

Abstract
Objective:
To investigate and characterize putative “loss-of-function” (LOF) adenosine triphosphate–binding cassette, subfamily A member 7 (ABCA7) mutations reported to associate with Alzheimer disease (AD) risk.
Methods:
We genotyped 6 previously reported ABCA7 putative LOF variants in 1,465 participants with AD, 381 participants with other neuropathologies (non-AD), and 1,043 controls and assessed the overall mutational burden for association with different diagnosis groups. We measured brain ABCA7 protein and messenger RNA (mRNA) levels using Western blot and quantitative PCR, respectively, in 11 carriers of the 3 most common variants, and sequenced all 47 ABCA7 exons in these participants to screen for other coding variants.
Results:
At least one of the investigated variants was identified in 45 participants with late-onset Alzheimer disease, 12 participants with other neuropathologies, and 11 elderly controls. Association analysis revealed a significantly higher burden of these variants in participants with AD (p = 5.00E-04) and those with other neuropathologies (p = 8.60E-03) when compared with controls. Concurrent analysis of brain ABCA7 mRNA and protein revealed lower protein but not mRNA in p.L1403fs carriers, lower mRNA but not protein in p.E709fs carriers, and additional deleterious mutations in some c.5570+5G>C carriers.
Conclusions:
Our results suggest that LOF may not be a common mechanism for these ABCA7 variants and expand the list of neurologic diseases enriched for them.
Enrichment of ABCA7 nonsense, frameshift, or splice-site variants was identified in Icelandic patients with Alzheimer disease (AD), and this association replicated in non-Icelandic cohorts.1 These AD risk variants were predicted to be loss-of-function (LOF) mutations that cause premature protein termination. In a Belgian cohort, 8 ABCA7 putative LOF mutations were identified,2 2 of which (E709fs, p.Trp1214X) showed reduced brain ABCA7 messenger RNA (mRNA). Additional putative LOF mutations were identified in North American Caucasian AD cases of which p.E1679X3 was statistically significant. These initial studies were followed by replications and/or identification of additional rare ABCA7 putative LOF mutations in other AD cohorts,4–6 in addition to one study in Parkinson disease.7
Although predicted to result in reduced ABCA7 protein, brain ABCA7 protein levels in putative LOF mutation carriers are unknown. Correlative investigations of ABCA7 brain protein and mRNA levels in putative LOF mutation carriers and controls are necessary to determine whether these variants lead to reduced protein levels via nonsense-mediated decay (NMD) of mRNA, as expected. Herein, we investigated the frequency of 6 previously reported ABCA7 putative LOF variants1–3 in our cohort of 1,465 participants with late-onset Alzheimer disease (LOAD) and 381 participants with other non-AD neuropathologies, in comparison with 1,043 clinically normal controls. We evaluated the corresponding brain ABCA7 protein and mRNA in 15 participants composed of carriers for 3 of the most common mutations (E709fs, L1403fs, and c.5570+5G>C) and noncarriers who lacked any predicted deleterious ABCA7 mutations. Our study provides a systematic investigation of downstream effects of ABCA7 predicted LOF mutations and has implications for potential mechanism of action of these variants.
METHODS
Standard protocol approvals, registrations, and patient consents.
This work was approved by the Mayo Clinic Institutional Review Board, and all participants and/or their proxies provided consent.
Participants and samples.
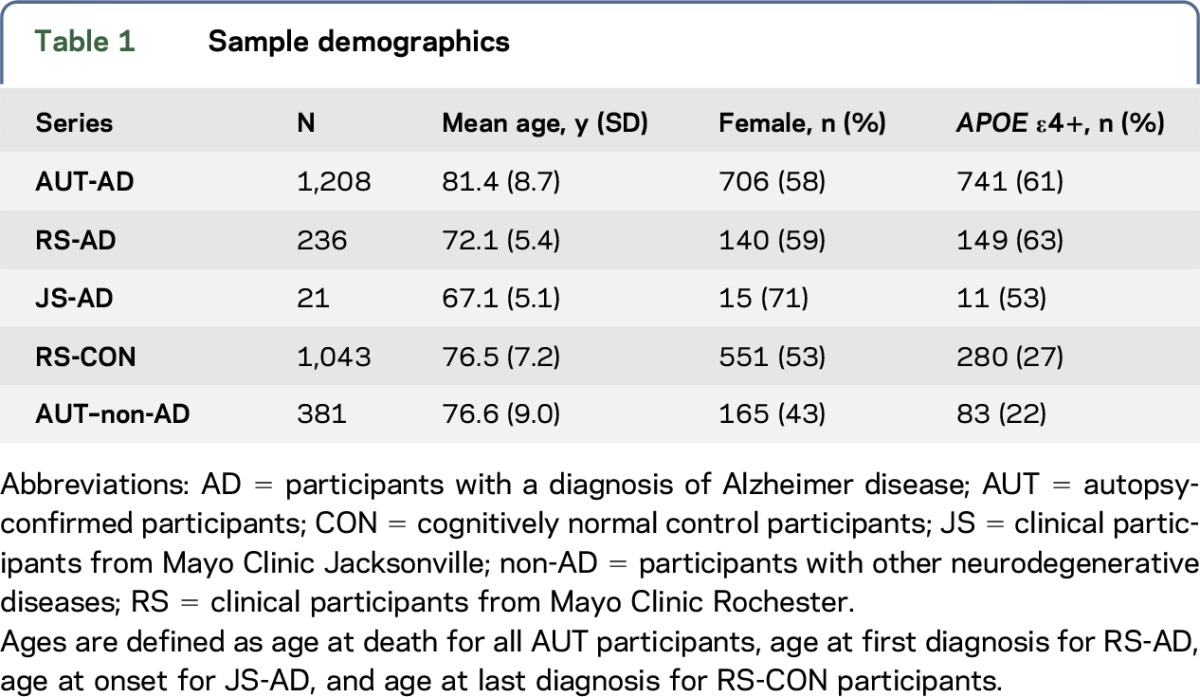
We screened 2,889 Caucasian American participants recruited at Mayo Clinic Rochester (RS), Jacksonville (JS), or Brain Bank (AUT) (table 1) for previously reported1–3 ABCA7 rare putative LOF variants. Of the 1,465 participants with AD, 1,208 were diagnosed at autopsy as definite AD and 257 as probable or possible AD clinically, according to NINCDS-ADRDA criteria.8 Autopsied participants who did not meet criteria for AD neuropathology, but who were enriched for other neurologic pathologies, such as vascular dementia (VaD), frontotemporal dementia, dementia with Lewy bodies, and progressive supranuclear palsy (PSP) were also screened (non-AD, n = 381), in addition to 1,043 elderly clinical control participants without cognitive impairment. The clinical control participants did not meet criteria for mild cognitive impairment, AD, or other dementias.
Table 1.
Sample demographics
Genotyping analysis.
Custom TaqMan SNP assays (Applied Biosystems, Foster City, CA) were designed and used to genotype ABCA7 mutations p.Trp1214X (rs201060968),2 L1403fs, c.4416+2T>G (rs113809142),1,2 p.E1679X,3 and c.5570+5G>C (rs200538373).1,2 Data were analyzed on ABI Prism 7900 Detection System using SDS version 2.2.2 software (Applied Biosystems). For genotyping E709fs 7-bp deletion mutation,1,2 fluorescently labeled PCR products were generated and run on an ABI3730xl Genetic Analyzer (Applied Biosystems). Allele sizes were determined using GeneMapper Software 5.0 (Applied Biosystems).
Sequencing analysis.
All ABCA7 mutations identified in our cohort were validated by sequence analysis. PCR primers were designed to amplify and sequence the genomic regions flanking the mutations. Primer sequences are provided in table e-1 at Neurology.org/ng. PCR products were purified using the Agencourt AMPure system (Beckman Coulter, Brea, CA) and then sequenced in both directions using a Big Dye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems). Sequencing reactions were purified using Agencourt CleanSEQ (Beckman Coulter) and run on an ABI3730xl Genetic Analyzer (Applied Biosystems). Sequence analysis was performed using Sequencher 4.8 software (Gene Codes Corporation, Ann Arbor, MI).
Quantitative PCR.
Brain ABCA7 mRNA levels were measured from the temporal cortex of autopsied participants with L1403fs (n = 3), E709fs (n = 4), and c.5570+5G>C (n = 4) mutations and 4 participants who lacked these mutations. These 15 participants were also sequenced for additional mutations in the 47 ABCA7 exons using Sanger sequencing as described above. Total RNA was extracted using TRIzol Reagent (Ambion Life Technology) followed by DNase RNA cleanup step using RNeasy (Qiagen, Germantown, MD). The quantity and quality of RNA samples were determined by the Agilent 2100 Bioanalyzer using an Agilent RNA 6000 Nano Chip. Complementary DNA (cDNA) synthesized from 1 μg of RNA with Applied Biosystems High-Capacity cDNA Archive Kit was used as a template for relative quantitative PCR using ABI Taqman chemistry (Applied Biosystems). ABCA7 mRNA expression was quantified using assay Hs01105117_m1 (exons 45–46). Hs00951083_m1 (TFRC), Hs02800695_m1 (HPRT), and Hs00355488_m1 (TXNL1) assays were used as endogenous controls for global normalization. Each sample was run in replicates of 4 on the QuantStudio 7 Real-Time PCR System, and analysis was performed using the applied biosystems App 3.0.0-PRC-build8 (Thermo Fisher Cloud).
Allele-specific expression in Glu709fs mutation carriers.
To further investigate allele-specific expression, semiquantitative PCR was performed on cDNA from the temporal cortex of Glu709fs carriers. Fluorescently labeled wild-type and/or Glu709fs mutant PCR products were generated with sizes expected at 281 and 274 bp, respectively. PCR amplification was stopped in the log phase after 25 cycles. PCR products were run on an ABI3730xl Genetic Analyzer (Applied Biosystems).
RNA sequencing.
We had RNA sequencing (RNAseq) data from temporal cortex tissue of a participant with c.5570+5G>C and another participant with c.4416+2T>G mutation. RNA was isolated as described above, and the TruSeq RNA Sample Prep Kit (Illumina, San Diego, CA) was used for library preparation. The library concentration and size distribution were determined on an Agilent Bioanalyzer DNA 1000 chip. Samples underwent 101-bp paired-end sequencing on Illumina HiSeq2000 sequencers. RNAseq measures were processed through the Mayo Clinic MAP-RSeq pipeline.9 Indexed bam files were used for visualizing alignments in Integrative Genomics Viewer.10
Western blot analysis.
Protein lysate was extracted from temporal cortex human brain tissue using a lysis buffer (150 mM NaCl, 50 mM Tris pH 7.5, 0.1% Triton). Protein concentration was determined using NanoDrop 2000 (ThermoFisher Scientific, Waltham, MA), and equal amounts of protein were separated on an NuPAGE 4%–12% Bis-Tris gel and transferred to an Immobilon P membrane (Millipore, Billerica, MA). Membranes were blocked in 5% milk in tris-buffered saline with 0.1% Tween 20 (TBST) and labeled overnight at 4°C with ABCA7 primary antibodies designed to epitope aa 2096-2146 (LS-C291064, LifeSpan BioSciences, Seattle, WA). We validated this antibody for specificity to ABCA7 as described in the supplemental results. Blots were incubated with rabbit IgG horseradish peroxidase–linked secondary antibody (Pierce, SA1-9510) for 1 hour, and proteins were detected using Western Lightning Plus-ECL (Perkin Elmer, Waltham, MA). Equivalent sample loading was confirmed by probing with anti–glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody. Protein bands were quantified using ImageJ, and relative amounts of ABCA7 protein were determined. In the Western blots from human brain lysates, both the full-length ABCA7 protein and the shorter 218 KDa ABCA7 product from its known isoform (ENSP00000465322) were observed. For Western blot quantifications, the sum of intensity of these 2 bands divided by the intensity of GAPDH was used.
Statistical analysis.
ABCA7 protein or mRNA levels in mutation carriers were compared with noncarriers using the nonparametric 1-sided Mann-Whitney U test. Combined counts for the putative LOF ABCA7 alleles were compared between different diagnostic groups using the Fisher exact 2-sided test. StatsDirect v2.7.8 was used for these analyses.
RESULTS
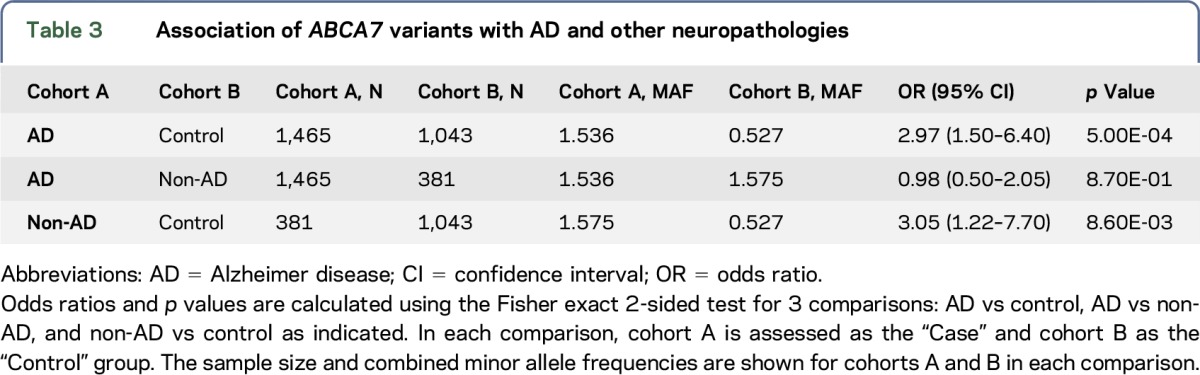
We evaluated 6 previously reported1–3 ABCA7 rare putative LOF variants predicted to lead to truncated protein due to the premature insertion of a stop codon into the transcript. Forty-five of 1,465 participants with LOAD vs 11 of 1,043 elderly cognitively normal controls (table 2) harbored one of these variants, resulting in AD risk association (odds ratio [OR] = 2.97, p = 5.00E-04) (table 3). In 381 autopsied non-AD participants who did not meet criteria for AD neuropathology, but who were enriched for other neurologic pathologies, 12 had one of these ABCA7 variants, leading to association (OR = 3.05, p = 8.60E-03) when compared with controls. These non-AD participants with ABCA7 variants consisted of 7 with PSP, 3 with VaD, 1 with diffuse Lewy body disease (DLBD), and 1 with pathologic aging (PA) and argyophilic grain disease (AGD). Three PSP participants had the secondary diagnoses of VaD, PA, or AGD. PSP participants had p.E709fs, p.E1679X, p.L1403fs, or c.5570+5G>C; and VaD participants had p.E1679X, c.4416+2T>G, or p.L1403fs variants. The p.E709fs variant was also observed in 2 participants with DLBD and PA/AGD. There was no difference in the frequency of putative ABCA7 LOF variants between participants with AD and non-AD neuropathologies (p = 8.70E-01).
Table 2.
ABCA7 loss-of-function mutations' frequency in patients with AD, controls, and participants with other non-AD neuropathologies
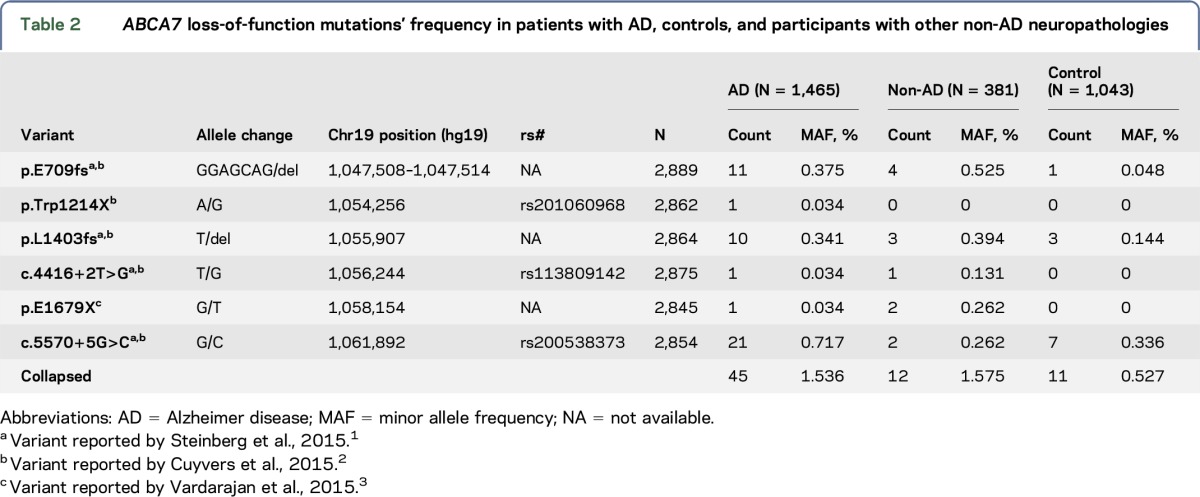
Table 3.
Association of ABCA7 variants with AD and other neuropathologies
Of the 6 variants evaluated, p.E709fs, p.L1403fs, and c.5570+5G>C had the highest frequencies, which enabled the evaluation of several mutation carriers for brain mRNA expression and protein levels (figure 1, A and B, table e-2). There was considerable variance in brain ABCA7 protein and mRNA levels, both in mutation carriers and noncarriers. Despite this, brain ABCA7 full-length protein (∼234 kD) levels were consistently lower in p.L1403fs carriers compared with noncarriers (p = 0.029) (figure 1A). However, p.L1403fs carriers did not have corresponding lower brain ABCA7 mRNA expression (figure 1B). In contrast, p.E709fs carriers had consistently lower brain ABCA7 mRNA expression (p = 0.029) (figure 1B), which was not reflected in their brain protein levels (figure 1A). Lower brain ABCA7 mRNA expression was observed in both the LOAD participants and a PSP participant who were p.E709fs carriers. Semiquantitative assessment of 4 p.E709fs carriers revealed lower brain levels of the shorter mutant allele (274 bp) in comparison with the wild-type allele PCR product (281 bp) (figure 2A). Similar assessment of p.L1403fs carriers did not support lower brain levels of the 1-bp deletion mutant vs wild-type allele (data not shown).
Figure 1. Brain ABCA7 protein and messenger RNA expression in mutation carriers and noncarriers.

(A) ABCA7 messenger RNA (mRNA) and (B) protein isolated from human brain tissue (temporal cortex) were assessed using quantitative PCR and Western blot, respectively, for carriers and noncarriers (nc) of the 3 most frequent ABCA7 putative loss-of-function mutations investigated (L1403fs, E709fs, and 5570+G>C). Mutation carriers with a pathologic diagnosis of either Alzheimer disease (AD) or progressive supranuclear palsy (PSP) were analyzed. Two clinically normal control (control.nc) and 2 pathologic AD participants (AD.nc) without any predicted deleterious ABCA7 mutations were analyzed as mutation noncarriers. *1 and *2 indicate 5570+G>C carriers with additional ABCA7 predicted deleterious mutations. The Western blots from the human brain lysates have 2 bands, 1 at 234 kDa corresponding to full-length ABCA7 protein (ENSP00000414062) and 1 at 218 kDa that is the expected molecular weight of a known alternative isoform ENSP00000465322 (ENST00000433129) starting in exon 7. *Denotes ABCA7 mutation carriers. GAPDH = glyceraldehyde 3-phosphate dehydrogenase; RQ = relative quantity.
Figure 2. Brain ABCA7 messenger RNA expression measurements with semiquantitative PCR or RNA sequencing in select mutation carriers and noncarriers.

(A) Relative messenger RNA (mRNA) levels of E709fs mutant (mt) and wild-type (wt) alleles in Alzheimer disease (AD) and progressive supranuclear palsy (PSP) carriers measured using semiquantitative PCR. A control noncarrier is shown for reference. Peak heights indicate relative mRNA levels and show reduced levels of the E709fs mutant allele. (B.b) RNAseq data for canonical splice-site mutation carriers for 5570+G>C and (C.b) 4416+2T>G are shown. B.a and C.a are from a participant without these splice-site mutations. RNAseq data are viewed using Integrative Genomics Viewer (IGV). Intronic reads for the mutant allele are included in the mRNA of both canonical splice-site mutation carriers, indicating that these mutations alter splicing. *Denotes ABCA7 mutation carriers.
Investigations of brain ABCA7 protein and mRNA expression levels in 4 c.5570+5G>C carriers vs 4 noncarriers did not reveal consistent differences (figure 1, A and B). RNAseq data from carriers of both splice-site mutations, c.5570+5G>C and 4416+2T>G, demonstrate that reads containing the mutant allele extend into the intronic sequence (figure 2, B and C). The inclusion of the mutant intronic sequence within mRNA indicates that these mutations lead to altered splicing. RNAseq data from a c.5570+5G>C carrier (figure 2B.b) and a noncarrier (figure 2B.a) illustrate the difference in reads mapped to the intron when compared with the exon. The gray bar chart at the top of each panel displays the depth of reads at the locus. In the c.5570+5G>C carrier, the read depth of the intron is approximately 50% of that of the exon. However, for the noncarrier, the read depth of the intron is substantially lower than that of the exon. This finding indicates that in the c.5570+5G>C carrier, splicing is affected in approximately 50% of the expressed mRNA molecules, thus refuting NMD of mRNA as its mechanism of action.
Of interest, a PSP c.5570+5G>C carrier had one of the lowest brain ABCA7 proteins and one of the highest mRNA levels (figure 1, A and B). Sequencing of the entire ABCA7 coding region for all participants in figure 1 revealed this PSP c.5570+5G>C carrier to have the highest variant burden with 6 missense mutations (table e-3), 2 of which had predicted deleterious effects with combined annotation dependent depletion (CADD) score >2011 (rs3752233:p.R463H and rs3752239:p.N718T). In addition, an AD c.5570+5G>C carrier with one of the lowest brain ABCA7 proteins had 4 missense mutations, 1 of which had predicted damaging effects (rs146191555: p.P1267L), with CADD score >20. None of the other ABCA7 putative LOF mutation carriers or noncarriers in figure 1 had any other mutations with predicted damaging effects.
DISCUSSION
Here, we report a systematic assessment of previously reported1,2 ABCA7 putative LOF variants on brain ABCA7 expression in corresponding mRNA and protein samples. Our findings demonstrate a lack of correlation between brain ABCA7 protein and mRNA levels and suggest distinct plausible mechanisms of action for the different ABCA7 putative LOF variants. It should be noted that the ABCA7 antibody recognizes only the full-length protein and none of the truncated species arising from the ABCA7 mutations. Brain ABCA7 protein is lower in p.L1403fs carriers, despite lack of corresponding lower brain ABCA7 mRNA. In contrast, lower brain ABCA7 mRNA in p.E709fs carriers does not translate to lower protein levels. These findings suggest that despite evidence for NMD, p.E709fs is unlikely to have a deleterious consequence simply by lowering full-length ABCA7 protein levels. The possibility that the mutant p.E709fs allele or the truncated protein could instead have toxic gain of function needs to be considered. Conversely, despite lack of evidence for NMD, p.L1403fs may be leading to ABCA7 LOF via inefficient translation or enhanced protein degradation.
We and others2 observed substantial variability in brain ABCA7 mRNA and protein levels. This may in part be due to the collective effect of multiple deleterious mutations in this gene and its interactions with other genes and/or the environment. The detection of additional predicted deleterious variants in 2 c.5570+5G>C carriers with unremarkable brain ABCA7 mRNA but lowest brain protein levels may suggest that the mutation burden in participants within this gene could influence its brain protein levels. One study investigated brain ABCA7 mRNA levels in 3 mutation carriers and 4 controls and identified lower mRNA levels in p.E709fs and p.Trp1214X, but not in p.L1403fs carriers,2 but did not report on brain ABCA7 protein levels.
Another novel finding is the statistically significant overall association of the tested ABCA7 variants in a cohort of mixed non-AD brain pathologies. The detection of ABCA7 putative functional variants in participants with PSP, VaD, DLBD, and PA/AGD in this study, in addition to their recent implication in Parkinson disease,7 suggests that this gene may be contributory to both AD and non-AD neuropathologies. Given evidence of its pleiotropic functions12,13 including phagocytosis, cell signaling, lipid homeostasis, and Aβ metabolism, it is likely that ABCA7 dysfunction may contribute to both AD-specific and non-AD neuropathologies.
Strengths of this study include sizable cohort of AD and control participants, expansion of ABCA7 mutation screen to non-AD participants, combined investigations of brain ABCA7 mRNA and protein levels in multiple mutation carriers and noncarriers who were screened for additional ABCA7 coding mutations, inclusion of allele-specific expression, and RNAseq as further technical approaches. Despite our systematic investigation of combined brain mRNA and protein levels in ABCA7 mutation carriers, this work needs to be expanded to include additional mutation carriers, larger sample sizes, and functional in vitro studies to uncover the precise mechanism of action. Although our study provides evidence for a role of ABCA7 variants in non-AD pathologies, because our non-AD cohort was not recruited to study a specific neurologic disease, ABCA7 mutation frequency estimates for this mixed diagnostic group may not be accurate. Finally, while our study replicated the risk association for the ABCA7 variants identified from the initial reports,1–3 continuation of variant identification studies is necessary to characterize the full spectrum of mutations in this gene. Our study demonstrates the distinct potential functional consequences of various ABCA7 AD-risk mutations, challenges the notion that they all act via LOF mechanism, and expands the list of neurologic diseases likely influenced by these mutations.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families; without their participation this study would not have been possible.
GLOSSARY
- AD
Alzheimer disease
- AGD
argyophilic grain disease
- CADD
combined annotation dependent depletion
- cDNA
complimentary DNA
- DLBD
diffuse Lewy body disease
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- LOAD
late-onset Alzheimer disease
- LOF
loss of function
- mRNA
messenger RNA
- NMD
nonsense-mediated decay
- OR
odds ratio
- PA
pathologic aging
- PSP
progressive supranuclear palsy
- RNAseq
RNA sequencing
- VaD
vascular dementia
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
M.A., S.J.L., and N.E.-T. contributed to conception and design of the study. M.A., S.J.L., M.C., J.O.W., M.M.C., J.S.R., J.D.B., T.N., K.M., R.C.P., N.R.G.-R., D.W.D., and N.E.-T. contributed to acquisition and analysis of data. M.A., S.J.L., J.O.W., and N.E.-T. contributed to drafting the text and preparing the figures.
STUDY FUNDING
This work was supported by the National Institute on Aging (U01 AG046139 and RF1 AG051504), National Institute of Neurological Disorders and Stroke (R01 NS080820), and Bright Focus Foundation (27J-01-03) awarded to N.E.-T.
DISCLOSURE
Dr. Allen has received research support from NIH. Ms. Lincoln is a coholder of patents for Identification of Mutations in PARK8, a Locus for Familial Parkinson's Disease; Transgenic Mouse Expressing P1 Artificial Chromosome (PAC) Containing the Entire Human Alpha- Synuclein (SNCA) Gene; LRRK2 BAC Transgenic Mice: A Model of Parkinsonism; LRRK2 Knock Out Mice; and iLRRK2 Wild-Type and Mutant (G2019S) Transgenic Mice. She has also received research support from Isis Pharmaceuticals, Inc.; Lundbeck; and Merck. Ms. Corda and Dr. Watzlawik report no disclosures. Dr. Carrasquillo has received research support from the Alzheimer's Association and Mayo Clinic. Dr. Reddy, Mr. Burgess, Ms. Nguyen, and Ms. Malphrus report no disclosures. Dr. Petersen has received publishing royalties from Oxford University Press; has been a consultant to Genentech, Inc., Merck, Inc., Biogen, Eli Lilly, and Roche, Inc.; has received research support from the National Institute on Aging; and has served on a data safety monitoring committee for Pfizer and Janssen Alzheimer Immunotherapy. Dr. Graff-Radford has served on the editorial board of Alzheimer Disease and Therapy; receives publishing royalties from UpToDate; has received research support from TauRx, Lilly, Biogen, Axovant, and NIA; and has consulted for Cytox. Dr. Dickson has received travel funding and a speaker honorarium from Novartis; has served on the editorial boards of Acta Neuropathologica, Brain, Brain Pathology, Neuropathology and Applied Neurobiology, Annals of Neurology, Neuropathology, International Journal of Clinical and Experimental Pathology, and American Journal of Neurodegenerative Disease; and has received research support from CurePSP, the Mangurian Foundation, and the following grants: P50AG016574, P50NS072187, P01NS084974, and P01AG003949. Dr. Ertekin-Taner has served on the editorial board of the American Journal of Neurodegenerative Disease; holds a patent for Human Monoclonal Antibodies Against Amyloid Beta Protein and Their Use as Therapeutic Agents Application (pending); has received research support from NIH, CurePSP, Bright Focus, and the Florida Health Ed and Ethel Moore Alzheimer's Disease Research Program Grant; and has consulted for Cytox. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Steinberg S, Stefansson H, Jonsson T, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet 2015;47:445–447. [DOI] [PubMed] [Google Scholar]
- 2.Cuyvers E, De Roeck A, Van den Bossche T, et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer's disease patients: a targeted resequencing study. Lancet Neurol 2015;14:814–822. [DOI] [PubMed] [Google Scholar]
- 3.Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann Neurol 2015;78:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Guennec K, Nicolas G, Quenez O, et al. ABCA7 rare variants and Alzheimer disease risk. Neurology 2016;86:2134–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Del-Aguila JL, Fernandez MV, Jimenez J, et al. Role of ABCA7 loss-of-function variant in Alzheimer's disease: a replication study in European-Americans. Alzheimers Res Ther 2015;7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cukier HN, Kunkle BW, Vardarajan BN, et al. ABCA7 frameshift deletion associated with Alzheimer disease in African Americans. Neurol Genet 2016;2:e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nuytemans K, Maldonado L, Ali A, et al. Overlap between Parkinson disease and Alzheimer disease in ABCA7 functional variants. Neurol Genet 2016;2:e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 9.Kalari KR, Nair AA, Bhavsar JD, et al. MAP-RSeq: mayo analysis pipeline for RNA sequencing. BMC Bioinformatics 2014;15:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jehle AW, Gardai SJ, Li S, et al. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol 2006;174:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakae N, Liu CC, Shinohara M, et al. ABCA7 deficiency accelerates amyloid-beta generation and Alzheimer's neuronal pathology. J Neurosci 2016;36:3848–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.