

Summary
STAT5, a member of the family of Signal Transducers and Activators of Transcription senses cytokines and controls the biology of cell lineages, including mammary, liver and T cells. Here we show that STAT5 activates lineage-specific and widely expressed genes through different mechanisms. STAT5 preferentially binds to promoter sequences of cytokine-responsive genes expressed across cell types and to putative enhancers of lineage-specific genes. While chromatin accessibility of STAT5-based enhancers was dependent on cytokine exposure, STAT5-responsive promoters of widely expressed target genes were generally constitutively accessible. While the contribution of STAT5 to enhancers is well established, its role on promoters is poorly understood. To address this we focused on Socs2, a widely expressed cytokine-sensing gene. Upon deletion of the STAT5 response elements from the Socs2 promoter, cytokine induction was abrogated, while basal activity remained intact. Our data suggest that promoter-bound STAT5 modulates cytokine responses and enhancer-bound STAT5 is mandatory for gene activation.
Keywords: Cytokine, STAT5, Mammary gland, SOCS2, Promoter, Enhancer
eTOC blurb
Zeng et al. find that the cytokine-sensing transcription factor STAT5 activates lineage-specific and widely expressed genes through different mechanisms. STAT5 controls lineage-specific genes through enhancers and widely expressed genes via promoters. While STAT5 is essential for the activation of cytokine-responsive enhancers, it merely modulates promoters upon cytokine stimulation.
Introduction
In mammals Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathways are central to cells responding to a wide array of cytokines and growth hormones (Baik et al., 2011; Hennighausen and Robinson, 2008; O’Shea et al., 2011). Signal Transducers and Activators of Transcription (STATs) are the principle transcription factors in these pathways, conveying extracellular cues to initialize lineage-specific and common transcription programs. Among STAT family members, STAT5A/B (referred to as STAT5) is essential for the establishment of milk secreting mammary alveoli, T cell differentiation and liver metabolism (Baik et al., 2011; Cui et al., 2004; Liu et al., 1996; Miyoshi et al., 2001; Udy et al., 1997; Yao et al., 2006). Prolactin (PRL)-mediated STAT5 phosphorylation rapidly increases during pregnancy, controlling proliferation of mammary epithelium and its differentiation into milk-secretory alveoli. Correspondingly, mammary genes are induced up to one-thousand-fold during pregnancy. Active STAT5 is present throughout lactation but rapidly vanishes at the interphase between lactation and involution, a dynamic process resulting in mammary remodeling (Li et al., 1997; Liu et al., 1996; Miyoshi et al., 2001). In the immune system, STAT5 is activated by interleukin-2 (IL-2) during CD4+ helper T cell differentiation and it controls differentiation of both type 1- and type 2- T helper cell (Th1 and Th2) (Liao et al., 2011; Liao et al., 2008; Villarino et al., 2016; Zhu et al., 2003). In liver, growth hormone (GH) activates JAK2, which in turn phosphorylates STAT5, and results in the activation of genes important for body growth and steroid metabolism (Engblom et al., 2007; Udy et al., 1997).
Given the wide range of programs controlled by STAT5, insight into the molecular mechanism underlying STAT5 regulation of target genes was provided by several genomic studies. Lineage-specific regulatory elements under the control of STAT5 have been identified in genes specifically activated in T cells, liver and mammary epithelium (Kang et al., 2013; Kang et al., 2014; O’Shea et al., 2011; Zhang et al., 2012). Recent studies have demonstrated that general transcription factors, including STATs, activate lineage-specific genes by guiding lineage-specific regulatory elements, likely enhancers that are distinct from gene promoters (Heinz et al., 2015; Metser et al., 2016; Shin et al., 2016; Vahedi et al., 2012). In addition to lineage restricted genes, STATs also bind to putative regulatory elements of genes that respond to cytokines across cell types (Kang et al., 2013). However, the significance of these JAK/STAT signature elements on regulating common cytokine target genes has not been clarified.
Traditionally in vitro reporter assays have been used to explore the function of transcription factor binding elements in controlling gene expression. However, such approaches do not provide an untainted understanding of the biological function of regulatory elements in the context of their native chromatin structure within their endogenous loci. In contrast, CRISPR/Cas9-mediated genome editing provides a suitable tool to investigate the biological significance of genetic elements in the context of the mouse genome (Metser et al., 2016). We have now identified lineage-restricted and common STAT5 regulatory elements from genomic data sets and investigated the in vivo function of one associated with the Socs2 gene that is activated by cytokines across cell types.
Results
Identification of shared and lineage-specific STAT5 binding sites
STAT5A is the major isoform of STAT5 in mammary tissue, while STAT5B is the major one in T cells. ChIP-seq of STAT5A in mammary tissue, ChIP-seq of STAT5B in T help cells, and ChIP-seq of total STAT5 in liver have been employed to study the genome-wide function of STAT5 (Kang et al., 2014; Liao et al., 2011; Zhang et al., 2012). Therefore, we identified STAT5 binding sites in mouse mammary tissue, T helper cells and liver. While 420 binding sites were shared between the three tissues, approximately 16,000 sites were specific to mammary tissue, 12,300 to liver and 12,400 to T cells (Figure 1A). Shared STAT5 binding sites, but not lineage-specific ones, were significantly enriched in promoter regions (Figure 1B). Out of 183 STAT5-positive common promoters, 135 (74%) contained at least one GAS motif (Table S1), suggesting STAT5 binding on these promoters is genuine (Jain et al., 2015). In contrast, GAS motifs were detected in 50%, 21% and 33% of lineages-specific non-promoter distal sites in mammary tissue, liver and T cells (Table S1). Approximately 5,700 mammary-specific binding sites were within 20 kb of transcriptional start sites (TSSs) and marked by H3K27ac, an indicator of active enhancers. Therefore we defined them as mammary-specific putative enhancers. Likewise we identified ~2,300 and ~4,300 lineage-specific putative enhancers in T cells and liver, respectively (Table S2). The genomic location of shared and lineage-specific binding sites implied that STAT5 regulates common and lineage-specific transcription programs via different genomic elements. We consequently focused on characterizing two categories of STAT5 binding elements, the 183 common STAT5 binding promoters and lineage-specific putative enhancers. Motif search on these two categories of elements indicated that lineage-specific transcription factors co-localized with STAT5 on putative enhancers but not on common promoters (Figure 1C and Table S3). For instance, ELF5, glucocorticoid receptor (GR) and NFIB motifs were significantly enriched in mammary specific STAT5-bound putative enhancers, while motifs for the liver factors HNF4A and FOXA1 were enriched in liver-specific ones (Figure 1C). Consistently, ChIP-seq data obtained from mammary tissue showed co-localization of STAT5, ELF5 and GR on putative enhancers but not on common promoters (Figure S1A). For instance, in mammary gland ELF5 and GR bind to three putative enhancers upstream of the Wap gene, while they are absent on the promoter of Socs2, a shared STAT5 target (Figure S1B). Similarly, HNF4A co-localized with STAT5 on putative enhancers instead of common promoters in liver, exemplified by enhancers at Ghr locus and the promoter of Socs2 (Figure S1C and S1D). Working with different partners, STAT5 bound to putative enhancers associated with cell identity genes, as suggested by gene ontology analysis (Figure 1D). For example, mammary specific enhancers were associated with genes linked to mammary gland development and lactation. In contrast, T cell specific enhancer-associated genes were linked to T cell activation. Taken together, our data suggest that STAT5 preferentially regulates common transcription program via promoters, but controls lineage-specific program by working synergistically with cofactors on enhancers.
Figure 1. Identification of shared and lineage-specific STAT5 binding sites.

ChIP-seq data of STAT5 in lactating mammary tissue (Mammary), liver from mice treated with growth hormone (Liver) and type I T helper cells (T cells) were used for identifying shared and lineage-specific STAT5 binding sites.
(A) Venn diagram depicts the common and lineage-specific STAT5 binding sites identified.
(B) Common STAT5 binding sites are enriched in promoter regions; lineage-specific ones are enriched in non-promoter regions. Pie charts depict the genomic distribution of common STAT5 binding sites and lineage-specific STAT5 binding sites. A promoter is defined as a 1kb region surrounding the transcriptional start site (TSS ± 500bp).
(C) Motif search of common promoter and putative enhancer (PE) sequences bound by STAT5. STAT5 co-localizes with lineage-specific factors on putative enhancers but not on common promoters. NS: Not Significant.
(D) Lineage-specific enhancers bound by STAT5 associate with lineage-specific genes. Bar charts depict gene ontology analysis of genes associated with lineage-specific enhancers.
Differential responses of shared and lineage-specific STAT5 binding sites to cytokines
Although it is undisputed that the JAK/STAT pathway executes cytokine stimuli, it is not known whether the STAT5-positive common promoters and lineage-specific enhancers are functionally equivalent and respond similarly to cytokines. To investigate this we monitored chromatin accessibility under different hormonal conditions using DNase-seq data. Prolactin (PRL) is the key activator of STAT5 in mammary tissue and its level rises during pregnancy followed by a sharp drop at the end of lactation. We integrated DNase-seq data obtained from mammary tissue at day 13 of pregnancy (p13), day 1 of lactation (L1) and day 1 after experimentally terminating lactation (involution day 1 (I1)). As expected, STAT5 effectively bound to common promoters and putative enhancers of mammary tissue at L1, and moderately at p13 and I1 (Figure S2). Concordantly, chromatin accessibility of enhancers, as well as H3K27ac coverage was high at L1 but low at p13 and I1. Surprisingly the chromatin accessibility of common promoters did not change with progressing differentiation. Similarly, histone H3K4 tri-methylation (H3K4me3) coverage, a marker for active promoters, was independent of the differentiation status (Figure S2). Pearson correlation of DNaseI hypersensitive site (DHS) signals also supported that chromatin accessibility of mammary specific STAT5-positive putative enhancers shifted in parallel with PRL levels. In contrast, the chromatin accessibility of common promoters almost persisted (blue squares in Figure 2A).
Figure 2. Differential cytokine response of STAT5-bound common promoters and lineage-specific enhancers.

Lineage-specific putative enhancers bound by STAT5 are sensitive to cytokines, while common promoters bound by STAT5 are insensitive.
(A) Heat maps depict Pearson correlation of DNase-seq data on indicated STAT5 binding elements from indicated tissues. The triangle with black gradient represents relative cytokine levels. Chromatin accessibility change of lineage-specific putative enhancers (PEs), but not common promoters coordinates with cytokine level variation in mammary tissue, T cells and liver. p13, pregnancy day 13; L1, lactation day 1; I1, Involution day 1; Naïve, naïve CD4+ T cells; Th1, type 1 T helper cells; +GH, growth hormone treatment.
(B) Representative gene loci with STAT5-bound promoter (Socs2 locus) and lineage-specific putative enhancers (Wap, Il2ra and Ghr loci). STAT5 ChIP-seq and DNase-seq (DHS) profiles (reads per 10 million) are shown. STAT5 binding to common promoter and lineage-specific putative enhancers with induced DHS signals are highlighted.
(C) Genes containing lineage-specific STAT5 binding putative enhancers (PEs) are induced to a significantly larger extent by cytokines compared to those with common promoters. Violin plots illustrate the expression change of genes associated with indicated STAT5 binding elements. P values of variance tests are shown on top.
Differential chromatin accessibility was observed at highly regulated genes with the progression of pregnancy. The Wap gene, which contains three mammary-specific putative enhancers, provides a clear example. While two of them, E2 and E3, were not visible at p13, they became exposed at L1, likely as the result of persistent PRL signaling (Figure 2B). Notably, accessibility of these two sites was restricted upon cessation of PRL signaling at the termination of lactation (Figure 2B upper right). In contrast, Socs2 and Cish, which encode universal negative regulators of the JAK/STAT pathway, were bound by STAT5 at their respective promoter sequences and chromatin accessibility in mammary tissue persisted independent of the developmental and hormonal status (Figure 2B upper left and Figure S3A). Consistently, genes containing mammary specific enhancers were induced to a significantly larger extent from p13 to L1. In contrast, genes with common promoters were less responsive (Figure 2C left panel). Twenty-six percent of enhancer-regulated genes vs. eighteen percent of promoter-regulated genes were induced over 2-fold from p13 to L1. And the proportion of enhancer-regulated genes showing significant induction was greater than that in genes with STAT5-bound promoters (Figure S3B left panel). Especially, genes in the vicinity of multiple enhancers were induced significantly higher than genes with common promoters (Figure S3C left), possibly due to accumulative effects of enhancers. These data suggest that STAT5-positive common promoters and lineage-specific enhancers display inherently distinct sensitivities to cytokines. To further test this hypothesis, we compared T cell- and liver-specific putative enhancers with common promoters. IL2-mediated STAT5 activation is important for naïve CD4+ T cells differentiating into type 1 T helper cells (Th1) (Liao et al., 2011). Chromatin accessibility of STAT5-positive putative enhancers was sensitive to IL2 in T cells, as exemplified by three enhancers at the Il2ra locus (Figure 2B lower left panel). In contrast, chromatin accessibility of STAT5-positive common promoters was equivalent in naïve T cells and Th1 cells (orange squares in Figure 2A), as exemplified by the Cish promoter (Figure S3A). In parallel with the emergence of DHS sites, genes adjacent to T cell-specific enhancers were induced at a significantly bigger range compared to those with common promoters (center panel of Figure 2C and Figure S3B). Genes with multiple enhancers were induced at a significantly higher level than those with common promoters (Figure S3C center panel). Similarly, liver specific enhancers and their associated genes responded to growth hormone (GH) stimulation more intensively than common promoters (green squares in Figure 2A and right panel of Figure 2C, S3B and S3C). Three representative putative enhancers were detected at the growth hormone receptor (Ghr) locus in liver, which were exposed upon GH treatment (Figure 2B). In contrast, chromatin accessibility at the Socs2 and Cish promoters in liver tissue remained constant and was independent of GH treatment (Figures 2B and S3A). Although Socs2 is considered a common STAT5 target gene, as evidenced by STAT5 binding to its promoter across cell types, DHS was restricted in naïve T cells and Th1 cells (Figure 2B) for unknown reasons. These data indicated that access of lineage-specific putative enhancers, but not common promoters relies on cytokine-mediated JAK/STAT5 activation in general.
Generation of mice lacking STAT5 binding sites in the Socs2 promoter using CRISPR/Cas9
Previous in vitro studies had suggested that STAT5 binding is important for the promoter activity (Barclay et al., 2009; Lin et al., 2014; Verdier et al., 1998). However, our global analysis indicated that the majority of STAT5-positive common promoters were still accessible with low levels of cytokines. Two scenarios would shed light on the biological significance of this observation. Either STAT5 preferentially binds to and activates these promoters even at low cytokine levels or STAT5 binding to common promoters has little significance on promoter activity. To investigate these possibilities and explore the bona fide biological relevance of STAT5 binding promoters in vivo, we focused on the Socs2 locus as a paradigm. ChIP-seq data demonstrated STAT5 binding at two adjacent GAS sites within the Socs2 promoter in mammary tissue, T helper cells and liver (Figure 3A). In mammary tissue, STAT5 occupancy was observed throughout pregnancy and lactation and coincided with active histone marks H3K4me3 and H3K27ac (Figure 3B). The data suggested that the two adjacent GAS sites are STAT5 regulatory elements shared between different cell types. To delete these sites, two individual guide RNAs were injected into zygotes to facilitate Cas9-mediated incisions at both sites (Figure 3C). Ten mutant founders were generated and screened, six of which carried genomic deletions covering only one GAS site, which may not entirely abolish STAT5 binding (Figure 3C). Deletions in two lines spanned both GAS sites (Figure 3C) and both lines were viable without obvious abnormalities on growth, feeding, reproducibility and lactation. The line with a 63 bp deletion (Socs2-Δs) was used for all subsequent studies.
Figure 3. Generation of mice lacking STAT5 binding sites in the Socs2 promoter.

(A) STAT5 binding at the Socs2 locus in indicated tissues as determined by ChIP-seq.
(B) ChIP-seq binding profiles of STAT5 and histone modifications in mammary tissue at day 13 of pregnancy (p13) and day 1 of lactation (L1).
(C) Scheme of Socs2-Δs mutant mouse lines generated using CRISPR/Cas9 genome editing. The arrows indicate the two Cas9-mediated incision positions. The targeted GAS motifs (shown in red) bound by STAT5 were deleted in lines A and B.
STAT5 binding is required for Socs2 induction but dispensable for basal expression
ChIP-seq assays validated the loss of STAT5 binding in Socs2-Δs mammary tissue. STAT5 binding was undisturbed on the Cish promoter indicating the specificity of genomic editing (Figure 4A). Socs2 expression was examined by qRT-PCR in mammary tissue at p13 and L1. In wild type tissue, Socs2 levels were elevated approximately 4-fold in response to increased PRL/STAT5 signaling in late pregnancy (Figure 4B). Socs2 mRNA levels in Socs2-Δs mutant tissue were reduced by ~70% at L1 and ~50% at p13, suggesting that Socs2 expression is not exclusively dependent on promoter-bound STAT5. Concordantly, H3K27ac coverage at the Socs2 promoter was only slightly reduced (Figure 4A). This is consistent with a previous report that Socs2 transcripts were attenuated but not absent in PRL receptor deficient mammary epithelium (Harris et al., 2006). Expression of Socs2 was also examined in liver tissue of mice treated with GH. Similarly, Socs2 expression was induced ~5 fold upon GH treatment and elimination of STAT5 binding impaired Socs2 expression by ~50% (Figure 4B). Two STAT5-bound putative enhancers were identified at the Socs2 locus in liver (Figure 3A), which may contribute to the maintenance of Socs2 expression. Although complete Socs2 deficiency leads to giantism in mice (Metcalf et al., 2000), such a pheontype was not observed in Socs2-Δs mutant mice, suggesting that loss of STAT5 binding does not affect basal Socs2 expression. Consequently, both lactation and body growth were normal in Socs2-Δs mutant mice. The structure of lactating mammary gland and size of alveoli in mutant mice was also normal (Figures 4C, S4B and D). Notably, while STAT5 binding is dispensable for Socs2 promoter activation, STAT5 binding to enhancers, such as the Wap super-enhancer, is essential for gene activation (Shin et al., 2016). These two pieces of genetic evidences provide an explanation for our global findings that lineage-specific enhancers are more sensitive to cytokines than promoters, namely that STAT5 is the central factor to shape lineage-specific enhancers but is not essential for basal promoter activity in general.
Figure 4. STAT5 binding is required for the cytokine-induced but not basal expression of Socs2.

(A) STAT5 binding is absent in mammary tissue of Socs2-Δs mutant mice (highlighted by red arrow). ChIP-seq binding profiles of STAT5 and H3K27ac in Socs2 and Cish loci are shown in wild type (WT) and Socs2-Δs mutant mammary tissue at indicated time points.
(B) qRT-PCR analysis of Socs2 in mammary tissue at indicated stages and male liver upon growth hormone treatment. Data were obtained from six mice per group. Asterisk indicates p < 0.05 in two sample t-test of indicated groups. N.S., Not Significant.
(C–F) JAK/STAT5 signaling is augmented in Socs2-Δs mutant mice.
(C) Mammary epithelium is precociously differentiated in Socs2-Δs mutant mice. Representative pictures of H&E staining sections from indicated mammary tissue are shown. “L” labeled normal lumen in WT tissue. Precociously developed alveoli with milk droplets are observed in mutant mammary tissue at p13 (black arrows).
(D) GSEA for STAT5 dependent and repressed genes in mammary tissue from WT and mutant samples. The analysis shows skewed distribution of the former gene set toward mutant and the latter one toward WT. (Nom P-value, normalized P-value; FDR, false discovery rate; NES, normalized enrichment score).
(E) Heat map depicts relative expression change of representative STAT5 target genes in WT and mutant mammary tissues.
(F) qRT-PCR analysis of indicated genes in WT and mutant p13 mammary gland verifies the RNA-seq data. Data were obtained from 6 mice per group. Asterisk indicates p < 0.05 in two sample t-test of WT and mutant groups.
Distinct genetic responses to hyperactive STAT5
Though basal activity of Socs2 was independent of STAT5, its induction was impaired in Socs2-Δs mutants due to loss of STAT5, and possibly other factors, binding to the promoter. SOCS2 suppresses PRL-mediated STAT5 activation and forms a negative feedback loop in mammary epithelium (Harris et al., 2006). Thus we asked whether the induction of Socs2 is important for modulating PRL-STAT5 signaling. Histological analysis showed that loss of Socs2 induction led to precocious mammary differentiation in Socs2-Δs mutants, evidenced by early milk secretion at day 13 of pregnancy (p13) (Figures 4C, S4A and S4C). Subsequently, RNA-seq was performed to reveal transcriptome change in the mutant at p13. In line with precocious mammary differentiation, a group of mammary genes were prematurely activated, including Wap, Cel, Glycam1, Lalba and Lao1. For instance, in wild type tissue Wap was barely detected at p13 and induced over 1,000 fold at lactation. In Socs2-Δs mutants, Wap was up-regulated over 25-fold compared to WT at p13. Previously, we identified 204 STAT5-dependent mammary genes and 260 STAT5-repressed ones by investigating mammary tissue expressing with different amounts of STAT5 (Yamaji et al., 2013). Gene set enrichment analysis of RNA-seq data demonstrated that STAT5-dependent mammary genes were significantly up-regulated in mutant p13 tissue, while STAT5-repressed ones were down-regulated (Figure 4D–E). The expression changes of representative genes, including Wap, Glycam1 and Slc28a3 were validated by qRT-PCR (Figure 4F). Taken together, our data demonstrated that although STAT5 does not control the basal expression of Socs2, STAT5-mediated Socs2 induction is critical for fine-tuning STAT5 activity in a temporary manner.
Disruption of the STAT5-SOCS2 negative regulatory loop enhanced STAT5 activity and therefore provides another system to investigate responses of STAT5 regulatory elements to cytokines. Towards this goal we conducted STAT5 ChIP-seq in WT and mutant p13 mammary tissue. Approximately 16,000 STAT5 binding peaks were identified in mutants, 4,827 of which were emerging sites compared to WT samples (Figure 5A). Among 460 up-regulated genes in p13 mutant tissue, 123 genes contained at least one emerging site within 20kb from their TSSs. In contrast, only 44 out of 489 down-regulated genes are associated with emerging STAT5 binding sites (Figure 5B). Of note, the majority (148 out of 159) of emerging binding sites next to up-regulated genes were part of putative enhancers. In comparison, only 3 genes with STAT5 binding common promoters, Cish, Clk1 and Slc25a42 were deregulated in the mutant (Table S5). For instance, at p13 STAT5 is poised at the proximal enhancer of Wap and subsequently expanded to all the upstream enhancers by L1 in WT mice (Figure 5C) (Shin et al., 2016). In mutant mice hyperactive STAT5 occupied at the proximal enhancer as early as at p13, resulting in precocious Wap expression. The GR functions as a cofactor of STAT5 during mammary development (Cella et al., 1998). ChIP-seq data demonstrated co-binding of GR and STAT5 at the two distal enhancers (Figure 5C). Given the fact that GR binding is dependent on STAT5 at the two sites (Shin et al., 2016), our data suggested that hyperactive STAT5 binding preceded GR binding on the enhancers. In addition, DNase-seq data demonstrated that hyperactive STAT5 uncovered the chromatin of those two sites (Figure 5C). Similar phenomena were observed at STAT5 enhancers in Glycam1 locus (Figure 5C). We further profiled GR and chromatin accessibility on all the 159 emerging STAT5 binding sites neighboring up-regulated genes. Overall, GR binding was elevated and chromatin accessibility was increased in Socs2-Δs mutant (Figure 5D). These data suggest that STAT5 plays a pioneer role in activating enhancers by opening chromatin and recruiting co-factors. However, neither GR binding, nor chromatin accessibility at STAT5-bound common promoters was altered in response to hyperactive STAT5 (Figure 5C–D). Consistent with our previous findings in T cells and liver, STAT5 binding common promoters, unlike lineage-specific enhancers, are generally insensitive to JAK/STAT5 activation.
Figure 5. Mammary-specific enhancers, but not promoter elements are sensitive to hyperactive STAT5.

(A) Pie chart depicts the distribution of STAT5 binding sites in Socs2-Δs mutant tissue compared with those identified in WT tissue.
(B) Volcano plot shows the fold change (log2 transformed) and variance for all transcripts relative to normal controls. Differentially expressed transcripts are highlighted in black. Neighboring emerging STAT5 binding sites in mutant tissue are highlighted in red (upper left) and in green (upper right). Numbers of differentially expressed transcripts are also indicated. Dotted red lines indicate 2-fold changes and 0.05 P-value.
(C–D) Hyperactive STAT5 facilitates GR binding and chromatin opening on mammary specific loci, but not on common promoters bound by STAT5.
(C) ChIP-seq binding profiles of STAT5, GR and DNase-seq (DHS) profiles in indicated samples. Red arrows and asterisks indicate common promoters and putative enhancers, respectively. Precocious STAT5 binding sites are highlighted. WT, wilde type; Mut, mutant; L1, lactating day 1; p13, pregnant day 13.
(D) Average profiles of GR binding and DHS (DNaseI hypersensitive sites) around the center of emerging STAT5 binding putative enhancers with up-regulated genes and common promoters in Socs2-Δs mutant mammary tissue at p13.
Distinct genetic responses upon reduction of STAT5
Next we explored the roles of STAT5-based enhancers upon loss of STAT5 activity during involution. DNase-seq assays were performed 24 hours after weaning, when phosphorylated STAT5 level have dropped significantly (Li et al., 1997; Liu et al., 1996). Concomitantly with loss of STAT5 activation, DNA access, as measured by DNase-seq, was rapidly restricted on mammary-specific enhancers, as exemplified by Wap and Glycam1 loci. However, no obvious change was detected on promoters of Socs3 and Fam109a (Figure 6A). Pearson correlation of chromatin accessibility of all mammary specific enhancers and common promoters was performed. The results indicated that chromatin accessibility of enhancers altered coordinated with JAK/STAT5 signaling activity (WT p13 vs. WT L1, WT p13 vs. Mut p13 and WT L1 vs. WT I1 in the left panel of Figure 6B). In contrast, DNA accessibility of STAT5-bound common promoters remained relatively constant (right panel in Figure 6B). The different sensitivities of these two groups of STAT5 binding elements to declining STAT5 activity were mirrored by their expression. In mammary tissue devoid of three copies of STAT5 genes, the expression of enhancer-associated genes reduced down to 0.01%; while that of genes with common promoters only reduced down to 13% (Figure 6C and Table S6). The genes with multiple enhancers were especially sensitive to the loss of STAT5 (Figure S5). Notably, the STAT5-SOCS2 regulatory loop was dispensable for mammary involution, as revealed by high correlation of DNase-seq signaling on STAT5 binding elements (WT I1 vs. Mut I1 in Figure 6B). Taken together, these data demonstrated again that STAT5 is central to the activation and maintenance of enhancers, but dispensable for the basal activity of common promoters for the most part.
Figure 6. Loss of STAT5 activation affects mammary specific enhancers but not common cytokine-responsive promoters.

(A) ChIP-seq profiles of STAT5 and DNase-seq (DHS) profiles in indicated samples. Sites are highlight as in Figure 5C. WT, wild type; Mut, mutant; L1, lactating day 1; p13, pregnant day 13; I1, involution day 1.
(B) Heat map depicts Pearson correlation of DNase-seq (DHS) signaling on indicated group of STAT5 binding sites.
(C) Expression of genes containing mammary specific STAT5 binding putative enhancers (PEs) is reduced to a significantly larger extent compared to those with common promoters in STAT5-deficient mice. Violin plot depicts gene expression change of indicated group of genes between wild type (AABB) and Stat5a−/−;Stat5b+/− (B) mammary tissue at L1. P value of variance test is shown on top.
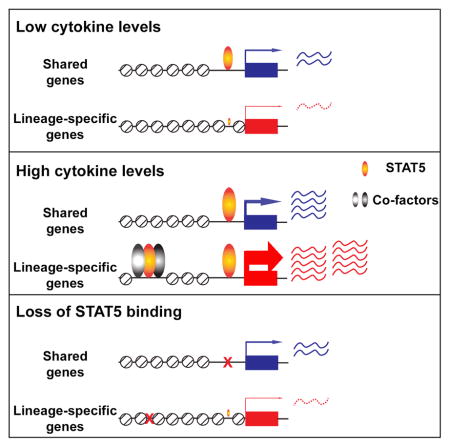
(D) Model depicts the differential mechanisms of STAT5 regulating shared and lineage-specific genes. STAT5 regulates shared target genes by binding to promoter sequences, while it regulates lineage-specific genes by activating enhancers together with cofactors. STAT5 induces lineage-specific genes at higher levels than shared genes in response to cytokine exposure. STAT5 is required for cytokine response but dispensable for chromatin structure and basal activity of promoters of shared genes. In contrast, STAT5 is critical for chromatin opening and activation of lineage-specific enhancers.
Discussion
STAT5 is critical for the activation of lineage-specific transcription programs in liver, T cells and mammary epithelium, yet also regulates genes responding to cytokines in many different cell types. Among these shared genes are Socs2, Socs3 and Cish that encode negative regulators of the JAK/STAT pathway. In general, activation of lineage-specific genes is higher than that of shared genes but the underlying mechanisms remained elusive. We now demonstrate that STAT5 regulates these two distinct gene categories through separate mechanisms, by binding to distal enhancers in lineage-specific genes and to promoters in shared genes (Figure 6D).
We have demonstrated that STAT5 binding to mammary-specific enhancers of the Wap gene is responsible for its more than one-thousand-fold activation during pregnancy (Shin et al., 2016). Here we demonstrate that STAT5 binding to promoter sequences of the shared target gene Socs2 does not affect basal activity but fine-tunes its expression during pregnancy. Genetic ablation of STAT5 binding results in excessive STAT5 signalling in mammary tissue, which leads to the precocious mammary gland development and the unscheduled epithelial differentiation and milk production. Recent studies have shown that lineage determining transcription factors can act as pioneers and recruit co-activators, histone modifiers and chromatin remodelers to shape lineage-specific enhancers (Heinz et al., 2015). Consistent with this, the master regulator STAT5 acts as a pioneer factor and facilitates chromatin opening and binding of cofactors, such as GR, to lineage-specific enhancers. Similar to mammary tissue, motif search analysis also suggested that lineage-specific transcription factors work with STAT5 on lineage-specific enhancers in T cells and liver. The sensitivity of STAT5-positive enhancers to IL2 implies that STAT5 is also critical for lineage-specific enhancer activation in T cells, which is in agreement with the critical role of STATs in shaping enhancers in T cells (Vahedi et al., 2012). In contrast, the role of STAT5 on promoters of shared genes, like Socs2, is likely reminiscent to co-activators.
The exceptional magnitude of gene activation observed in lineage-specific STAT5 targets can likely be explained by STAT5-based enhancers, while shared genes are characterized by STAT5-based promoters. In lineage-specific genes STAT5 frequently binds to super-enhancers, with the constituent enhancers functioning synergistically and activating genes up to one thousand-fold (Shin et al., 2016). Although there has been no genetic validation of putative super-enhancers in T cells and liver, several presumed enhancers were detected within the Il2ra gene in T cells and the Ghr gene in liver. In contrast, STAT5 binds exclusively to the promoters of Socs2 and other shared genes under cytokine control. Without additive or synergistic contributions from enhancers such shared genes are induced less than 10-fold upon STAT5 activation (Figure 6D). Of note, some genes are under STAT5 control asserted through both promoter- and enhancer-bound STAT5.
Socs2 is not only a shared STAT5 target gene, but also a member of a family of repressors that modulate the JAK/STAT pathway. The eight members of the SOCS family, CISH and SOCS 1–7 can be grouped into four pairs according to their similarity on structure and function. The pair of CISH and SOCS2 competes with STATs on binding to receptor tyrosine kinase, while the pair of SOCS1 and 3 inhibit JAK kinase activity. Moreover, all the SOCS proteins facilitate phosphorylated STAT degradation leading to the repression of JAK/STAT signaling across different tissues (Trengove and Ward, 2013; Yoshimura et al., 2007). Previous studies have suggested that Socs genes are regulated by cytokines, forming a classic negative feedback loop (Cohney et al., 1999; Davey et al., 1999; Naka et al., 1997; Pezet et al., 1999; Starr et al., 1997; Verdier et al., 1998). Although STAT binding elements were identified by in vitro assays (Barclay et al., 2009; Lin et al., 2014; Verdier et al., 1998), direct genetic evidence that they are targets of STATs has been lacking. Through CRISPR/Cas9-mediated genome editing, we have now, for the first time, identified a STAT5 control element in a Socs locus. Without STAT5 binding at the promoter, Socs2 cannot be induced during pregnancy consistent with increased STAT5 phosphorylation, which in turn resulted in precocious activation of target genes and mammary epithelial differentiation. Besides Socs2, STAT5, and likely other STAT members, also binds to the promoters of Cish, Socs1 and Socs3 across cell types. We propose that STAT proteins, through binding to promoter-based regulatory elements, are at the core of cytokine-mediated autoregulatory loops that modulate temporal and cell-specific genetic programs.
Experimental Procedures
Generation of mutant mice by CRIPSR-Cas9
CRISPR sgRNA constructs were designed based on their proximity to the mutation sites and their off-target scores (calculated by the online tool at crispr.mit.edu). The 5′-CCGCTTTCCAGGAACTTTCCAGG-3′ and 5′-CCAGGAATCCGCCTCACGTGACC-3′ sequences were cloned into the pDR274 vector (Addgene #42250) separately, and injectable RNAs were in vitro transcribed using the MEGAshortscript T7 kit (Life Technologies). Cas9 mRNA was in vitro transcribed from plasmid MLM3613 (Addgene #42251) using the mMESSAGE mMACHINE T7 kit (Life Technologies).
Mice used in this study were handled and housed in accordance with NIH guidelines. Animal experiments were approved by the NIDDK Animal Care and Use Committee. Zygotes preparation and microinjection were performed as previously described (Yang et al., 2014). Superovulated B6CBAF1/J female mice (JAX) were mated with B6CBAF1/J males, and fertilized eggs were collected from their oviducts. For microinjection, 100ng/μl of Cas9 mRNA and 50 ng/μl of each sgRNA in nuclease-free microinjection buffer (10 mM Tris, pH 7.5, 0.1 mM EDTA) were microinjected into the cytoplasm of fertilized eggs. Injected zygotes were cultured overnight in M16 medium at 37°C in 5%CO2. The next morning, those embryos that had reached the 2-cell stage were implanted into oviducts of pseudopregnant fosters (Swiss Webster, Taconic Farm). Genotyping was done to get the homozygous mice.
Histological Examination
Harvested p13 and L1 mammary tissues were fixed in 10% formalin, dehydrated through ethanol and xylene, embedded in paraffin and sectioned. Sections were stained with hematoxylin and eosin (H&E) by standard methods, and examined by light microscopy. Area size of alveoli were measured using ImageJ.
qRT-PCR
Mouse recombinant growth hormone (2μg/kg) was injected to 8–10 weeks male mice through intraperitoneal injection. After 45 minutes, liver tissues were collected for RNA extraction. For mammary gland, total tissues were harvested at p13 followed by RNA extraction. RNA was extracted using the PureLink RNA Mini Kit (Ambion) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from total RNA using Superscript II (Invitrogen) and quantitative PCR was performed with the following primers.
Socs2, Forward: 5′-TCAGCTGGACCGACTAACCT-3′; Reverse: 5′-CAGGTGAACAGTCCCATTCC-3′. Stat5a, Forward: 5′-ATTACACTCCTGTACTTGCGA-3′; Reverse: 5′-GGTCAAACTCGCCATCTTGG-3′. Stat5b, Forward: 5′-TCCCCTGTGAGCCCGCAAC-3′; Reverse: 5′-GGTGAGGTCTGGTCATGACT-3′. Wap, Forward: 5′-CCAGAATGCCATGTGCTGTC-3′; Reverse: 5′-CGGTCGCTGGAGCATTCTAT-3′. Gapdh, Forward: 5′-TTGTCAAGCTCATTTCCTGGT-3′; Reverse: 5′-TTACTCCTTGGAGGCCATGTA-3′. Glycam1, Forward: 5′-CCACCAGCTACACCAGTGAG-3′; Reverse:5′-CCTGGGCCTCTTGATTCTCTG-3′. Slc28a3, Forward: 5′-GACCTTGAACGGCAGAACACT-3′; Reverse: 5′-CTTTGTTTCCTAGAGGCTCCTG-3′. Real-time PCR was carried out using the BioRad CFX96 Real-Time PCR Detection System (185-5196; BioRad). Individual PCRs were performed in triplicate using the reference gene GAPDH for normalization. The threshold cycle (Cq) was determined by default settings. Relative gene expression (as a fold change) was calculated with the 2-ΔΔCq method. Data were presented as standard deviation in each group and were evaluated with a two-tailed, unpaired Student’s t-test using PRISM GraphPad. Statistical significance was obtained by comparing the measures from wild type group and mutant group at each time point. A value of p < 0.05 was considered statistically significant.
RNA-seq
RNA-seq was done as described (Metser et al., 2016). Total RNA from 4 WT and 3 Socs2-ΔS mammary glands was isolated from mouse mammary tissues using RNeasy Plus Mini Kit (Qiagen). One microgram of total RNA was used to generate sequencing library using TruSeq RNA Library Preparation Kit v2 (Illumina) according to manufacture’s protocol. RNA-seq library was sequenced with HiSeq 2500 system (Illumina).
ChIP-seq and DNase-seq
ChIP-seq and DNase-seq was done as described, following ENCODE guidelines (Metser et al., 2016). Replicates were done for each assay (see also Figure S6). For ChIP-seq frozen-stored mammary tissues were ground into powder and then crosslinked with 1% formaldehyde (Sigma-Aldrich) for 10 min. Fixed nuclei were isolated, followed by chromatin fragmentation using sonicator 3000 (25 cycles; 30 sec pulse/30 sec rest, Misonix Sonicators). One milligram of chromatin was immunoprecipitated with Dynabeads Protein A (Novex) coated with anti-H3K4me3 (Millipore, 17-614), anti-H3K27ac (Abcam, ab4729), anti-GR (ThermoFisher, PA1-511A) and anti-STAT5A (Santa Cruz, sc-1081). After serial bead washes, ChIPed DNA was reverse-crosslinked and purified. The DNA fragments were blunt-ended using End-it DNA End-Repair Kit (Epicentre Biotechnology), ligated to the Illumina Indexed DNA adaptors, and sequenced with HiSeq 2500 (Illumina). For DNase-seq, frozen tissue was ground into powder and homogenized with buffer A (15mM Tris-HCl PH 8.0, 15 mM NaCl, 60 mM KCl, 1mM EDTA, 0.5mM EGTA, 0.15mM Spermine, 0.5mM Spermidine, 0.5mM DTT, 1mM PMSF with proteinase inhibitors). After cells were lysated, nuclei were collected, counted and re-suspended in DNase buffer. 10U DNase I (New England Biolab) was used to digest 10 million nuclei at 37°C for 5 minutes followed by proteinase K digestion. Genomic DNA was then purified. DNA fragments between 50bp and 100bp were selected for further library construction and sequencing.
All the experiments in this study were conducted in accordance with NIH guidelines.
Data analysis
ChIP-seq and DNase-seq data were mapped to the reference genome mm9 using Bowtie aligner (Langmead and Salzberg, 2012) and visualized using HOMER (http://homer.salk.edu/homer/) and IGV (Thorvaldsdottir et al., 2013). For visualization, the total reads number of mapped result in each sample was normalized to 10 million and background signals of less than 2 were eliminated. Peak calling of STAT5 ChIP-seq data was done to identify STAT5 binding sites using MACS2 with qvalue-0.05 and t-size30 (Zhang et al., 2008).. H3K27ac peak calling was done using SICER with FDR 1e-3 (Zang et al., 2009). STAT5 binding sites were assigned to nearest gene using HOMER. STAT5 binding sites in the vinicity of 500 bp of TSSs were defined as promoter-binding sites. Non-pomoter STAT5 binding sites within 20 kb of TSSs that overlapped with H3K27ac peaks were defined as putative enhancers. Motif search was done using HOMER. Gene ontology analysis was performed using DAVID (https://david.ncifcrf.gov/summary.jsp) (Huang da et al., 2009). DeepTools were used to caculate Pearson correlation of DNase-seq signaling (Ramirez et al., 2014) and Spearman correlation of replicated experiments (Figure S6).
RNA-seq data was mapped to the reference genome mm9 using STAR aligner (Dobin et al., 2013). And for comparing the expression levels of all samples, FPKM, fold change and variance were calculated using DESeq2 package in R (http://www.R-project.org) (Love et al., 2014). Genes with FPKM less than 5 in all samples were removed in the downstream analysis. Transcripts with a fold change over 2 and significant pairwise variance (p <0.05) were classified as differentially expressed genes. GSEA analysis was performed as described (Subramanian et al., 2005). Briefly, unabridged RNA-seq datasets were used to compute a ranked list of genes that were differentially expressed in mutant relative to wild type controls. Subsequent enrichment was calculated against user generated gene sets (See Result part). Enrichment score curves and member ranks were generated by the GSEA software package (Broad Institute, Cambridge, MA). Volcano plot, heat maps, violin plots and box plots were generated in R.
Supplementary Material
Highlights.
STAT5 binds to distinct elements in common and lineage-specific target genes
STAT5-responsive enhancers are more sensitive to cytokines than promoters
STAT5 is required for inducible, but not basal activity of Socs2
Promoter-bound STAT5 regulates SOCS2-STAT5 feedback loop and mammary development
Acknowledgments
We thank Dr. Harold Smith from the NIDDK genomics core for never-ending help with NGS and Dr. Chengyu Liu from the NHLBI transgenic core for generating the CRISPR/Cas9-based mouse mutants. M.W. is a graduate student of the Individual Graduate Partnership Program (GPP) between NIH/NIDDK and the Medical University of Innsbruck (Innsbruck, Austria). This research was funded by the IPR of the NIDDK/NIH.
Footnotes
Accession Numbers
All the ChIP-seq, DNase-seq and RNA-seq data generated in this study were deposited in GEO database (accession number GSE82275). RNA-seq datasets in wild type and STAT5 deficient mammary gland were obtained from GSE37646. ChIP-seq and DNase-seq datasets of wild type mammary gland were obtained from GSE74826 and GSE37646. STAT5 ChIP-seq datasets of T cells and liver were obtained from GSE27158 and GSE31578 respectively. H3K27ac ChIP-seq datasets of T cells and liver were obtained from GSE60353 and GSE31039. HNF4A ChIP-seq of liver were obtained from GSE77670. DNase-seq datasets of T cells and liver were obtained from GSE33802 and GSE21777 respectively. RNA-seq datasets of T cells and liver were obtained from GSE48138 and GSE66140 respectively.
Competing Financial Interests
The authors declare no competing financial interests.
Author Contributions
Conceptualization, L.H. and C.W.; Investigation, X.Z. and C.W.; Formal Analysis, X.Z., M.W. and C.W.; Data Curation, M.W. and C.W.; Resources, X.Z. and H.Y.S.; Writing – Original Draft, X.Z. and C.W.; Writing – Review & Editing, M.W., H.Y.S. and L.H.; Supervision, L.H. and C.W.; Project Administration, C.W. and L.H.; Funding Acquisition, L.H..
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baik M, Yu JH, Hennighausen L. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann N Y Acad Sci. 2011;1229:29–37. doi: 10.1111/j.1749-6632.2011.06100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay JL, Anderson ST, Waters MJ, Curlewis JD. SOCS3 as a tumor suppressor in breast cancer cells, and its regulation by PRL. Int J Cancer. 2009;124:1756–1766. doi: 10.1002/ijc.24172. [DOI] [PubMed] [Google Scholar]
- Cella N, Groner B, Hynes NE. Characterization of Stat5a and Stat5b homodimers and heterodimers and their association with the glucocortiocoid receptor in mammary cells. Mol Cell Biol. 1998;18:1783–1792. doi: 10.1128/mcb.18.4.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohney SJ, Sanden D, Cacalano NA, Yoshimura A, Mui A, Migone TS, Johnston JA. SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol Cell Biol. 1999;19:4980–4988. doi: 10.1128/mcb.19.7.4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey HW, McLachlan MJ, Wilkins RJ, Hilton DJ, Adams TE. STAT5b mediates the GH-induced expression of SOCS-2 and SOCS-3 mRNA in the liver. Mol Cell Endocrinol. 1999;158:111–116. doi: 10.1016/s0303-7207(99)00175-6. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom D, Kornfeld JW, Schwake L, Tronche F, Reimann A, Beug H, Hennighausen L, Moriggl R, Schutz G. Direct glucocorticoid receptor-Stat5 interaction in hepatocytes controls body size and maturation-related gene expression. Genes Dev. 2007;21:1157–1162. doi: 10.1101/gad.426007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J, Stanford PM, Sutherland K, Oakes SR, Naylor MJ, Robertson FG, Blazek KD, Kazlauskas M, Hilton HN, Wittlin S, et al. Socs2 and elf5 mediate prolactin-induced mammary gland development. Mol Endocrinol. 2006;20:1177–1187. doi: 10.1210/me.2005-0473. [DOI] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22:711–721. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jain D, Baldi S, Zabel A, Straub T, Becker PB. Active promoters give rise to false positive ‘Phantom Peaks’ in ChIP-seq experiments. Nucleic Acids Res. 2015;43:6959–6968. doi: 10.1093/nar/gkv637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang K, Robinson GW, Hennighausen L. Comprehensive meta-analysis of Signal Transducers and Activators of Transcription (STAT) genomic binding patterns discerns cell-specific cis-regulatory modules. BMC Genomics. 2013;14:4. doi: 10.1186/1471-2164-14-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang K, Yamaji D, Yoo KH, Robinson GW, Hennighausen L. Mammary-specific gene activation is defined by progressive recruitment of STAT5 during pregnancy and the establishment of H3K4me3 marks. Mol Cell Biol. 2014;34:464–473. doi: 10.1128/MCB.00988-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Liu X, Robinson G, Bar-Peled U, Wagner KU, Young WS, Hennighausen L, Furth PA. Mammary-derived signals activate programmed cell death during the first stage of mammary gland involution. Proc Natl Acad Sci U S A. 1997;94:3425–3430. doi: 10.1073/pnas.94.7.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol. 2011;12:551–559. doi: 10.1038/ni.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY, Zhao K, Leonard WJ. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor alpha-chain expression. Nat Immunol. 2008;9:1288–1296. doi: 10.1038/ni.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, LaPensee CR, Qin ZS, Schwartz J. Reciprocal occupancy of BCL6 and STAT5 on Growth Hormone target genes: contrasting transcriptional outcomes and promoter-specific roles of p300 and HDAC3. Mol Cell Endocrinol. 2014;395:19–31. doi: 10.1016/j.mce.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Robinson GW, Hennighausen L. Activation of Stat5a and Stat5b by tyrosine phosphorylation is tightly linked to mammary gland differentiation. Mol Endocrinol. 1996;10:1496–1506. doi: 10.1210/mend.10.12.8961260. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA, Hilton DJ, Alexander WS. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature. 2000;405:1069–1073. doi: 10.1038/35016611. [DOI] [PubMed] [Google Scholar]
- Metser G, Shin HY, Wang C, Yoo KH, Oh S, Villarino AV, O’Shea JJ, Kang K, Hennighausen L. An autoregulatory enhancer controls mammary-specific STAT5 functions. Nucleic Acids Res. 2016;44:1052–1063. doi: 10.1093/nar/gkv999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi K, Shillingford JM, Smith GH, Grimm SL, Wagner KU, Oka T, Rosen JM, Robinson GW, Hennighausen L. Signal transducer and activator of transcription (Stat) 5 controls the proliferation and differentiation of mammary alveolar epithelium. J Cell Biol. 2001;155:531–542. doi: 10.1083/jcb.200107065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat Rev Immunol. 2011;11:239–250. doi: 10.1038/nri2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezet A, Favre H, Kelly PA, Edery M. Inhibition and restoration of prolactin signal transduction by suppressors of cytokine signaling. J Biol Chem. 1999;274:24497–24502. doi: 10.1074/jbc.274.35.24497. [DOI] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HY, Willi M, Yoo KH, Zeng X, Wang C, Metser G, Hennighausen L. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat Genet. 2016;48:904–911. doi: 10.1038/ng.3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trengove MC, Ward AC. SOCS proteins in development and disease. Am J Clin Exp Immunol. 2013;2:1–29. [PMC free article] [PubMed] [Google Scholar]
- Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A. 1997;94:7239–7244. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, O’Shea JJ. STATs shape the active enhancer landscape of T cell populations. Cell. 2012;151:981–993. doi: 10.1016/j.cell.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdier F, Rabionet R, Gouilleux F, Beisenherz-Huss C, Varlet P, Muller O, Mayeux P, Lacombe C, Gisselbrecht S, Chretien S. A sequence of the CIS gene promoter interacts preferentially with two associated STAT5A dimers: a distinct biochemical difference between STAT5A and STAT5B. Mol Cell Biol. 1998;18:5852–5860. doi: 10.1128/mcb.18.10.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino A, Laurence A, Robinson GW, Bonelli M, Dema B, Afzali B, Shih HY, Sun HW, Brooks SR, Hennighausen L, et al. Signal transducer and activator of transcription 5 (STAT5) paralog dose governs T cell effector and regulatory functions. Elife. 2016;5 doi: 10.7554/eLife.08384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji D, Kang K, Robinson GW, Hennighausen L. Sequential activation of genetic programs in mouse mammary epithelium during pregnancy depends on STAT5A/B concentration. Nucleic Acids Res. 2013;41:1622–1636. doi: 10.1093/nar/gks1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 2014;9:1956–1968. doi: 10.1038/nprot.2014.134. [DOI] [PubMed] [Google Scholar]
- Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci U S A. 2006;103:1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–1958. doi: 10.1093/bioinformatics/btp340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Laz EV, Waxman DJ. Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Mol Cell Biol. 2012;32:880–896. doi: 10.1128/MCB.06312-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. 2003;19:739–748. doi: 10.1016/s1074-7613(03)00292-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.