transgenic mice exhibit high levels of EREG.72 In EGFR-mutant NSCLC cells, both shRNA-mediated EREG knockdown and an anti-EREG antibody inhibit cell proliferation and invasion and induce apoptosis.43 These findings indicate that targeting EREG is a good therapeutic option for EGFR-mutant NSCLC cells with resistance to EGFR-TKIs.
BRAF mutations occur in 2%–3% of NSCLCs (predominantly in lung adenocarcinomas)73,74 and are potential therapeutic targets. BRAF-mutant NSCLC cells were found to have high EREG expression at similar levels to those in KRAS-mutant NSCLC cells.30,49 In BRAF-mutant NSCLC cells, siRNAs targeting BRAF and the inhibitor of MEK or ERK reduce EREG expression, showing that oncogenic BRAF upregulates EREG expression via activation of the MEK/ERK pathway. Thus, targeting EREG might also be effective for BRAF-mutant NSCLC cells.
Conclusion
Accumulated evidences suggest that oncogenic mutations in the EGFR, KRAS, or BRAF genes induce EREG overexpression via activation of the MEK/ERK signaling pathway. Overproduced EREG can stimulate the EGFR/ErbB receptors to activate multiple downstream signaling pathways, including the MEK/ERK and PI3K/Akt pathways through an autocrine loop mechanism (Figure 2). Thus, EREG is likely to play diverse oncogenic roles, including the regulation of cell proliferation, invasion, and metastasis as a potent pan-ErbB ligand; therefore, it may contribute to the acquisition of highly malignant phenotypes and the development of human cancers, including NSCLC. Considering that half of the lung adenocarcinomas have mutations in EGFR, BRAF, or KRAS in a mutually exclusive manner7,9,10 and that tumors with such driver mutations overexpress EREG, it is plausible that the majority of NSCLCs could benefit from EREG-targeted therapy. Although the exact mechanism behind the regulation of EREG is still unclear, EREG may be an excellent target for anticancer therapies, especially for NSCLCs. Furthermore, in vivo studies and clinical trials are warranted to clarify the effectiveness of EREG-targeted therapy for NSCLCs.
Figure 2.
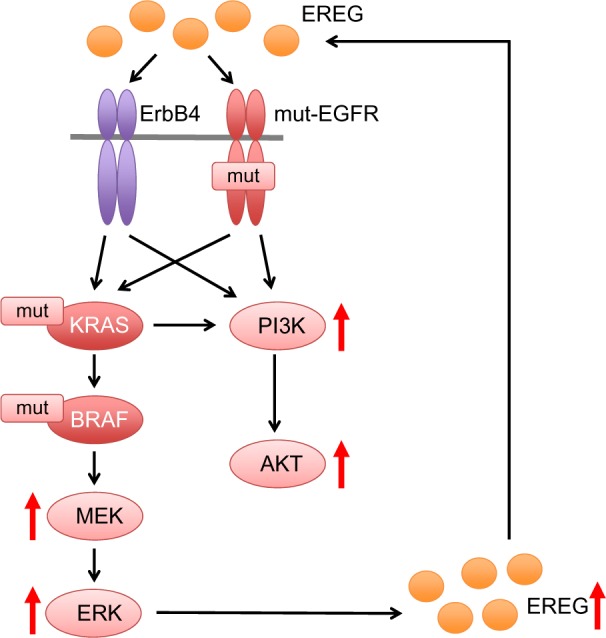
Oncogenic mutations for the upregulation of EREG expression.
Notes: Oncogenic mutations in the EGFR, KRAS, or BRAF genes result in activation of the MEK/ERK signaling pathway, which in turn upregulates EREG expression. Overproduced EREG can stimulate the EGFR/ErbB receptors to activate multiple downstream signaling pathways, including the PI3K/AKT pathway through an autocrine loop mechanism.
Abbreviations: EREG, epiregulin; MEK, mitogen-activated protein kinase/extracellular signal-regulated kinase; ERK, extracellular signal-regulated kinase; PI3K, Phosphoinositide 3-kinase; AKT, AKT8 virus oncogene cellular homolog.
Acknowledgments
The authors apologize to other investigators for the omission of any references. This work was supported by Grants-in-Aid for Scientific Research (C) (grant number 23591134) from the Japan Society for the Promotion of Science.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Bender E. Epidemiology: the dominant malignancy. Nature. 2014;513(7517):S2–S3. doi: 10.1038/513S2a. [DOI] [PubMed] [Google Scholar]
- 2.Larsen JE, Minna JD. Molecular biology of lung cancer: clinical implications. Clin Chest Med. 2011;32(4):703–740. doi: 10.1016/j.ccm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Houston KA, Henley SJ, Li J, White MC, Richards TB. Patterns in lung cancer incidence rates and trends by histologic type in the United States, 2004–2009. Lung Cancer. 2014;86(1):22–28. doi: 10.1016/j.lungcan.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramalingam S, Belani C. Systemic chemotherapy for advanced non-small cell lung cancer: recent advances and future directions. Oncologist. 2008;13(Suppl 1):5–13. doi: 10.1634/theoncologist.13-S1-5. [DOI] [PubMed] [Google Scholar]
- 5.Weinstein IB. Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science. 2002;297(5578):63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 6.Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311(19):1998–2006. doi: 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sholl LM, Aisner DL, Varella-Garcia M, et al. LCMC Investigators Multi-institutional oncogenic driver mutation analysis in lung adenocarcinoma: the lung cancer mutation consortium experience. J Thorac Oncol. 2015;10(5):768–777. doi: 10.1097/JTO.0000000000000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 12.Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 13.Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18(3):375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18(3):382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 16.Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863–870. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carnio S, Novello S, Mele T, Levra MG, Scagliotti GV. Extending survival of stage IV non-small cell lung cancer. Semin Oncol. 2014;41(1):69–92. doi: 10.1053/j.seminoncol.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 18.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 19.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 20.Maemondo M, Inoue A, Kobayashi K, et al. North-East Japan Study Group Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 21.Mitsudomi T, Morita S, Yatabe Y, et al. West Japan Oncology Group Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 22.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 23.Rosell R, Carcereny E, Gervais R, et al. Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 24.Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 25.Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013;14(7):590–598. doi: 10.1016/S1470-2045(13)70142-6. [DOI] [PubMed] [Google Scholar]
- 26.O’Brien CP, Taylor SE, O’Leary JJ, Finn SP. Molecular testing in oncology: problems, pitfalls and progress. Lung Cancer. 2014;83(3):309–315. doi: 10.1016/j.lungcan.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 27.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 28.Vasan N, Boyer JL, Herbst RS. A RAS renaissance: emerging targeted therapies for KRAS-mutated non-small cell lung cancer. Clin Cancer Res. 2014;20(15):3921–3930. doi: 10.1158/1078-0432.CCR-13-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sunaga N, Shames DS, Girard L, et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther. 2011;10(2):336–346. doi: 10.1158/1535-7163.MCT-10-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sunaga N, Kaira K, Imai H, et al. Oncogenic KRAS-induced epiregulin overexpression contributes to aggressive phenotype and is a promising therapeutic target in non-small-cell lung cancer. Oncogene. 2013;32(34):4034–4042. doi: 10.1038/onc.2012.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toyoda H, Komurasaki T, Uchida D, et al. Epiregulin. A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J Biol Chem. 1995;270(13):7495–7500. doi: 10.1074/jbc.270.13.7495. [DOI] [PubMed] [Google Scholar]
- 32.Conti M, Hsieh M, Park JY, Su YQ. Role of the epidermal growth factor network in ovarian follicles. Mol Endocrinol. 2006;20(4):715–723. doi: 10.1210/me.2005-0185. [DOI] [PubMed] [Google Scholar]
- 33.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 34.Shelly M, Pinkas-Kramarski R, Guarino BC, et al. Epiregulin is a potent pan-ErbB ligand that preferentially activates heterodimeric receptor complexes. J Biol Chem. 1998;273(17):10496–10505. doi: 10.1074/jbc.273.17.10496. [DOI] [PubMed] [Google Scholar]
- 35.Vermeer PD, Panko L, Karp P, Lee JH, Zabner J. Differentiation of human airway epithelia is dependent on erbB2. Am J Physiol Lung Cell Mol Physiol. 2006;291(2):L175–L180. doi: 10.1152/ajplung.00547.2005. [DOI] [PubMed] [Google Scholar]
- 36.Chu EK, Foley JS, Cheng J, Patel AS, Drazen JM, Tschumperlin DJ. Bronchial epithelial compression regulates epidermal growth factor receptor family ligand expression in an autocrine manner. Am J Respir Cell Mol Biol. 2005;32(5):373–380. doi: 10.1165/rcmb.2004-0266OC. [DOI] [PubMed] [Google Scholar]
- 37.Riese DJ, 2nd, Cullum RL. Epiregulin: roles in normal physiology and cancer. Semin Cell Dev Biol. 2014;28:49–56. doi: 10.1016/j.semcdb.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toyoda H, Komurasaki T, Uchida D, Morimoto S. Distribution of mRNA for human epiregulin, a differentially expressed member of the epidermal growth factor family. Biochem J. 1997;326(pt 1):69–75. doi: 10.1042/bj3260069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Z, Kleeff J, Friess H, et al. Epiregulin is up-regulated in pancreatic cancer and stimulates pancreatic cancer cell growth. Biochem Biophys Res Commun. 2000;273(3):1019–1024. doi: 10.1006/bbrc.2000.3033. [DOI] [PubMed] [Google Scholar]
- 40.Torring N, Jorgensen PE, Sorensen BS, Nexo E. Increased expression of heparin binding EGF (HB-EGF), amphiregulin, TGF alpha and epiregulin in androgen-independent prostate cancer cell lines. Anticancer Res. 2000;20(1A):91–95. [PubMed] [Google Scholar]
- 41.Thogersen VB, Sorensen BS, Poulsen SS, Orntoft TF, Wolf H, Nexo E. A subclass of HER1 ligands are prognostic markers for survival in bladder cancer patients. Cancer Res. 2001;61(16):6227–6233. [PubMed] [Google Scholar]
- 42.Nicholson BE, Frierson HF, Conaway MR, et al. Profiling the evolution of human metastatic bladder cancer. Cancer Res. 2004;64(21):7813–7821. doi: 10.1158/0008-5472.CAN-04-0826. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Iwanaga K, Choi KC, et al. Intratumoral epiregulin is a marker of advanced disease in non-small cell lung cancer patients and confers invasive properties on EGFR-mutant cells. Cancer Prev Res. 2008;1(3):201–207. doi: 10.1158/1940-6207.CAPR-08-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Revillion F, Lhotellier V, Hornez L, Bonneterre J, Peyrat JP. ErbB/HER ligands in human breast cancer, and relationships with their receptors, the biopathological features and prognosis. Ann Oncol. 2008;19(1):73–80. doi: 10.1093/annonc/mdm431. [DOI] [PubMed] [Google Scholar]
- 45.Shigeishi H, Higashikawa K, Hiraoka M, et al. Expression of epiregulin, a novel epidermal growth factor ligand associated with prognosis in human oral squamous cell carcinomas. Oncol Rep. 2008;19(6):1557–1564. [PubMed] [Google Scholar]
- 46.Hu K, Li SL, Gan YH, Wang CY, Yu GY. Epiregulin promotes migration and invasion of salivary adenoid cystic carcinoma cell line SACC-83 through activation of ERK and Akt. Oral Oncol. 2009;45(2):156–163. doi: 10.1016/j.oraloncology.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 47.Zhao M, He HW, Sun HX, Ren KH, Shao RG. Dual knockdown of N-ras and epiregulin synergistically suppressed the growth of human hepatoma cells. Biochem Biophys Res Commun. 2009;387(2):239–244. doi: 10.1016/j.bbrc.2009.06.128. [DOI] [PubMed] [Google Scholar]
- 48.Amsterdam A, Shezen E, Raanan C, et al. Epiregulin as a marker for the initial steps of ovarian cancer development. Int J Oncol. 2011;39(5):1165–1172. doi: 10.3892/ijo.2011.1123. [DOI] [PubMed] [Google Scholar]
- 49.Sunaga N, Tomizawa Y, Shimizu K, et al. Clinicopathological and biological significance of epiregulin expression in non-small cell lung cancer. J Thorac Oncol. 2013;8(Suppl 2):S426–S427. [Google Scholar]
- 50.Weissferdt A, Lin H, Woods D, et al. HER family receptor and ligand status in thymic carcinoma. Lung Cancer. 2012;77(3):515–521. doi: 10.1016/j.lungcan.2012.05.108. [DOI] [PubMed] [Google Scholar]
- 51.Neufert C, Becker C, Türeci Ö, et al. Tumor fibroblast-derived epi-regulin promotes growth of colitis-associated neoplasms through ERK. J Clin Invest. 2013;123(4):1428–1443. doi: 10.1172/JCI63748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Auf G, Jabouille A, Delugin M, et al. High epiregulin expression in human U87 glioma cells relies on IRE1alpha and promotes autocrine growth through EGF receptor. BMC Cancer. 2013;13:597. doi: 10.1186/1471-2407-13-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Missiaglia E, Jacobs B, D’Ario G, et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann Oncol. 2014;25(10):1995–2001. doi: 10.1093/annonc/mdu275. [DOI] [PubMed] [Google Scholar]
- 54.Kohsaka S, Hinohara K, Wang L, et al. Epiregulin enhances tumorigenicity by activating the ERK/MAPK pathway in glioblastoma. Neuro Oncol. 2014;16(7):960–970. doi: 10.1093/neuonc/not315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lindvall C, Hou M, Komurasaki T, et al. Molecular characterization of human telomerase reverse transcriptase-immortalized human fibroblasts by gene expression profiling: activation of the epiregulin gene. Cancer Res. 2003;63(8):1743–1747. [PubMed] [Google Scholar]
- 56.Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Colby JK, Rengel RC, Fischer SM, Clinton SK, Klein RD. Overexpression of cyclooxygenase-2 (COX-2) in the mouse urinary bladder induces the expression of immune- and cell proliferation-related genes. Mol Carcinog. 2009;48(1):1–13. doi: 10.1002/mc.20449. [DOI] [PubMed] [Google Scholar]
- 58.Dapito DH, Mencin A, Gwak GY, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21(4):504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gupta GP, Nguyen DX, Chiang AC, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446(7137):765–770. doi: 10.1038/nature05760. [DOI] [PubMed] [Google Scholar]
- 61.Watanabe T, Kobunai T, Yamamoto Y, et al. Prediction of liver metastasis after colorectal cancer using reverse transcription-polymerase chain reaction analysis of 10 genes. Eur J Cancer. 2010;46(11):2119–2126. doi: 10.1016/j.ejca.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 62.Addison JB, Koontz C, Fugett JH, et al. KAP1 promotes proliferation and metastatic progression of breast cancer cells. Cancer Res. 2015;75(2):344–355. doi: 10.1158/0008-5472.CAN-14-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kobayashi S, Yamada-Okabe H, Suzuki M, et al. LGR5-positive colon cancer stem cells interconvert with drug-resistant LGR5-negative cells and are capable of tumor reconstitution. Stem Cells. 2012;30(12):2631–2644. doi: 10.1002/stem.1257. [DOI] [PubMed] [Google Scholar]
- 64.Ryuge S, Sato Y, Jiang SX, et al. The clinicopathological significance of Lgr5 expression in lung adenocarcinoma. Lung Cancer. 2013;82(1):143–148. doi: 10.1016/j.lungcan.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 65.Yun J, Song SH, Park J, et al. Gene silencing of EREG mediated by DNA methylation and histone modification in human gastric cancers. Lab Invest. 2012;92(7):1033–1044. doi: 10.1038/labinvest.2012.61. [DOI] [PubMed] [Google Scholar]
- 66.Castellano E, Santos E. Functional specificity of ras isoforms: so similar but so different. Genes Cancer. 2011;2(3):216–231. doi: 10.1177/1947601911408081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baba I, Shirasawa S, Iwamoto R, et al. Involvement of deregulated epiregulin expression in tumorigenesis in vivo through activated Ki-Ras signaling pathway in human colon cancer cells. Cancer Res. 2000;60(24):6886–6889. [PubMed] [Google Scholar]
- 68.Cho MC, Choi HS, Lee S, et al. Epiregulin expression by Ets-1 and ERK signaling pathway in Ki-ras-transformed cells. Biochem Biophys Res Commun. 2008;377(3):832–837. doi: 10.1016/j.bbrc.2008.10.053. [DOI] [PubMed] [Google Scholar]
- 69.Fujimoto N, Wislez M, Zhang J, et al. High expression of ErbB family members and their ligands in lung adenocarcinomas that are sensitive to inhibition of epidermal growth factor receptor. Cancer Res. 2005;65(24):11478–11485. doi: 10.1158/0008-5472.CAN-05-1977. [DOI] [PubMed] [Google Scholar]
- 70.Soh J, Okumura N, Lockwood WW, et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One. 2009;4(10):e7464. doi: 10.1371/journal.pone.0007464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li XD, Miao SY, Wang GL, Yang L, Shu YQ, Yin YM. Amphiregulin and epiregulin expression in colorectal carcinoma and the correlation with clinicopathological characteristics. Onkologie. 2010;33(7):353–358. doi: 10.1159/000315380. [DOI] [PubMed] [Google Scholar]
- 72.Regales L, Gong Y, Shen R, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. 2009;119(10):3000–3010. doi: 10.1172/JCI38746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen D, Zhang LQ, Huang JF, et al. BRAF mutations in patients with non-small cell lung cancer: a systematic review and meta-analysis. PLoS One. 2014;9(6):e101354. doi: 10.1371/journal.pone.0101354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the lung cancer mutation consortium. Cancer. 2015;121(3):448–456. doi: 10.1002/cncr.29042. [DOI] [PMC free article] [PubMed] [Google Scholar]