

Abstract
Astrocytes respond to CNS insults through reactive astrogliosis, but the underlying mechanisms are unclear. In this study, we show that inactivation of mechanistic target of rapamycin complex (mTORC1) signaling in postnatal neurons induces reactive astrogliosis in mice. Ablation of Raptor (an mTORC1-specific component) in postmitotic neurons abolished mTORC1 activity and produced neurons with smaller soma and fewer dendrites, resulting in microcephaly and aberrant behavior in adult mice. Interestingly, extensive astrogliosis without significant astrocyte proliferation and glial scar formation was observed in these mice. The inhibition of neuronal mTORC1 may activate astrogliosis by reducing neuron-derived fibroblast growth factor 2 (FGF-2), which might trigger FGF receptor signaling in astrocytes to maintain their nonreactive state, and FGF-2 injection successfully prevented astrogliosis in Raptor knock-out mice. This study demonstrates that neuronal mTORC1 inhibits reactive astrogliosis and plays an important role in CNS pathologies.
Keywords: astrocyte, fibroblast growth factor (FGF), mammalian target of rapamycin (mTOR), mTOR complex (mTORC), neuron, Raptor, fibroblast growth factor 2, reactive astrogliosis
Introduction
Reactive astrogliosis occurs in response to all forms of CNS insults including trauma, infections, stroke, neurodegeneration, and epilepsy (1–3). In most cases, astrocytes become transiently hypertrophic and express high levels of intermediate filament proteins such as glial fibrillary acid protein (GFAP)3 and vimentin (4, 5). In cases of severe damage, astrocyte proliferation increases, causing scar formation (6, 7). Reactive astrogliosis can be both beneficial and detrimental to CNS recovery. It is crucial for minimizing the spread of damage and inflammation, but excessive or sustained astrogliosis inhibits axonal and cellular regeneration. There is increasing evidence that the consequences of reactive astrogliosis contribute to CNS disorders (8), although the specific mechanisms underlying these pathologies are not fully understood.
Neurons and astrocytes are generated sequentially from the same pool of neural stem cells (9). Neuron-astrocyte interactions play crucial roles in the physiology and pathology of the mammalian CNS (10–12). Previous studies have focused primarily on the housekeeping function of astrocytes in supporting and regulating neurons; they maintain a viable environment for neurons, such as providing metabolic support, buffering excess potassium and neurotransmitters, promoting neuronal maturation, synaptogenesis, and angiogenesis, and maintaining the blood-brain barrier (8, 13–15). Not surprisingly, impaired astrocyte function contributes to neuronal dysfunction. Astrocytes are able to sense neuronal activity and can respond to neuronal changes (16, 17), but how neurons influence astrocytes and in particular how injured neurons induce reactive astrogliosis remain poorly understood.
The mechanistic target of rapamycin complex 1 (mTORC1) integrates intracellular and extracellular signals, (including growth factors, nutrients, energy levels, neurotransmitters, and cellular stress) to regulate cell metabolism, growth, and proliferation (18, 19). Importantly, mTORC1 plays a crucial role in cerebral development and functional disorders (20, 21). Disrupted mTORC1 signaling has been linked to epilepsy, neurodegenerative disorders, and mental disorders in tuberous sclerosis complex (TSC) (20, 22–24). However, whether altered mTORC1 signaling is a cause or consequence of these pathologies has not been determined. Deletion of the mTORC1-specific component Raptor in neural progenitors in mice reduced the size and number of neurons and astrocytes and led to a deficit in glial differentiation and microcephaly in mice (25). Therefore, the role of mTORC1 in adult neurons is not known. The effects of neuron-specific mTORC1 inactivation on astrocytes have not been investigated either.
To investigate the postnatal role of mTORC1 in the brain, we inactivated mTORC1 specifically in postmitotic neurons by deleting Raptor. In addition to reducing the size of the brain and neurons, mTORC1 inactivation also induced reactive astrogliosis. Neuronal mTORC1 inhibition also reduced levels of fibroblast growth factor 2 (FGF-2), which is required to maintain astrocytes in a nonreactive state. These findings reveal a novel role for mTORC1 in reactive astrogliosis of CNS disorders.
Results
Deletion of Raptor in Mature Neurons Induces Reactive Astrogliosis in the CNS
The functions of mTORC1 in neural progenitors and glial cells are well established. To investigate the role of mTORC1 in postnatal neurons, we generated mice with a conditionally ablated Raptor gene in mature neurons using a Cre expression cassette under the control of the CaMKIIα-Cre promoter (Raptor nKO). Raptor nKO mice were born at the expected Mendelian frequency, and the specific recombination and deletion of Raptor alleles in neurons were confirmed by allele-specific PCR (Fig. 1, A and B). Regional deletion of Raptor was examined by immunofluorescence staining (supplemental Fig. S1). P-S6 (Ser-235/236) (a marker for mTORC1 activity) expression was examined by Western blotting, and these findings revealed a reduced expression of P-S6 in the neurons of Raptor nKO mice (Fig. 1C), indicating that mTORC1 was inactivated by Raptor deletion.
FIGURE 1.

Raptor deletion in mature neurons results in inactivated mTORC1 signaling and decreased brain size and lifespan. A, genotyping of the offspring after mating transgenic Cre and LoxP mice. The expected band of mutant carrying a Cre-recombinase gene is nearly 250 bp. M, molecular weight markers. B, genotyping of the offspring after mating transgenic Cre and loxp mice. The expected band of mutant carrying floxed Raptor allele is nearly 180 bp, and bands of ∼180 and ∼150 bp were detected in heterozygote mice. C, expression of total P-S6, Raptor, and GFAP was measured by Western blotting in the forebrain of control and nRaKO mice. mTORC1 inhibition, as reflected by P-S6 and Raptor expression, is decreased when compared with controls. Astrogliosis, as reflected by GFAP expression, was increased when compared with controls. Tubulin was included as an internal control for protein loading. (n = 5). D, decreased body size and brain size in adult nRaKO mice. No difference in brain size of newborn nRaKO mice was detected when compared with wild type. E, continuously decreasing body weight of nRaKO mice (n = 18) when compared with controls (n = 22). Body weight of mice was weighed every week after P1. F, decreased weight of P90 brains from nRaKO mice (n = 12) when compared with controls (n = 20). Weight of other organs (liver, spleen, kidney) has no variation. G, nRaKO mice (n = 12) died earlier than wild type mice (n = 18) (lifespan up to 12–15 months). All data are represented as mean ± S.E. (*, p < 0.05); ns means no significant difference.
We examined the general appearance of Raptor nKO mice; although brain size (t test, p = 0.1012) and body weight were normal at birth, a significant reduction in brain size (t test, p = 0.0316) and a slight decrease in body weight were observed in adult Raptor nKO mice when compared with normal littermate controls (Fig. 1, D–F). We performed histology analyses of Raptor nKO brains and observed significant histological changes when compared with normal brains. Golgi-Cox staining of brain sections showed a significant decrease in the size of the hippocampus and the soma of cortical neurons (t test, p < 0.001; Fig. 2A), and fewer hippocampal dendrites (Fig. 2B) in Raptor nKO mice. To determine whether these morphological changes had behavioral consequences, we performed RotaRod and Open-Field Tests to measure locomotor activity and motor learning. We found that Raptor nKO mice performed worse and barely maintained balance on the RotaRod assay after 12 consecutive training days (Fig. 2C). Moreover, we observed that Raptor nKO mice exhibited less movement and were less interested in exploring their environment (t test, time spent in center: p = 0.022; time spent in margin: p = 0.028; total distance: p = 0.0076; number of vertical movements: p = 0.0051; Fig. 2D).
FIGURE 2.

Raptor deletion in mature neurons results in morphological changes in the brain and causes behavioral consequences. A, Golgi-Cox staining of 120-μm sagittal sections showed an obvious minimized soma size of cortical neurons (n = 30 per brain section). Slices were imaged by 40× objectives. B, Golgi-Cox staining of 120-μm sagittal sections showed hippocampal architecture in groups of mice (n = 6). Minimized size and sparse dendrites were observed in hippocampus of nRaKO mice (n = 6). Slices were imaged by 10× objectives. C, RotaRod examination detected motor coordination and motor learning deficits of nRaKO mice (n = 12) when compared with control (n = 15). Mice performed trials for 12 days continuously. D, Open-Field examination detected decreased locomotor activity of nRaKO mice (n = 12) when compared with control (n = 15). All data are represented as mean ± S.E. (***, p < 0.001, **, p < 0.01, *, p < 0.05). VMOVNO, number of vertical movements.
The hyperactivation of mTORC1 in neurons or astrocytes by TSC mutation/deletion has been shown to induce epilepsy and reactive astrogliosis (26–28). Our findings revealed that the inactivation of mTORC1 in postnatal neurons can also induce reactive astrogliosis. Our immunofluorescence study showed that P-S6 expression was reduced in Raptor nKO hippocampal neurons, and that it was accompanied by a marked increase in GFAP- and vimentin-positive cells at postnatal days P21 and P90 (Fig. 3, A–D). Hypertrophy of astrocyte cell bodies and processes was also observed (Fig. 3E). Furthermore, GFAP expression was increased in brain tissue lysates of Raptor nKO mice (Fig. 1C). Besides, the microglia population increased in Raptor nKO mice (supplemental Fig. S2). We did not observe any significant change in astrocyte proliferation in the cortex and hippocampus of Raptor nKO mice at P90 (t test, p = 0.2663), as evident by comparable numbers of Ki67- and GFAP-positive cells in Raptor nKO and wild type mice (Fig. 3F), indicating that neuronal mTORC1 deficiency induces reactive astrogliosis without affecting astrocyte proliferation. Nevertheless, these lesions reduced the lifespan of Raptor nKO mice to 12–15 months (Fig. 1G). Collectively, these findings demonstrate that mTORC1 deficiency in mature neuron reduces neuronal growth and dendrite extension and stimulates reactive astrogliosis.
FIGURE 3.

Raptor deletion in mature neuron results in reactive astrogliosis. A, confocal image montage of sagittal sections from nRaKO mice (n = 6). Green signal indicates decreased P-S6 expression in CA1 and dentate gyrus regions; red signal indicates increased GFAP expression both in P21 and in P90 mice. Quantification on the right shows the intensity of GFAP immunostaining per image. AU, arbitrary units. B, confocal images were enlarged, and P-S6-positive or GFAP-positive cells are shown compared with wild type mouse brains. C, co-staining of GFAP (red) and vimentin (green) both showed increased expression (merge, yellow) in astrocytes of P21 nRaKO mice. Slices were imaged with 10× objectives. D, confocal images were enlarged, and GFAP-positive vimentin-positive cells exhibit significantly greater expression when compared with P21 and P90 wild type mouse brains. Quantification on the right shows the intensity of vimentin immunostaining per image. E, high power fields of confocal image montage demonstrate increased GFAP and vimentin expression in astrocytes of nRaKO mice. Slices were imaged using 60× objectives with 3-fold amplification. F, immunofluorescence co-staining of GFAP and Ki67 indicates no astrocyte proliferation in the brain. Confocal images were obtained using 20× and 60× objectives. GFAP-positive cell numbers between mutant and control mice (n = 6) were analyzed by cell counting. All data are represented as mean ± S.E. (***, p < 0.001).
Neuron mTORC1 Inhibition Induces Reactive Astrogliosis by Regulating Neuron-derived Secretory Factors
Next, we investigated how loss of mTORC1 in neurons stimulates astrocyte activation. Astrocytes cultured in neuronal culture supernatant exhibited a reduced GFAP expression when compared with cells cultured in normal neuron or astrocyte culture medium without serum (Fig. 4A). This suggests that neurons are able to suppress astrocyte reactivation in paracrine in normal brain. We hypothesized that neuronal mTORC1 deficiency stimulates astrocyte activation by regulating the production of signaling molecules. Primary wild type astrocytes were cultured in medium conditioned by wild type and Raptor KO neurons (Fig. 4B). Interestingly, media conditioned by Raptor KO neurons were able to stimulate astrocyte activation in vitro, as observed by a dramatic increase in GFAP and vimentin expression when compared with astrocytes cultured in medium conditioned by wild type neurons (t test, GFAP: p = 0.0002, vimentin: p = 0.0002) (Fig. 4, C and D). To investigate which neuron-derived factors were regulated by mTORC1, we performed mouse cytokine array analysis on the neuron culture supernatants from mutant and wild type mice. Eight out of ninety-six factors were up-regulated, whereas three were down-regulated in neurons with mTORC1 inactivation (Table 1). These regulated factors included growth factors and inflammatory factors. Among the three factors with reduced expression, only FGF-2 shows marked changes (t test, FGF-2: p = 0.0079; SDF-1: p = 0.1431; VEGF: p = 0.0901) (Fig. 4E). It has recently been found that FGF signaling is required to maintain the nonreactive state of astrocytes (29) and that mTORC1 is involved in the regulation of FGF-2 expression in other cells (30); therefore, we predicted that mTORC1 suppresses astrocyte activation by activating the production of neuron-derived FGF-2.
FIGURE 4.

Neuron mTORC1 inhibition induces reactive astrogliosis via regulation of neuron-derived molecules. A, neurons are capable of suppressing astrogliosis. Panel a, GFAP expression (red) in primary astrocytes cultured in neuron culture supernatant for 48 h. Panel b, GFAP expression in primary astrocytes cultured in Neurobasal Medium (without AraC and B27 supplement). Panel c, GFAP expression in primary astrocytes cultured in DMEM/F12 medium without serum. B, schematic model of conditional medium culture experiments. primary AST, primary astrocytes. C, increased GFAP and vimentin expression levels in primary astrocytes cultured on coverslips in nRaKO neuron-conditioned medium (n = 5). Multiple experiments were performed with a single control experiment. D, qPCR detection of both elevated GFAP and vimentin mRNA levels (n = 5) in astrocytes cultured with conditional medium (nRaKO neurons). Their expression was normalized over GAPDH. E, cytokine array assay showed different expression value of decreased factors. All data are represented as mean ± S.E. (***, p < 0.001, **, p < 0.01, ns means no significant difference).
TABLE 1.
Differential expression of factors derived from neurons in RaptorCaMKIIαCKO mice
Summary of mean value acquired from primary neurons culture medium from WT and nRaKO neonatal mice (female, n = 3). bFGF, basic FGF.
| Cytokines | Value |
-Fold change | |
|---|---|---|---|
| WT | RaKO | ||
| bFGF | 28738 | 12248 | 0.42↓a |
| SDF-1 | 17086 | 15203 | 0.89↓ |
| VEGF | 24892 | 16328 | 0.66↓ |
| Eotaxin-2 | 17810 | 27577 | 1.55↑ |
| Fractalkine | 10793 | 20235 | 1.87↑a |
| IL-13 | 8559 | 10300 | 1.20↑ |
| IL-1α | 15564 | 20153 | 1.29↑ |
| IL-1β | 10897 | 22401 | 2.06↑a |
| IGF-1 | 16035 | 23268 | 1.45↑ |
| IL-3 | 10832 | 16528 | 1.53↑ |
| IL-4 | 9540 | 10500 | 1.10↑ |
a Data with statistical significance.
mTORC1 Regulates Neuron-derived FGF-2 to Suppress Reactive Astrogliosis
Because it has been suggested that FGF signaling may be necessary for controlling astrocyte morphology and activation (31), one possibility is that extracellular FGF-2 reacts with the FGF-2 receptor on astrocytes in Raptor nKO mice. To confirm the regulation of FGF-2 by mTORC1, we verified the transcriptional levels of FGF-2 and other crucial FGF family members (32) in neurons cultured from mutant or wild type mice. We found that the level of FGF-2 mRNA was significantly reduced in primary neurons (t test, FGF-2: p = 0.0005; FGF-1: p = 0.1197; FGF-4: p = 0.8639; FGF-9: p = 0.0790; FGF-11: p = 0.0966; FGF-13: p = 0.0758; FGF-14:p = 0.3311; FGF-23: p = 0.0583) and in brain tissue (t test: p = 0.0178) from Raptor nKO mice (Fig. 5, A and B). Mice with neuronal specific deletion of TSC1 (TSC1 nKO) demonstrated macroencephaly, severe cerebral damage, and premature death (supplemental Fig. S3, A−C), but exhibited milder reactive astrogliosis than Raptor nKO mice (supplemental Fig. S3D).
FIGURE 5.

mTORC1 regulates FGF-2 expression in neurons. A, qPCR detection of decreased FGF-2 mRNA level in primary neurons lacking raptor from mutant mice (n = 5). FGF-1, -4, and -9 showed no differences between groups. B, qPCR detection of decreased FGF-2 mRNA level in forebrain tissue from mutant mice (n = 5). C, qPCR detection of decreased FGF-2 mRNA level in primary neurons treated with 5 nm rapamycin for 12 h. D, protein levels of FGF-2 were determined by Western blotting analyses after treatment with 5 nm rapamycin (RAP) for 12 h. CON, control. E, increased GFAP expression levels in primary astrocytes cultured on coverslips in neuron-conditioned medium treated with rapamycin (n = 5). Multiple experiments were performed with a single control experiment. F, luciferase activity in HEK293 cells upon transfection with Tsc1 or Raptor siRNA (indicated by iTSC1 and iRapor) after transfection of indicated FGF-2 reporter constructs (n = 5). All data are represented as mean ± S.E. (**, p < 0.01, *, p < 0.05, ns means no significant difference).
Moreover, FGF-2 mRNA and protein levels were significantly decreased in rapamycin-treated neurons (Fig. 5, C and D). Besides, GFAP expression was increased in astrocytes incubated with medium conditioned by rapamycin (Fig. 5E). To assess whether the FGF-2 transcription activity could be regulated by mTORC1, a luciferase assay was performed in Raptor or TSC1 siRNA-transfected HEK293 cells. We found that FGF-2 transcriptional activity in targeted Raptor cells was decreased significantly (t test, p = 0.0126; Fig. 5F), and in targeted TSC1 cells, it was increased to a minor extent (t test, p = 0.0757). Together, these results suggest that mTORC1 signaling promotes FGF-2 expression in mature neurons, and solely based on mTORC1 inhibition.
To further elucidate the inhibitory effect of FGF-2 on astrocyte activation in vivo, we administered FGF-2 into the lateral ventricle of Raptor nKO brains via intracerebroventricular (i.c.v.) microinjections. Interestingly, FGF-2 treatment reversed astrogliosis in Raptor nKO neurons, as evidenced by decreased GFAP and vimentin expression and inhibition of cell hypotrophy in astrocytes when compared with untreated mice (Fig. 6, A–C). We further administered FGFR3 blocking antibody into the lateral ventricle of wild type brains and found that FGFR3 blocking antibody increased GFAP expression (one-way analysis of variance (ANOVA) with Bonferroni's multiple comparison, control versus FGFR3: p = 0.0003; Fig. 6D). Besides, GFAP and vimentin mRNA levels in hippocampus and cortex were also increased (one-way ANOVA with Bonferroni's multiple comparison; hippocampus: p = 0.0021; cortex: p = 0.0259 in Fig. 6E; hippocampus: p = 0.0174; cortex: p = 0.0406 in Fig. 6F). Taken together, these observations indicate that FGF signaling may contribute to the activation of astrocytes due to the inactivation of neuronal mTORC1.
FIGURE 6.

FGF-2 reduces astrogliosis in nRaKO mice. A, confocal image montage of coronal sections from nRaKO (RaptorCaMKIIαCKO) mice with or without FGF-2 i.c.v. treatment (n = 14). Sections stained with P-S6 (green) indicate the absence of mTORC1 activity; GFAP-stained sections (red) indicate weakened astrogliosis in FGF-2-treated mice. Slices were imaged with 20× objectives (left); images are enlarged when compared with control mice (right). Quantification in the lower right shows the intensity of GFAP immunostaining per image. con, control; AU, arbitrary units. B, high power fields of images showed changes of hypertrophic astrocytes in FGF-2 treatment groups. GFAP and vimentin expression were significantly reduced, suggesting a suppressive role of external FGF-2. C, protein levels of GFAP and vimentin were determined by Western blotting analyses after treatment with or without FGF-2. D, Confocal image montage of sagittal sections from wild type mice with FGFR3 blocking antibody (FGFR3Ab) i.c.v. treatment (n = 10). Sections stained with GFAP (green) indicate astrocyte activation in the different groups. E, qPCR detection of mRNA levels of GFAP in brains treated with FGFR3Ab (n = 8). FGFR3Ab single treatment remarkably increased GFAP mRNA expression. HIPPO, hippocampus. F, qPCR detection of mRNA levels of vimentin in brains treated with FGFR3Ab (n = 8). All data are represented as mean ± S.E.(***, p < 0.001, **, p < 0.01, *, p < 0.05, ns means no significant difference).
Discussion
Reactive astrogliosis is a dynamic process regulated by specific signaling events. These signaling events have been investigated by the generation of mice deficient for specific genes (26, 33–35). The induction of reactive astrogliosis by injured neurons and the specific pathways by which neurons control the activation of astrocytes remain poorly understood. This study has revealed that the inactivation of mTORC1 in postmitotic neurons causes moderate reactive astrogliosis. The loss of neural mTORC1 activity may induce astrogliosis by reducing the neuronal secretion of FGF-2, thereby inhibiting FGF receptor signaling in astrocytes, which is required to maintain their nonreactive state (36) (Fig. 7). Although our present data could not identify the exact role of FGF-2 in this process, and the underlying mechanism needs to be further investigated, our findings have uncovered a novel mechanism for the regulation of astrocytes by dysfunctional neurons and have established a potential important link between mTORC1 signaling and CNS pathologies.
FIGURE 7.
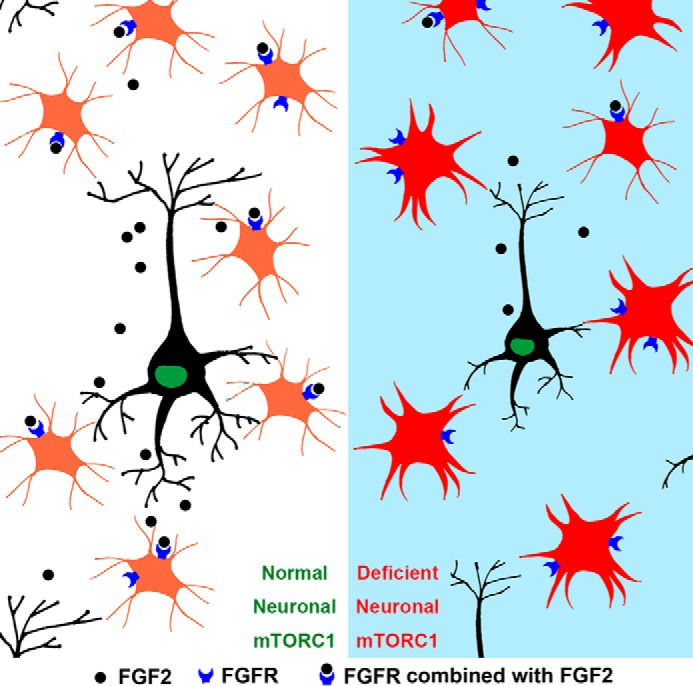
The schematic model of reactive astrogliosis regulated by neuron mTORC1 activity.
The function of mTORC1 in neurons and astrocytes has been extensively studied in conditional knock-out mice (26, 37–39). Hyperactivation of mTORC1 by TSC1/2 deletion induces aberrant growth, proliferation, and differentiation of neurons and astrocytes, resulting in neuronal dysplasia, abnormal neuronal architecture, reactive astrogliosis, and seizures (27, 40–42). Inactivation of mTORC1 in neuronal progenitors impairs the growth and proliferation of neurons and astrocytes, resulting in a smaller brain and in death shortly after birth (25, 43).
Zou et al. (43) have shown that Rheb1 deletion in neural progenitors causes a milder phenotype than Raptor deletion, suggesting either a degree of functional redundancy in the GTPases that activate mTORC1 during brain development or alternative pathways that can activate mTORC1 in neural progenitors independent of Rheb1. In this study, Raptor deletion in postmitotic neurons causes abnormal size and morphology, suggesting a lack of redundancy and alternative pathways in mature neurons. Notably, inhibition of mTORC1 in both neurons and astrocytes, as in rapamycin-treated mice (supplemental Fig. S4), does not induce astrogliosis, suggesting that mTORC1 activity in astrocytes is also essential for astrogliosis.
Although the importance of FGF signaling in CNS development is well established, the role of FGF-2 in astrogliosis remains controversial (17, 36, 44). FGF-2 was shown to induce astroglial and microglial reactivity in the rat brain (45) and mediate the morphogenesis of Drosophila astrocytes during development (17). However, FGF-2 also inhibited the TGF-β1-mediated increase of GFAP expression in astrocytes (44). A recent study demonstrated that the loss of the FGF-2 receptor in astrocytes induced astrocyte activation in unperturbed mice, suggesting that FGF signaling in astrocytes is required to maintain their nonreactive state (36). In this study, we have confirmed the activated effects of neurons on astrocyte and the related signaling (supplemental Fig. S5). Importantly, we have confirmed the inhibitory effect of FGF-2 on astrocyte activation using a much lower dose than was previously reported (48) and have further demonstrated that FGF-2 may be derived from neuronal cells and modulated by mTORC1. Evidence that other factors may participate in the induction of astrogliosis in response to CNS damage comes from TSC1 nKO mice, which produce more FGF-2 but still have active astrocytes. Furthermore, activated astrocytes produce FGF-2, which may reduce astrogliosis in a negative feedback loop (46). The generation of mice with conditional ablation of FGF-2 in postmitotic neurons would help to better understand FGF-2-induced astrogliosis in response to neuronal injury.
In summary, this study has demonstrated that the inactivation of mTORC1 in postmitotic neurons induces reactive astrogliosis, possibly by inhibiting FGF-2 secretion. mTORC1 activity in postmitotic neurons is required for maintaining astrocytes in a nonreactive state. Astrogliosis is likely to be regulated by various signaling pathways and various cell types in different nuclei on the CNS. In our studies, neuronal mTORC1 activity regulates astrocyte activation, possibly via multiple potential signals directly or indirectly. Further investigation is required to define the pathological consequences of astrogliosis induced by the loss of neuronal mTORC1 and its association with CNS disease. Manipulating mTORC1 in neurons or FGF-2 signaling in astrocytes may represent a novel therapeutic mechanism for treating CNS disorders and improving functional recovery in neuropathological conditions.
Experimental Procedures
Mice
CaMKIIα-Cre transgenic mice were a gift from (and generated by) Dr. Günther Schütz (47). Cre recombinase expression is driven by the calcium/calmodulin-dependent protein kinase II α (Camk2a) promoter. Mice with floxed Raptor (Raptorfl/fl) alleles and Tsc1 (Tsc1fl/fl) alleles were obtained from The Jackson Laboratory (stock numbers: 013188 and 005680). Conditional knock-out mice were generated by mating Cre mice with floxed mice. Care and use of animals conformed to a protocol approved by the Animal Studies Committee of Southern Medical University. We used a balanced ratio of male and female mice in this experiment.
Tissue Preparation and Immunofluorescence
Animals were deeply anesthetized by intraperitoneal injection of solution of thiopental (Sigma-Aldrich T1200000, 0.64 mg/g of body weight), and then intracardially perfused by 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PBS; pH 6.5). After perfusion, the brains were dissected and postfixed in the 4% PFA for 24 h at 4 °C. The fixed brains were washed with flowing water for 2 h at room temperature and then dehydrated in 30% sucrose solution in PBS for at least 24 h at 4 °C. For immunofluorescence, the brains were sectioned coronally or sagittally with a vibratome at a thickness of 60 μm to prepare free-floating brain slices. The groups of brain slices were treated with blocking buffer (0.1% Triton X-100 and 1% bovine serum albumin in PBS) for 1 h at room temperature, and then primary antibodies were diluted in the blocking buffer and brain slices were incubated overnight at 4 °C. Brain slices were washed with PBS after the incubation and sequentially incubated with fluorescent secondary antibodies for 1 h at room temperature; ProLong® Gold Antifade Mountant (Thermo Fisher) with DAPI was used to detect the nuclei and mounting. Spread and labeling efficiency were scored by hand examination of every section for the presence of mCherry fluorescence using a confocal microscope (Olympus, LV1000, Japan). For quantification of co-labeling, confocal images were acquired and individual cells were identified independently for each of the two fluorescent channels. The slices were imaged using 10× or 20× objectives or 600× oil-immersion objective (NA1.2, Olympus).
The following primary antibodies and dilutions were used: GFAP (Cell Signaling Technology, Beverly, MA, 12389 and 3670, 1:400), NeuN (Cell Signaling Technology 12943; Millipore, Billerica, MA, MAB377, 1:500), vimentin (Bioworld Technology, St. Louis Park, MN, BS1491, 1:200), SMI-311 (Calbiochem, NE1017, 1:500), and phospho-S6 ribosomal protein (Cell Signaling Technology 2211, 1:200). Secondary fluorescent antibodies and dilutions were Alexa Fluor 488 and Alexa Fluor 594 (Invitrogen, A21203 and A21206, 1:500).
Astrocyte Cultures
Neonatal astrocyte cultures derived from neocortex and hippocampus were established from postnatal day 1 (P1) mice. Cultures were maintained in DMEM/F-12 supplemented with 10% fetal calf serum, 50 units/ml penicillin, and 50 μg/ml streptomycin. On the 4th culture day, culture vessels were knocked on the desktop vigorously to remove microglia, oligodendroglia, and some neurons. Prior to experiments, astrocytes were maintained in culture for 7–14 days.
Neuron Cultures
Neonatal neuronal cultures were derived from neocortex and hippocampus of P0-specific knock-out mice. Cultures were maintained in Neurobasal Medium (Gibco 10888022) with 2% B27 supplement and 25 μm glutamate (1% GlutaMAX-I 100×). Culture medium was replaced with Neurobasal Medium with 2% B27 supplement and 1% GlutaMAX-I 100×. Before dissection, 24-well culture dishes were coated with 0.1 mg/ml poly-d-lysine and were left overnight in a cell culture incubator (37 °C). On the following day, dishes were washed twice with sterile water.
Conditional Cell Culture and Detection
Prior to dissection, astrocyte cultures were covered with 24-well coverslips in 24-well dishes. Neuronal cultures were maintained for 14 days and made ready for the conditional culture experiments. At day 7 after inhibiting overgrowth of glia cells with AraC, neurons, cultured in a medium containing AraC, were changed to fresh medium without AraC. Prior to transferring the neuron culture medium to astrocyte cultures, fresh Neurobasal Medium (without AraC and B27 supplement) was replaced in neuronal cultures 48 h earlier, and astrocytes were maintained in conditional culture medium for 72 h. For the conditional rapamycin treatment experiments, neurons were cultured with 10 nm rapamycin for 12 h, with fresh medium provided for the 12-h culture. The final neuronal culture medium was transferred to astrocyte cultures for 12 h. At the end of the culture period, coverslips were collected and gently washed with PBS, and then fixed in 4% PFA for 10 min at room temperature. Coverslips were used for double-immunofluorescence detection of GFAP and vimentin.
Behavior
The standard RotaRod tests were performed with the accelerating rotarod apparatus. Mice were placed on the apparatus for 12 trials (three trials a day on 4 consecutive days) with a 30–60-min rest interval between trials. Each trial lasted for a maximum of 5 min, during which the rod accelerated linearly from 10 to 40 rpm. The amount of time for each mouse to fall from the rod was recorded for each trial. After trials, mice were placed on a designated rotation speed from 10 to 40 rpm. The amount of time for each mouse to fall from the rod was also recorded for each test.
For the Open-Field locomotor test, mice were placed in the center of an open-field arena (40 × 40 × 30 cm3). Normal animals typically acclimate to the arena and start exploring the surrounding area as well as the center area. Activity was quantified by a computer-operated Digiscan optical animal activity system (RXYZCM, Accuscan). Each test session lasted 30 min, and data were analyzed as three blocks of 10 min each.
Histology
In P90 mice, histological analysis was performed to assess neuronal organization with standard methods. Fixed brains were embedded in paraffin and then sectioned sagittally with a vibratome at a thickness of 10 μm. For cresyl violet staining, deparaffinized and rehydrated sections were mounted and immersed in 0.5% cresyl violet for 5 min. For Golgi-Cox staining, mice were anesthetized, and then the brains were quickly removed and placed in distilled water. Golgi staining was performed in Golgi-Cox staining buffer for 14 days: sections of 120 μm were cut and treated with developing solution and fixing solution, and then rehydrated with gradient ethanol and chloroform:xylene:ethanol (CXA) solution. Slices were observed and recorded by using a light microscope (Axio Scope.A1), and were imaged by 10× and 40× objectives.
Cytokine Array Assay
Primary neurons from wild type and Raptor nKO neonatal mice (n = 3) were cultured and maintained as introduced previously. Before collecting, fresh culture medium was replaced and maintained for 48 h. At day 14, supernatant culture medium was collected for cytokine array detection. Data collection was performed conforming to the standard protocol (AAM-CYT-1000, RayBiotech) and analyzed using RayBio® Analysis Tool.
mRNA Analysis
Mice were euthanized, and then brains were rapidly removed, dissected, and flash-frozen in liquid nitrogen. Total neocortex or hippocampus RNA from P90 mice for quantitative PCR (qPCR) was isolated using TRIzol (Invitrogen) and reverse-transcribed into cDNA using the PrimeScriptTM RT reagent Kit (TaKaRa). Real-time quantitative PCR (SYBR Green, TaKaRa) analysis was performed with primers for genes (Table 2) using an ABI 9700 Thermal Cycler (Applied Biosystems). Quantification of RNA expression levels was analyzed in triplicate, and GAPDH was used as a positive control.
TABLE 2.
Sequences of primers used in this study
| PCR primers | |
| CaMKIIαcre F | TGCCCAAGAAGAAGAGGAA |
| CaMKIIαcre R | TTGCAGGTACAGGAGGTAGTC |
| Tsc1loxp/loxp F | GTCACGACCGTAGGAGAAGC |
| Tsc1loxp/loxp R | GAATCAACCCCACAGAGCAT |
| Raptorloxp/loxp F | CTCAGTAGTGGTATGTGCTCAG |
| Raptorloxp/loxp R | GGGTACGTATGTCAGCACAG |
| FGF2 F | GAGATGTAGAAGATGTGACGCC |
| FGF2 R | CTGTGAGGGTCGCTCTTCTC |
| q-PCR primers | |
| GAPDH F | TGTGTCCGTCGTGGATCTGA |
| GAPDH R | TTGCTGTTGAAGTCGCAGGAG |
| FGF-2 F | TCAGTAGCTGCCTGGAAACA |
| FGF-2 R | AAGCCCTGCACCAAGTACAC |
| FGF-1 F | ACCGAGAGGTTCAACCTGCC |
| FGF-1 R | GCCATAGTGAGTCCGAGGACC |
| FGF-4 F | CAAGCTCTTCGGTGTGCCTT |
| FGF-4 R | GTCCGCCCGTTCTTACTGAG |
| FGF-9 F | ATGCCTGTCTTCCTGTCCTG |
| FGF-9 R | CGTCTGGCTCCTCTCTCTGT |
| FGF-11 F | CTGTCCAAGGTGCGACTGTG |
| FGF-11 R | GAACGACGCTGACGGTAGAGA |
| FGF-13 F | GGCAATGAACAGCGAGGGATACTTGTACAC |
| FGF-13 R | CGGATTGCTGCTGACGGTAGATCATTGATG |
| FGF-14 F | GGCAACCTGGTGGATATCTT |
| FGF-14 R | CCTTGCCTGCAATATAACCT |
| FGF-23 F | GGTGATAACAGGAGCCATGAC |
| FGF-23 R | TGCTTCTGCGACAAGTAGAC |
| GFAP F | CGTTAAGCTAGCCCTGGACATC |
| GFAP R | GGATCTGGAGGTTGGAGAAAGTC |
| Vimentin F | TGCTTCTCTGGCACGTCTTG |
| Vimentin R | GGACATGCTGTTCCTGAATCTG |
Western Blotting
Brain tissues were rapidly removed and collected from P90 male and female mice, and then the neocortex and hippocampus were dissected and sonicated in lysis buffer supplemented with protease inhibitors. Western blotting was performed to assay for the expression of GFAP (Cell Signaling Technology 3670, 1:1000), phospho-S6 (Cell Signaling Technology 2211, 1:2000), and β-tubulin (Epitomics 1879-1, 1:1500) as an internal control for protein loading.
Intracerebroventricular Microinjections
Under aseptic conditions, the i.c.v. injections were performed using the following stereotaxic coordinates: 0.58 mm caudal from bregma, 2.00 mm ventral from dura mater, and 1.25 mm lateral to midline (unilateral injections on the right ventricle). The lateral ventricle injections were made with a Hamilton syringe (5-μl capacity) coupled with polyethylene tubing connected to a needle (0.3 mm in diameter and 12 mm in length). Each injection was administered into the lateral ventricle at a speed of 0.33 μl/min. The injection was left in place for an additional 5 min to allow the reagent to diffuse in the cerebral spinal fluid, followed by removal of the injector and suture.
P90 mice were divided into vehicle control groups and reagent injection groups (n = 6/group). Single injection with 1 μl of FGF-2 (recombinant murine basic fibroblast growth factor, PrimeGene 124-02, 10 μg/ml) or vehicle (saline) was performed. At 24 h after injection, brain tissues were dissected carefully and frozen at −80 °C for subsequent molecular assays or fixed for histological detection. Mice were injected with 1 μl of FGFR3 antibody (R&D Systems, MAB710, 50 μg/ml) into the left lateral ventricle to block FGFR3.
Reporter Construction
A fragment relative to the transcriptional start site of human FGF-2 genomic sequences was produced by PCR. This fragment was fused to the promoterless firefly luciferase gene of pGL-3 luciferase vector (Promega) to generate FGF-2 luciferase reporter plasmid.
Transient Transfection and Luciferase Assays
HEK293 cells were co-transfected with pFGF-2 luciferase construct and β-gal normalization control before being transfected with TSC1 or Raptor siRNA 6 h later, respectively. Lysates were collected 36 h after transfection, and β-gal and firefly luciferase activities were measured with β-gal and the Luciferase Reporter System (Promega).
Statistical Analysis
Statistical differences were analyzed using the unpaired t test for two-group comparison or one-way ANOVA with multiple comparison for four-group comparison. The levels of significance were set as *, p < 0.05; **, p < 0.01; ***, p < 0.001. Data are shown as means ± S.E.
Author Contributions
Y. Z. and S. X. performed most of the experiments and data analysis. K. Liang helped to perform the behavioral and supplemental experiments and data analysis. K. Liang and K. Li contributed to mouse generation and genotype detection. Z. Z., C. Y., K. T., X. C., Y. J., and T. G. helped to analyze datasets, and X. B. supervised the project and wrote the paper.
Supplementary Material
This work was supported by the State Key Development Program for Basic Research of China (Grants 2013CB945203 and 2015CB55360), National Natural Sciences Foundation of China (Grants U1301222, 81530070, 31600964, and 81270080), The National Science Fund for Distinguished Young Scholars (Grant 81625015), and GDHVPS (2011). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S4.
- GFAP
- glial fibrillary acid protein
- mTOR
- mechanistic target of rapamycin
- TSC
- tuberous sclerosis complex
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- Cre
- cAMP-responsive element
- FGFR
- FGF receptor
- nKO
- neuronal knock-out
- nRaKO
- neuronal Raptor knock-out
- P
- postnatal day
- i.c.v.
- intracerebroventricular
- AraC
- arabinosylcytosine
- ANOVA
- analysis of variance
- PFA
- paraformaldehyde
- qPCR
- quantitative PCR.
References
- 1. Myer D. J., Gurkoff G. G., Lee S. M., Hovda D. A., and Sofroniew M. V. (2006) Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129, 2761–2772 [DOI] [PubMed] [Google Scholar]
- 2. de Lanerolle N. C., Lee T. S., and Spencer D. D. (2010) Astrocytes and epilepsy. Neurotherapeutics 7, 424–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Phatnani H., and Maniatis T. (2015) Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 7, a020628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pekny M., and Nilsson M. (2005) Astrocyte activation and reactive gliosis. Glia 50, 427–434 [DOI] [PubMed] [Google Scholar]
- 5. Hol E. M., and Pekny M. (2015) Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 32, 121–130 [DOI] [PubMed] [Google Scholar]
- 6. Silver J., and Miller J. H. (2004) Regeneration beyond the glial scar. Nat. Rev. Neurosci. 5, 146–156 [DOI] [PubMed] [Google Scholar]
- 7. Windle W. F., and Chambers W. W. (1950) Regeneration in the spinal cord of the cat and dog. J. Comp. Neurol. 93, 241–257 [DOI] [PubMed] [Google Scholar]
- 8. Sofroniew M. V., and Vinters H. V. (2010) Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doetsch F. (2003) The glial identity of neural stem cells. Nat. Neurosci. 6, 1127–1134 [DOI] [PubMed] [Google Scholar]
- 10. Allaman I., Bélanger M., and Magistretti P. J. (2011) Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 34, 76–87 [DOI] [PubMed] [Google Scholar]
- 11. Bélanger M., Allaman I., and Magistretti P. J. (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738 [DOI] [PubMed] [Google Scholar]
- 12. Perea G., Sur M., and Araque A. (2014) Neuron-glia networks: integral gear of brain function. Front. Cell. Neurosci. 8, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Volterra A., and Meldolesi J. (2005) Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 6, 626–640 [DOI] [PubMed] [Google Scholar]
- 14. Walz W. (2000) Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 36, 291–300 [DOI] [PubMed] [Google Scholar]
- 15. Attwell D., Buchan A. M., Charpak S., Lauritzen M., Macvicar B. A., and Newman E. A. (2010) Glial and neuronal control of brain blood flow. Nature 468, 232–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Habas A., Hahn J., Wang X., and Margeta M. (2013) Neuronal activity regulates astrocytic Nrf2 signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 18291–18296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stork T., Sheehan A., Tasdemir-Yilmaz O. E., and Freeman M. R. (2014) Neuron-glia interactions through the Heartless FGF receptor signaling pathway mediate morphogenesis of Drosophila astrocytes. Neuron 83, 388–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shaw R. J., and Cantley L. C. (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441, 424–430 [DOI] [PubMed] [Google Scholar]
- 20. Costa-Mattioli M., and Monteggia L. M. (2013) mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat. Neurosci. 16, 1537–1543 [DOI] [PubMed] [Google Scholar]
- 21. Lipton J. O., and Sahin M. (2014) The neurology of mTOR. Neuron 84, 275–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crino P. B., Nathanson K. L., and Henske E. P. (2006) The tuberous sclerosis complex. N. Engl. J. Med. 355, 1345–1356 [DOI] [PubMed] [Google Scholar]
- 23. Lee J. H., Tecedor L., Chen Y. H., Monteys A. M., Sowada M. J., Thompson L. M., and Davidson B. L. (2015) Reinstating aberrant mTORC1 activity in Huntington's disease mice improves disease phenotypes. Neuron 85, 303–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Russo E., Citraro R., Constanti A., and De Sarro G. (2012) The mTOR signaling pathway in the brain: focus on epilepsy and epileptogenesis. Mol. Neurobiol. 46, 662–681 [DOI] [PubMed] [Google Scholar]
- 25. Cloëtta D., Thomanetz V., Baranek C., Lustenberger R. M., Lin S., Oliveri F., Atanasoski S., and Rüegg M. A. (2013) Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J. Neurosci. 33, 7799–7810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uhlmann E. J., Wong M., Baldwin R. L., Bajenaru M. L., Onda H., Kwiatkowski D. J., Yamada K., and Gutmann D. H. (2002) Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann. Neurol. 52, 285–296 [DOI] [PubMed] [Google Scholar]
- 27. Meikle L., Talos D. M., Onda H., Pollizzi K., Rotenberg A., Sahin M., Jensen F. E., and Kwiatkowski D. J. (2007) A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J. Neurosci. 27, 5546–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Magri L., Cambiaghi M., Cominelli M., Alfaro-Cervello C., Cursi M., Pala M., Bulfone A., Garcìa-Verdugo J. M., Leocani L., Minicucci F., Poliani P. L., and Galli R. (2011) Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 9, 447–462 [DOI] [PubMed] [Google Scholar]
- 29. Agarwal A., and Bergles D. E. (2014) Astrocyte morphology is controlled by neuron-derived FGF. Neuron 83, 255–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang B., Cao H., and Rao G. N. (2006) Fibroblast growth factor-2 is a downstream mediator of phosphatidylinositol 3-kinase-Akt signaling in 14,15-epoxyeicosatrienoic acid-induced angiogenesis. The Journal of biological chemistry 281, 905–914 [DOI] [PubMed] [Google Scholar]
- 31. Kang K., Lee S. W., Han J. E., Choi J. W., and Song M. R. (2014) The complex morphology of reactive astrocytes controlled by fibroblast growth factor signaling. Glia 62, 1328–1344 [DOI] [PubMed] [Google Scholar]
- 32. Reuss B., and von Bohlen und Halbach O. (2003) Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 313, 139–157 [DOI] [PubMed] [Google Scholar]
- 33. McLean J. R., Sanelli T. R., Leystra-Lantz C., He B. P., and Strong M. J. (2005) Temporal profiles of neuronal degeneration, glial proliferation, and cell death in hNFL+/+ and NFL−/− mice. Glia 52, 59–69 [DOI] [PubMed] [Google Scholar]
- 34. Nobuta H., Ghiani C. A., Paez P. M., Spreuer V., Dong H., Korsak R. A., Manukyan A., Li J., Vinters H. V., Huang E. J., Rowitch D. H., Sofroniew M. V., Campagnoni A. T., de Vellis J., and Waschek J. A. (2012) STAT3-mediated astrogliosis protects myelin development in neonatal brain injury. Ann. Neurol. 72, 750–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. LeComte M. D., Shimada I. S., Sherwin C., and Spees J. L. (2015) Notch1-STAT3-ETBR signaling axis controls reactive astrocyte proliferation after brain injury. Proc. Natl. Acad. Sci. U.S.A. 112, 8726–8731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kang W., Balordi F., Su N., Chen L., Fishell G., and Hébert J. M. (2014) Astrocyte activation is suppressed in both normal and injured brain by FGF signaling. Proc. Natl. Acad. Sci. U.S.A. 111, E2987–E2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoeffer C. A., Tang W., Wong H., Santillan A., Patterson R. J., Martinez L. A., Tejada-Simon M. V., Paylor R., Hamilton S. L., and Klann E. (2008) Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron 60, 832–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pun R. Y., Rolle I. J., Lasarge C. L., Hosford B. E., Rosen J. M., Uhl J. D., Schmeltzer S. N., Faulkner C., Bronson S. L., Murphy B. L., Richards D. A., Holland K. D., and Danzer S. C. (2012) Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 75, 1022–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chuang J. H., Tung L. C., Yin Y., and Lin Y. (2013) Differentiation of glutamatergic neurons from mouse embryonic stem cells requires raptor S6K signaling. Stem Cell Res. 11, 1117–1128 [DOI] [PubMed] [Google Scholar]
- 40. Ehninger D., Han S., Shilyansky C., Zhou Y., Li W., Kwiatkowski D. J., Ramesh V., and Silva A. J. (2008) Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 14, 843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nie D., Di Nardo A., Han J. M., Baharanyi H., Kramvis I., Huynh T., Dabora S., Codeluppi S., Pandolfi P. P., Pasquale E. B., and Sahin M. (2010) Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat. Neurosci. 13, 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kassai H., Sugaya Y., Noda S., Nakao K., Maeda T., Kano M., and Aiba A. (2014) Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 7, 1626–1639 [DOI] [PubMed] [Google Scholar]
- 43. Zou J., Zhou L., Du X. X., Ji Y., Xu J., Tian J., Jiang W., Zou Y., Yu S., Gan L., Luo M., Yang Q., Cui Y., Yang W., Xia X., et al. (2011) Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev. Cell 20, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reilly J. F., Maher P. A., and Kumari V. G. (1998) Regulation of astrocyte GFAP expression by TGF-β1 and FGF-2. Glia 22, 202–210 [DOI] [PubMed] [Google Scholar]
- 45. Goddard D. R., Berry M., Kirvell S. L., and Butt A. M. (2002) Fibroblast growth factor-2 induces astroglial and microglial reactivity in vivo. J. Anat. 200, 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Neary J. T., Kang Y., Shi Y. F., Tran M. D., and Wanner I. B. (2006) P2 receptor signalling, proliferation of astrocytes, and expression of molecules involved in cell-cell interactions. Novartis Found. Symp. 276, 131–143; discussion 143–147, 233–237, 275–281 [PubMed] [Google Scholar]
- 47. Casanova E., Fehsenfeld S., Mantamadiotis T., Lemberger T., Greiner E., Stewart A. F., and Schütz G. (2001) A CamKIIα iCre BAC allows brain-specific gene inactivation. Genesis 31, 37–42 [DOI] [PubMed] [Google Scholar]
- 48. Goddard D. R., Berry M., Kirvell S. L., and Butt A. M. (2002) Fibroblast growth factor-2 induced astroglial and microglial reactivity in vivo. J. Anat. 200, 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.