

Abstract
Nuclear factor erythroid 2-related factor 2 (NRF2) is a transcription factor that plays a crucial role in protection of cells from electrophile-induced toxicity through up-regulating phase II detoxifying enzymes and phase III transporters. We previously reported that oxidative stress induces up-regulation of interleukin-11 (IL-11), a member of the IL-6 family that ameliorates acetaminophen-induced liver toxicity. However, a role for IL-11 in protection of cells from electrophile-induced toxicity remains unclear. Here we show that an environmental electrophile, 1,2-naphthoquinone (1,2-NQ), but not 15d-prostaglandin J2 (PGJ2) or tert-butylhydroxyquinone (tBHQ), induced IL-11 production. Consistent with a crucial role for prolonged ERK activation in H2O2-induced IL-11 production, 1,2-NQ, but not 15d-PGJ2 or tBHQ, elicited prolonged ERK activation. Conversely, inhibition of the ERK pathway by a MEK inhibitor completely blocked 1,2-NQ-induced IL-11 production at both protein and mRNA levels, further substantiating an intimate cross-talk between ERK activation and 1,2-NQ-induced IL-11 production. Promoter analysis of the Il11 gene revealed that two AP-1 sites were essential for 1,2-NQ-induced promoter activities. Among various members of the AP-1 family, Fra-1 was up-regulated by 1,2-NQ, and its up-regulation was blocked by a MEK inhibitor. Although NRF2 was not required for H2O2-induced IL11 up-regulation, NRF2 was essential for 1,2-NQ-induced IL11 up-regulation by increasing Fra-1 proteins possibly through promoting mRNA translation of FOSL1. Finally, intraperitoneal administration of 1,2-NQ induced body weight loss in wild-type mice, which was further exacerbated in Il11ra1−/− mice compared with Il11ra1+/− mice. Together, both Fra-1 and NRF2 play crucial roles in IL-11 production that protects cells from 1,2-NQ intestinal toxicity.
Keywords: AP1 transcription factor (AP-1), extracellular signal-regulated kinase (ERK), interleukin, nuclear factor 2 (erythroid-derived 2-like factor) (NFE2L2) (Nrf2), oxidative stress, Fra-1, electrophile
Introduction
Electrophiles are electron-deficient compounds that can accept an electron pair to form a covalent bond with its reaction partner (nucleophile), resulting in adducts of macromolecules (e.g. proteins, lipids, and DNA) (1). Such reactive species have been shown to be involved in stress, aging, and cancer through modification of various signaling molecules. It is well recognized that endogenous electrophiles, such as 8-nitro-cGMP, 15-deoxy-prostaglandin J2 (15d-PGJ2),3 and nitrated fatty acids, are produced during oxidative stress and inflammation (2, 3) and then activate redox signal transduction pathways associated with up-regulation of antioxidant and anti-inflammation genes (1–3). In addition, there are a variety of electrophiles in the environment (4, 5). For example, particles with an aerodynamic diameter of 2.5 μm or less (PM2.5) and tobacco smoke (6) contain an atmospheric electrophile, 1,2-naphtoquinone (1,2-NQ), that activates epidermal growth factor receptor (EGFR) through S-arylation of Cys-121 of protein-tyrosine phosphatase 1B (PTP1B) (7). A transcription factor, NF-E2-related factor 2 (NRF2), is activated and attenuates electrophile-induced toxicity through inducing phase II-detoxifying enzymes and phase III transporters (8, 9). Under normal conditions, NRF2 is sequestered by an E3 ligase, Keap1, and is constitutively degraded by the proteasome. Once the cysteine residues on Keap1 are modified by electrophiles, Cul3-dependent ubiquitination of NRF2 is attenuated, thereby rescuing newly synthesized NRF2 from proteasomal degradation and allowing translocation of NRF2 into the nucleus. NRF2 binds to a cognate DNA regulatory element termed the antioxidant response element and up-regulates its target genes. Of interest, 1,2-NQ also activates NRF2 through covalent modification of Keap1 (10). Collectively, it seems likely that there are different adaptive responses against environmental electrophiles, such as 1,2-NQ, via activating signal transduction pathways.
Interleukin-11 (IL-11) is a member of the IL-6 family cytokines and controls various cellular responses, including hematopoiesis, bone development, tissue repair, and carcinogenesis (11). IL-11 binds to the IL-11 receptor α1 (IL-11Rα1) and gp130 complex and activates the family of signal transducer and activator of transcription (STAT) proteins. We previously reported that IL-11 is produced by hepatocytes in an oxidative stress-dependent manner and ameliorates acetaminophen-induced liver injury (12). In addition, IL-11 treatment prevents cardiac dysfunction and attenuates oxidative stress after ischemia and reperfusion injury in heart (13). Together, these results suggest that IL-11 acts as a protective cytokine against oxidative stress-induced tissue injury. However, it is unclear whether IL-11 contributes to protection of cells against electrophile-induced toxicity. We previously reported that 1,2-NQ activates the MEK/ERK pathway through modification and subsequent activation of epidermal growth factor receptor (14). Given that expression of Il11 is regulated by the MEK/ERK pathway (12), we surmised that 1,2-NQ might induce IL-11 production.
We found that 1,2-NQ, but not tBHQ or 15d-PGJ2, induced IL-11 expression at both mRNA and protein levels. We showed that 1,2-NQ-induced up-regulation of Il11 mRNA largely depended on phosphorylation of ERK and subsequent induction of Fra-1. Moreover, knockdown of NRF2 by siRNA completely abolished 1,2-NQ-induced IL11 induction. Conversely, overexpression of NRF2 induced Il11 reporter gene activation. Unexpectedly, we found that knockdown of NRF2 abolished expression of Fra-1 proteins before or after 1,2-NQ stimulation, whereas overexpression of NRF2 alone induced an increase in Fra-1 proteins. Finally, 1,2-NQ-induced body weight loss and intestinal toxicity were exacerbated in Il11ra1−/− mice compared with control Il11ra1+/− mice. Together, these data suggest that NRF2 plays a crucial role in IL11 up-regulation through increasing Fra-1 proteins.
Results
An Electrophile Induces IL-11 Expression
To test whether electrophiles increase Il11 expression, we treated a murine colon cancer cell line, CT26, and a human hepatoma cell line, HepG2, with several electrophiles, including 15d-PGJ2, tBHQ, and 1,2-NQ. To be precise, tBHQ is not an electrophile but is easily converted to an electrophilic quinone through autoxidation. As expected, all three electrophiles induced accumulation of NRF2 (Fig. 1A and supplemental Fig. 1) and significantly increased expression of an NRF2 target gene, Hmox1, in CT26 and HepG2 cells (Fig. 1, B and C). Intriguingly, 1,2-NQ, but not 15d-PGJ2 or tBHQ, increased expression of Il11 mRNA in CT26 and HepG2 cells (Fig. 1, B and C). Moreover, we found that 1,2-NQ induced the production of IL-11 protein in CT26 and HepG2 cells (Fig. 1D).
FIGURE 1.

IL-11 is induced by 1,2-NQ. A–C, CT26 and HepG2 cells were stimulated with 15d-PGJ2 (10 μm), tBHQ (100 μm), or 1,2-NQ (50 μm) for the indicated times. A, cell lysates were analyzed by immunoblotting with the indicated antibodies. B and C, RNAs were extracted from CT26 (B) or HepG2 (C) cells. Relative amounts of Hmox1 and Il11 mRNAs were determined by qPCR. Results are mean ± S.D. (error bars) of triplicate samples. D, CT26 and HepG2 cells were stimulated with the indicated concentrations of 1,2-NQ (μm) for 18 h. Concentrations of IL-11 in the culture supernatants were determined by ELISA. Results are mean ± S.D. of triplicate samples. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant versus untreated cells. All results are representative of two or three independent experiments.
Activation of the ERK Pathway Is Required for 1,2-NQ-induced Il11 Up-regulation
To investigate the molecular mechanisms underlying 1,2-NQ-induced Il11 mRNA expression, we sought to identify the signaling pathways leading to Il11 expression. We previously reported that H2O2-induced IL-11 production largely depends on the ERK pathway (12). We also reported that 1,2-NQ induces ERK activation by inactivating PTP1B through modification of a cysteine residue of PTP1B (7). As shown in Fig. 2A, 1,2-NQ induced phosphorylation of JNK, ERK, AKT, and to a lesser extent p38 in CT26 cells, whereas 1,2-NQ only induced phosphorylation of ERK in HepG2 cells. We next tested the effects of various MAP kinase inhibitors on 1,2-NQ-induced Il11 induction in these cells. Inhibition of the MEK/ERK pathway with a MEK inhibitor, U0126, almost completely abolished 1,2-NQ-induced Il11 up-regulation in both cells (Fig. 2B). Surprisingly, inhibition of the p38MAPK pathway by SB230580 strongly up-regulated expression of Il11 mRNA in HepG2, but not CT26 cells, although the mechanism was not investigated in this study. We also found that strong and sustained ERK activation was only observed in 1,2-NQ-stimulated, and not 15d-PGJ2- or tBHQ-stimulated, CT26 and HepG2 cells (Fig. 2C).
FIGURE 2.

The ERK pathway is crucial for 1,2-NQ-induced IL-11 production. A, CT26 and HepG2 cells were stimulated with 1,2-NQ for the indicated times, and cell lysates were analyzed by immunoblotting with the indicated antibodies. B, CT26 and HepG2 cells were stimulated with 1,2-NQ in the absence or presence (final concentration, 20 μm) of SP600125 (SP; a JNK inhibitor), SB203580 (SB; a p38 inhibitor), U0126 (U01; a MEK inhibitor), or LY294002 (LY; a PI3K inhibitor) for 2 h. RNAs were extracted from CT26 and HepG2 cells, and expression of Il11 was determined by qPCR. Results are mean ± S.D. (error bars) of triplicate samples. *, p < 0.05; ***, p < 0.001; ns, not significant versus 1,2-NQ-treated cells in the absence of inhibitors. C, CT26 and HepG2 cells were stimulated with 15d-PGJ2, tBHQ, or 1,2-NQ as in Fig. 1A for the indicated time points, and cell lysates were analyzed by immunoblotting with the indicated antibodies.
Two AP-1 Sites Are Essential for 1,2-NQ-induced Il11 Gene Promoter Activity
To further clarify the mechanisms underlying 1,2-NQ-dependent Il11 expression, we tested whether 1,2-NQ could activate a luciferase reporter under the control of the −1.3 kb fragment of murine Il11 promoter (Fig. 3A). Il11 gene promoter activity was increased by 1,2-NQ treatment, and this increase was almost completely abolished in the presence of U0126 (Fig. 3B). Given that the Il11 promoter contains two AP-1 binding sites and the MAPK pathway induces activation of AP-1 (15), we introduced mutation of each element alone or a combination of them (Fig. 3A). As expected, mutation of each AP-1 element alone partially reduced, and both mutations of two AP-1 sites completely abolished, a 1,2-NQ-induced increase in promoter activities (Fig. 3C). Moreover, 1,2-NQ could activate a luciferase reporter containing the −0.8 kb fragment of the murine Il11 promoter containing two wild-type AP-1 sites but not two AP-1 mutants (Fig. 3D). These data suggest that two AP-1 sites are required for 1,2-NQ-induced Il11 induction.
FIGURE 3.
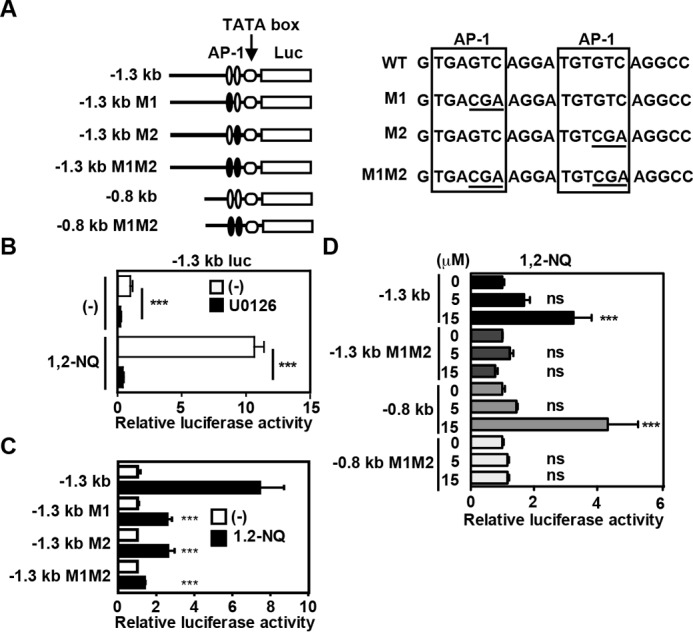
Two AP1 sites are essential for 1,2-NQ-induced IL-11 production. A, schematic diagrams of reporter vectors, sequences of two AP-1 sites, and their mutants. B, HepG2 cells were transfected with a −1.3 kb reporter vector and then untreated or treated with 1,2-NQ in the absence or presence of U0126 (20 μm) for 18 h. Luciferase activities were calculated, and relative luciferase activities are expressed as -fold activation compared with unstimulated cells. Results are mean ± S.D. (error bars) of triplicate samples. ***, p < 0.001 versus 1,2-NQ-treated cells in the absence of U0126. C, HepG2 cells were transfected with the indicated reporter vectors and then stimulated and analyzed as in B. ***, p < 0.001 versus 1,2-NQ-treated cells transfected with the −1.3 kb reporter vector. D, HepG2 cells were transfected with the indicated reporter vectors and stimulated with the indicated concentrations of 1,2-NQ and then analyzed as in B. ***, p < 0.001; ns, not significant versus untreated cells transfected with each reporter vector.
Fra-1 Is Required for Induction of Il11
The AP-1 is a dimeric transcriptional factor complex that is composed of Fos and Jun family proteins (15). Moreover, the MEK/ERK pathway regulates Fos and Jun family protein levels. We examined whether 1,2-NQ treatment might affect protein levels of members of the Fos and Jun family in CT-26 and HepG2 cells in the absence or presence of U0126. Whereas c-Fos, FosB, c-Jun, and JunD protein levels remained constant after 1,2-NQ treatment, expression of Fra-1 was increased by treatment with 1,2-NQ, and this increase was suppressed by U0126 pretreatment (Fig. 4A and supplemental Fig. 2). In addition, quantitative PCR showed that 1,2-NQ treatment increased expression of Fosl1 encoding Fra-1 protein along with Il11 expression, and this increase was suppressed by U0126 treatment in CT26 and HepG2 cells (Fig. 4B). These results suggest that 1,2-NQ up-regulated Fra-1 at both mRNA and protein levels in a MEK/ERK-dependent manner.
FIGURE 4.

Fra-1 is essential for 1,2-NQ-induced IL-11 expression. A and B, CT26 and HepG2 cells were stimulated with 1,2-NQ (50 μm) in the absence or presence of U0126 (10 μm) for the indicated times. A, cell lysates were analyzed by immunoblotting with the indicated antibodies. B, relative amounts of Il11 and Fosl1 mRNAs were determined by qPCR. Results are mean ± S.D. (error bars) of triplicate samples. *, p < 0.05; ***, p < 0.001; ns, not significant versus DMSO-treated cells. C, HepG2 cells were stimulated as in A. Nuclear extracts were prepared and preincubated with the indicated antibodies and then incubated with 32P-labeled AP-1 oligonucleotides. The DNA-protein complexes were subjected to EMSA. C and F, oligonucleotide-protein complexes and free oligonucleotides, respectively. *, nonspecific complex. D, HepG2 cells were transfected with control or FOSL1 siRNAs. At 48 h after transfection, cells were unstimulated or stimulated with 1,2-NQ (50 μm) for 4 h. Expression of Fra-1 in control or FOSL1 siRNAs-treated cells was analyzed by immunoblotting with the indicated antibodies. Relative amounts of FOSL1 and IL11 mRNAs were determined by qPCR and analyzed as in B. *, p < 0.05; ***, p < 0.001 versus 1,2-NQ-treated control siRNA-transfected cells. E, Fra-1 is recruited to the Il11 promoter following H2O2 or 1,2-NQ stimulation. CT26 cells were stimulated with H2O2 (1 mm) or 1,2-NQ (15 μm) for 2 h, and then cells were subjected to ChIP assays using control Ig or anti-Fra-1 antibody. Immunoprecipitated fragments of the Il11 promoters were determined by qPCR. Enrichment of the promoter fragments was expressed as a percentage of input. Results are mean ± S.D. of triplicate samples. **, p < 0.01; ***, p < 0.001 compared with untreated cells. C and F, control Ig and anti-FLAG antibody, respectively. All results are representative of two independent experiments.
We next performed an electrophoretic mobility shift assay (EMSA) using nuclear extracts from 1,2-NQ-treated HepG2 cells and an oligonucleotide probe containing two AP-1 sites of the Il11 promoter. 1,2-NQ induced a retarded complex containing labeled AP-1 oligonucleotides that disappeared in the presence of U0126 (Fig. 4C). Moreover, we found that the complex was supershifted with the addition of antibodies against Fra-1, JunB, or JunD, suggesting that the 1,2-NQ-induced nuclear complex is at least composed of Fra-1, JunB, and JunD. To further investigate whether these transcriptional factors are involved in Il11 expression, we knocked down expressions of each gene by respective siRNA. Whereas knockdown of Junb or Jund by the respective siRNA did not suppress, but rather enhanced, 1,2-NQ-induced Il11 expression (supplemental Fig. 3), knockdown of FOSL1 by siRNA significantly suppressed 1,2-NQ-induced FOSL1 and IL11 mRNA expression (Fig. 4D). Furthermore, a chromatin immunoprecipitation (ChIP) assay revealed that Fra-1 was recruited to the Il11 promoter following 1,2-NQ as well as H2O2 stimulation (Fig. 4E).
We finally compared expression of phosphorylated and total Fra-1 in CT-26 and HepG2 cells after treatment with 15d-PGJ2, tBHQ, and 1,2-NQ. As expected, 1,2-NQ, but not 15d-PGJ2 or tBHQ, increased both phosphorylated and total Fra-1 in CT26 cells and, to a lesser extent, HepG2 cells (supplemental Fig. 4).
NRF2 Is Required for 1,2-NQ-induced IL11 Expression
A previous study has shown that the murine glutathione S-transferase Ya subunit gene promoter contains the AP-1-like sites that partially overlapped with the antioxidant response element, and Gst Ya expression is induced by PMA and electrophiles (16). Given that one of the two AP-1 sites in the Il11 promoter overlapped with the consensus sequence of NRF2 binding sites (17) (Fig. 5A), we tested whether NRF2 could activate a luciferase reporter under the control of the Il11 promoter. Overexpression of NRF2 activated reporter vectors containing −1.3 or −0.8 kb fragments of the Il11 promoter in a dose-dependent manner (Fig. 5B). Moreover, NRF2-dependent promoter activity was abolished in a reporter vector, in which mutations of two AP-1 sites were introduced (Fig. 5B). We finally knocked down expression of NRF2 by siRNA in HepG2 cells (Fig. 5C). Knockdown of NRF2 by two different siRNAs significantly decreased expression of NRF2 mRNA and blocked 1,2-NQ-induced up-regulation of NRF2 at protein levels (Fig. 5C). Moreover, knockdown of NRF2 abolished 1,2-NQ-induced IL11 induction. In sharp contrast, H2O2-induced IL11 expression was not blocked in NRF2-knocked down cells (Fig. 5D). These results suggest that NRF2 is essential for 1,2-NQ but not H2O2-induced Il11 expression.
FIGURE 5.

NRF2 is essential for 1,2-NQ-induced IL-11 production. A, a putative NRF2-binding site is partially overlapped with the 5′ AP-1 site. B, HepG2 cells were transfected with an empty vector or graded amounts (0.1, 0.3, and 1.0 μg/sample) of an expression vector for Myc-tagged NRF2 along with the indicated reporter vectors. Luciferase activities were calculated 18 h after transfection, and relative luciferase activities are expressed as -fold activation compared with empty vector-transfected cells. Results are mean ± S.D. (error bars) of triplicate samples. ***, p < 0.001; ns, not significant versus control vector-transfected cells. C, knockdown of NRF2 by siRNA suppresses 1,2-NQ-induced Il11 mRNA induction. HepG2 cells were transfected with control siRNA or two different siRNAs against NRF2. At 48 h after transfection, cells were unstimulated or stimulated with 1,2-NQ (50 μm) for 4 h. Relative amounts of NRF2 or IL11 mRNAs were determined by qPCR. Results are mean ± S.D. of triplicate samples. ***, p < 0.001 versus control siRNA-transfected cells. Cell lysates were analyzed by immunoblotting with the indicated antibodies. D, knockdown of NRF2 by siRNA does not suppress H2O2-induced Il11 mRNA induction. HepG2 cells were transfected with siRNAs as in C and stimulated with H2O2 (1 mm) for 4 h. Relative amounts of NRF2 or IL11 mRNAs were determined by qPCR. Results are mean ± S.D. of triplicate samples. **, p < 0.01; ns, not significant versus control siRNA-transfected cells. All results are representative of two or three independent experiments.
NRF2 Is Not Recruited to the IL11 Promoter and Does Not Interact with Fra-1
The fact that NRF2 activated transcription of IL11 mRNA prompted us to tested whether NRF2 was recruited to the IL11 promoter after 1,2-NQ treatment by a ChIP assay. Anti-NRF2 antibody efficiently precipitated the promoter, but not an unrelated region of NAD(P)H:quinone oxidoreductase 1 (NQO1), a canonical target gene of NRF2, after 1,2-NQ stimulation (Fig. 6A). However, under these experimental conditions, the promoter of IL11 was not precipitated with anti-NRF2 antibody after 1,2-NQ stimulation (Fig. 6A).
FIGURE 6.

NRF2 is neither recruited to the IL11 promoter nor interacts with Fra-1. A, NRF2 is not recruited to the IL11 promoter after 1,2-NQ stimulation. HepG2 cells were stimulated with the indicated concentrations of 1,2-NQ (50 μm) for 2 h, and then cells were subjected to ChIP assays using control Ig or anti-NRF2 antibody. Immunoprecipitated fragments of the NQO1 and IL11 promoters or an unrelated region of NQO1 were analyzed by qPCR. Enrichment of the promoter fragments was expressed as a percentage of input. Results are mean ± S.D. (error bars) of triplicate samples. *, p < 0.05; ***, p < 0.001; ns, not significant versus untreated cells. Results are representative of four independent experiments. B, Fra-1 interacts with ERK2, but not NRF2. HEK293T cells were transiently transfected with the indicated expression vectors, and lysates were immunoprecipitated (IP) with the indicated antibodies. Co-immunoprecipitated proteins were analyzed by immunoblotting with the indicated antibodies. Expression of transfected proteins was verified by immunoblotting with the indicated antibodies using total cell lysates. Arrowheads and asterisks, phosphorylated and unphosphorylated Fra-1, respectively. C and F, control Ig and anti-FLAG antibody, respectively. Results are representative of three independent experiments.
We next tested the possibility that NRF2 might be recruited to the IL11 promoter via interaction with Fra-1. To test this possibility, we transiently transfected an expression vector for Myc-tagged NRF2 along with FLAG-tagged Fra-1 into HEK293T cells. Moreover, to test a possibility that phosphorylation of Fra-1 might affect its binding to NRF2, we co-transfected expression vectors for Myc-tagged NRF2, FLAG-tagged Fra-1, HA-tagged constitutively active MEKK1 (MEKK1ΔN), and HA-tagged ERK2. Consistent with our previous study (12), HA-tagged ERK2 was efficiently immunoprecipitated with FLAG-tagged Fra-1 (Fig. 6B). In sharp contrast, anti-FLAG antibody could not immunoprecipitate Myc-tagged NRF2, suggesting that NRF2 could not interact with either phosphorylated or unphosphorylated Fra-1 at least under our experimental conditions. These results suggest that NRF2 does not appear to be directly or indirectly recruited to the IL11 promoter.
Knockdown of NRF2 Abolishes Expression of Fra-1 Proteins but Not FOSL1 mRNAs
The fact that NRF2 was not recruited to the IL11 promoter following 1,2-NQ stimulation or did not interact with Fra-1 (Fig. 6) prompted us to test whether NRF2 might regulate expression of Fra-1 at mRNA or protein levels. Intriguingly, a very recent study has shown that NRF2 promotes mRNA translation of a particular set of genes (18). Although knockdown of NRF2 by siRNA did not affect FOSL1 mRNA expression (Fig. 7A), knockdown of NRF2 abolished Fra-1 expression at protein levels (Fig. 7B). In sharp contrast, expressions of c-Fos, STAT3, pERK, or total ERK were not decreased in NRF2-knockdown cells. This suggests that NRF2 might increase Fra-1 proteins through preventing degradation of Fra-1 by the ubiquitin-proteasome pathway or enhance mRNA translation of FOSL1.
FIGURE 7.
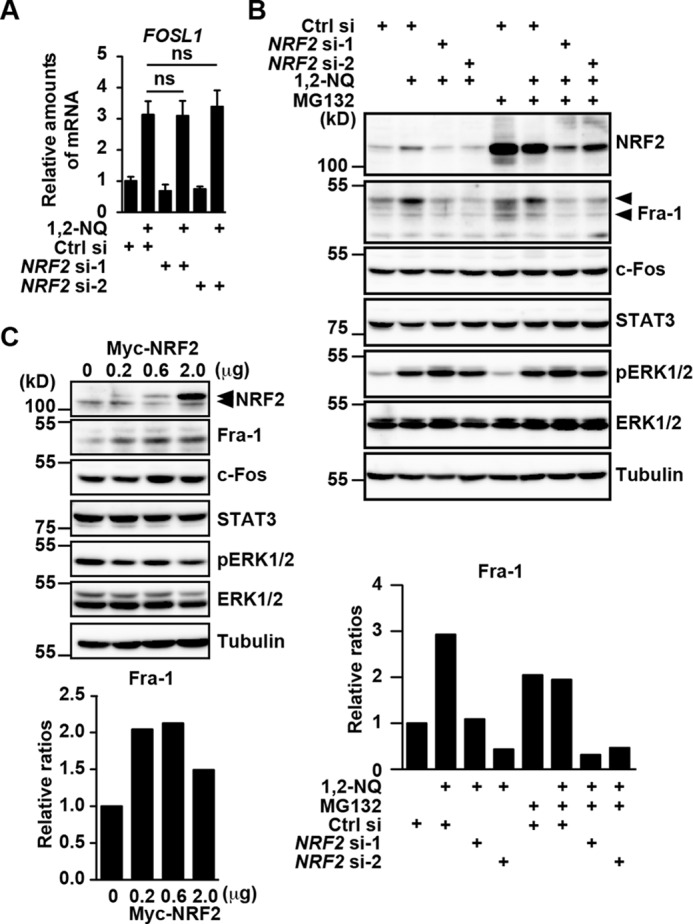
Knockdown of NRF2 abolishes expression of Fra-1 at the protein level, whereas expression of NRF2 induces up-regulation of Fra-1 proteins. A and B, HepG2 cells were transfected with siRNAs as in Fig. 5C and stimulated with 1,2-NQ (50 μm) in the absence or presence of a proteasome inhibitor, MG132 (10 μm), for 3 h. A, knockdown of NRF2 does not affect mRNA expression of FOSL1. Relative amounts of FOSL1 mRNAs were determined by qPCR. Results are mean ± S.D. (error bars) of triplicate samples. ns, not significant versus control siRNA-transfected cells. B, knockdown of NRF2 abolishes expression of Fra-1 at the protein level, but MG132 does not block a decrease in Fra-1 proteins. Cell lysates were analyzed by immunoblotting with the indicated antibodies. Top and bottom arrowheads, phosphorylated and hypophosphorylated Fra-1, respectively. Relative ratios of Fra-1/tubulin after the indicated treatment were calculated and are plotted as in supplemental Fig. 2. C, overexpression of NRF2 induces up-regulation of Fra-1 proteins. HepG2 cells were transfected with an empty vector or graded amounts (0.2, 0.6, and 2.0 μg/sample) of an expression vector for Myc-tagged NRF2, and then cell lysates were analyzed by immunoblotting with the indicated antibodies. Relative ratios of Fra-1/tubulin in cells transfected with the indicated doses of Myc-tagged NRF2 were calculated and are plotted as in supplemental Fig. 2. Top and bottom arrowheads, transfected Myc-tagged NRF2 and endogenous NRF2, respectively. All results are representative of two or three independent experiments.
To discriminate between these two possibilities, we next tested the effect of knockdown of NRF2 on expression of Fra-1 proteins in the absence or presence of a proteasome inhibitor, MG132. Because unphosphorylated Fra-1 is constitutively degraded by the ubiquitin-proteasome pathway (12), hypophosphorylated Fra-1 accumulated in the presence of MG132 (Fig. 7B). Notably, even in the presence of MG132, Fra-1 was decreased in NRF2-knockdown cells, suggesting that NRF2 did not block degradation of Fra-1 by the ubiquitin-proteasome pathway. Conversely, overexpression of NRF2 increased Fra-1, but not c-Fos, STAT3, pERK, or ERK proteins (Fig. 7C). Together, NRF2 increases expression of Fra-1, possibly through up-regulating its translation, thereby inducing IL11 expression.
Administration of 1,2-NQ Induces Il11 Expression and Enhances Proliferation of Intestinal Epithelial Cells (IECs) in the Cecum
To investigate the biological consequences of 1,2-NQ-dependent IL-11 production in vivo, wild-type mice were injected intraperitoneally with 1,2-NQ. Compared with oil-treated mice, 1,2-NQ-treated mice exhibited body weight loss 72 h after injection (Fig. 8A). To our surprise, histology of small intestine and colon appeared to be normal in 1,2-NQ-treated mice (data not shown). In sharp contrast, 1,2-NQ administration induced dilatation of the cecum and increased numbers of apoptotic IECs (Fig. 8, B–D). Moreover, numbers of Ki67- and cyclin D1-positive proliferating IECs were significantly increased in the cecum of mice compared with untreated mice (Fig. 8, E and F). Under these experimental conditions, we examined whether 1,2-NQ induced expression of Il11 and Hmox1 mRNAs in the cecum. Consistent with in vitro data, administration of 1,2-NQ into wild-type mice resulted in up-regulation of Il11 and Hmox1 mRNAs (Fig. 8G). These results suggest that 1,2-NQ administrations may enhance proliferation of IECs through up-regulation of IL-11.
FIGURE 8.

Administration of 1,2-NQ enhances apoptosis and proliferation of IECs along with Il11 up-regulation in the cecum of wild-type mice. Eight-week-old male mice were injected with corn oil or 1,2-NQ (2.5 or 5 mg/kg). In some experiments (A, B, C, and E), mice were injected with 1,2-NQ (5 mg/kg). Mice were sacrificed, and body weight was measured 72 h after injection. A, changes of body weight are expressed as percentages of initial body weight. Results are mean ± S.E. (error bars) (n = 8–11 mice). ***, p < 0.001 versus corn oil-treated mice. B, H&E-stained cecum sections (n = 7–11 mice). Arrowheads, apoptotic cells. Scale bar, 100 μm. C, cecum sections were stained with anti-CC3. Arrows, apoptotic cells. Scale bar, 100 μm. D, numbers of CC3-positive apoptotic cells were calculated and are expressed as numbers of CC3-positive cells per villus (n = 3–5 mice). *, p < 0.05; ns, not significant versus corn oil-treated mice. E, cecum sections were stained with anti-Ki67 and anti-cyclin D1 antibodies (n = 5 mice). Scale bar, 100 μm. F, numbers of Ki67-positive proliferating cells were calculated, and the percentage of Ki67-positive cells among total epithelial cells is expressed as the labeling index. *, p < 0.05 versus corn oil-treated mice. G, cecum RNAs were prepared, and relative amounts of Hmox1 and Il11 mRNAs were determined by qPCR. Results are means ± S.E. (n = 5 mice). *, p < 0.05; **, p < 0.01; ns, not significant versus corn oil-treated mice.
Exacerbation of 1,2-NQ-induced Intestinal Toxicity in Il11ra1−/− Mice
To test whether production of IL-11 by 1,2-NQ might attenuate 1,2-NQ-induced intestinal injury, we next administered 1,2-NQ into Il11ra1−/− mice. We first confirmed that numbers of apoptotic cells and proliferating cells of IECs were not different between Il11ra1−/− and Il11ra1+/− mice under normal conditions (Fig. 9, D and F). Body weight loss was significantly exacerbated in 1,2-NQ-treated Il11ra1−/− mice compared with Il11ra1+/− mice (Fig. 9A). Whereas numbers of apoptotic cells were decreased in Il11ra1−/− mice compared with Il11Ra1+/− mice (Fig. 8, B–D), numbers of Ki67- and cyclin D1-positive proliferating cells were severely decreased in the cecum of Il11ra1−/− mice compared with Il11ra1+/− mice (Fig. 9, E and F). Given that 1,2-NQ increased the number of both apoptotic and proliferating cells in wild-type mice, together these results suggest that 1,2-NQ promotes turnover of IECs, resulting in apoptosis of IECs through up-regulation of IL-11.
FIGURE 9.
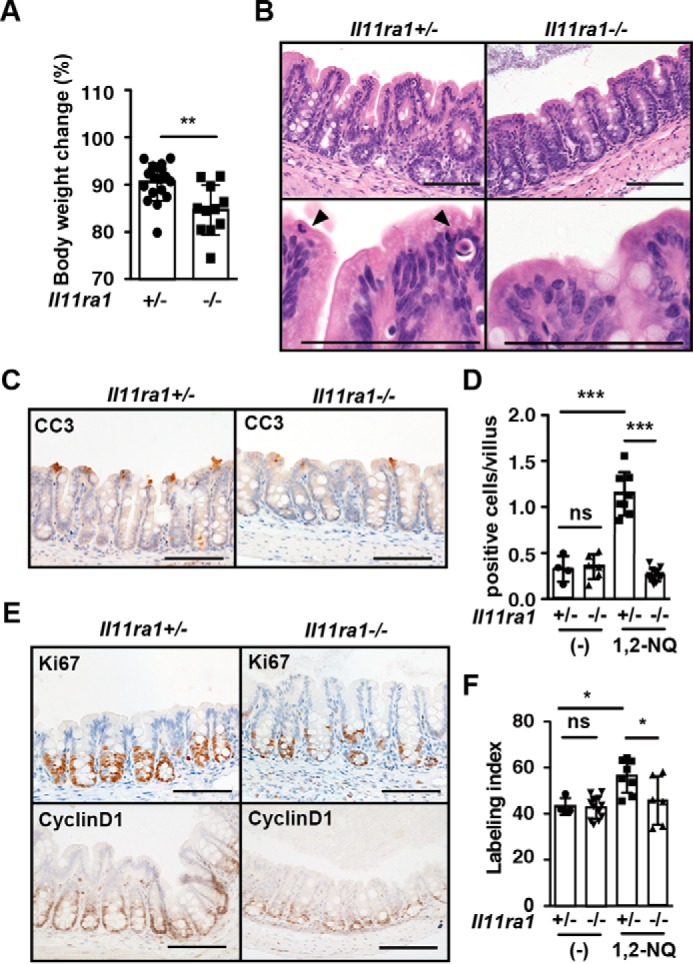
Exacerbation of 1,2-NQ-induced intestinal toxicity in I111ra1−/− mice. A, 10–12-week-old Il11ra1+/− and Il11ra1−/− mice were injected with 1,2-NQ (5 mg/kg) and analyzed as in Fig. 8. Results are means ± S.E. (error bars) (n = 9–16 mice). ***, p < 0.001; ns, not significant versus Il11ra1+/− mice. B, H&E-stained cecum section. Scale bar, 100 μm. C, cecum sections were stained with anti-CC3 antibody before (data not shown) and after 1,2-NQ injection (n = 4–12 mice/group). Scale bar, 100 μm. D, numbers of CC3-positive cells were quantified and expressed as in Fig. 8D. E, cecum sections were stained with anti-Ki67 or anti-cyclin D1 antibodies before (data not shown) and after 1,2-NQ injection. F, numbers of positive cells were quantified and expressed as in Fig. 8D. Results are mean ± S.E. (n = 4–12 mice/group). *, p < 0.05; ns, not significant versus Il11ra1+/− mice. Scale bar, 100 μm.
Discussion
In the present study, we showed that 1,2-NQ induced IL-11 expression in an MEK/ERK-dependent manner. Both Fra-1 and NRF2 were essential for 1,2-NQ-induced IL-11 production, although the mechanisms underlying Fra-1- and NRF2-dependent up-regulation of IL11 mRNA appeared to be different. Upon stimulation of cells with 1,2-NQ, Fra-1 was recruited to the Il11 promoter and acted as a transcription factor. In sharp contrast, NRF2 was not recruited to the IL11 promoter but increased translation of the FOSL1 transcript, which subsequently increased expression of Fra-1 proteins. Moreover, given that electrophile-induced intestinal toxicity was exacerbated in Il11ra1−/− mice compared with Il11ra1+/− mice, IL-11 protects cells against electrophile-induced as well as oxidative stress-induced tissue injury.
We previously reported that 1,2-NQ induces ERK activation through modification of PTP1B and subsequent activation of EGFR (14). Although 1,2-NQ, 15d-PGJ2, and tBHQ activated the NRF2 pathway, only 1,2-NQ induced IL-11 production. Although tBHQ induced prolonged ERK activation in HepG2 cells, levels of ERK phosphorylation induced by tBHQ were very low compared with those induced by 1,2-NQ (Fig. 2C). Thus, we surmise that levels of ERK phosphorylation were not sufficient to induce IL-11 production, at least under our experimental conditions. Notably, inhibition of the MEK/ERK pathway blocked 1,2-NQ-induced up-regulation of Fra-1 and IL-11 at both mRNA and protein levels. Given that knockdown of Fosl1 inhibited 1,2-NQ-induced IL-11 production, the MEK/ERK/Fra-1 pathway is crucial for 1,2-NQ- as well as H2O2-induced IL-11 production (12).
The mechanisms underlying prolonged ERK activation by 1,2-NQ, but not other agents tested here, are not fully investigated in the present study. In this respect, quinones exhibit two chemical properties, including those of an electrophile and electron transfer agent. Thus, quinones transfer electrons from a reducing agent, such as NADPH, to oxygen, resulting in generation of superoxides that are finally converted to hydrogen peroxide. Consistently, a previous study has reported that 1,2-NQ induces hydrogen peroxide-dependent oxidation of protein thiols corresponding to sulfenic acid in the cells (19). Given that hydrogen peroxide is responsible for prolonged ERK activation (12), 1,2-NQ-dependent production of hydrogen peroxide, in concert with activation of EGFR, might be responsible for 1,2-NQ-induced ERK activation. Further study is required to address this issue.
One of the most important findings of this study is that NRF2 contributes to 1,2-NQ-induced IL-11 production. An essential role for NRF2 in 1,2-NQ-induced IL-11 production is supported by the following findings. First, 1,2-NQ induced accumulation of NRF2. Second, overexpression of NRF2 activated the IL11 gene promoter activities via AP-1 sites, which contain a putative NRF2 consensus motif. Third, knockdown of NRF2 by siRNAs blocked 1,2-NQ-induced IL11 up-regulation. However, we could not detect either the recruitment of NRF2 to the IL11 promoter by a ChIP assay or interaction of NRF2 with Fra-1. To our surprise, we found that knockdown of NRF2 abolished expression of Fra-1 proteins but not FOSL1 mRNA levels. Moreover, a proteasome inhibitor did not suppress a decrease in Fra-1 in NRF2-knockdown cells, suggesting that NRF2 did not appear to up-regulate Fra-1 through preventing the degradation of Fra-1 by the ubiquitin-proteasome. A recent study has shown that NRF2 promotes mRNA translation of several genes through preventing oxidative stress-dependent blockade of assembly of the translation initiation complex in pancreatic cancer (18). Thus, it is reasonable to hypothesize that NRF2 promotes mRNA translation of FOSL1, thereby increasing protein expression of Fra-1. Further study will be required to elucidate the detailed mechanism whereby NRF2 regulates Fra-1 expression.
Constitutive activation of the NRF2 pathway and elevated expression of IL-11 are tightly associated with the development of cancer (11, 20), IL-11 might contribute, at least in part, to NRF2-dependent oncogenesis in a context-dependent manner. Intriguingly, knockdown of NRF2 could not suppress H2O2-induced IL11 up-regulation. Although Fra-1 is essential for both 1,2-NQ- and H2O2-induced IL-11 production (12) (in this study), different transcription factors induced by H2O2 or NRF2 in concert with Fra-1 might mediate IL-11 production in a stimulus-dependent manner.
Intraperitoneal injection of 1,2-NQ resulted in dilatation of the cecum along with severe weight loss of wild-type mice. We found that the number of apoptotic IECs was increased in the cecum of 1,2-NQ-treated wild-type mice compared with untreated mice. Under normal conditions, IECs move toward the tip of the villi and undergo apoptosis, followed by shedding into the lumen of the intestine. Numbers of Ki67- and cyclin D1-positive proliferating IECs were significantly increased in 1,2-NQ-treated wild-type mice compared with untreated mice, suggesting that an increase in apoptotic IECs might not be a primary event but rather a secondary event induced by an increased turnover of IECs after 1,2-NQ treatment. Consistent with this idea, numbers of both apoptotic and proliferating IECs were reduced in Il11ra1−/− mice compared with Il11ra1+/− mice. Our preliminary experiments showed that injection of recombinant IL-11 up-regulated expression of Reg3b and Reg3g in the intestines (data not shown). Given that Reg3β and Reg3γ act as antimicrobial proteins and promote tissue repair (21, 22), IL-11 might attenuate 1,2-NQ-induced intestinal toxicity through up-regulation of Reg3b and Reg3g. It is currently unknown why intestinal toxicity was relatively restricted to the cecum and not the small intestine or colon. Further study is required to address this issue.
Experimental Procedures
Reagents
U0126 (Merck), SB230580 and SP600125 (Calbiochem), 15d-PGJ2 (Cayman Chemical), tBHQ (WAKO), 1,2-NQ (Tokyo Kasei Industries), MG132 (Biomol), and LY294002 (Biomol) were purchased from the indicated sources. Antibodies used were against NRF2 (sc-13032, Santa Cruz Biotechnology, Inc.), phospho-ERK (catalog no. 9101), total ERK (catalog no. 9102), phospho-JNK (catalog no. 9251), total JNK (catalog no. 9252), phospho-p38 (catalog no. 9211), total p38 (catalog no. 9212), phospho-AKT (catalog no. 9271), total AKT (catalog no. 9272), c-Fos (sc-52, Santa Cruz Biotechnology), c-Jun (catalog no. 9162), phospho-Fra-1 (catalog no. 3880), Fra-1 (sc-605 and sc-183, Santa Cruz Biotechnology), FosB (sc-7203, Santa Cruz Biotechnology), JunB (sc-8051, Santa Cruz Biotechnology), JunD (sc-74, Santa Cruz Biotechnology), cleaved caspase-3 (catalog no. 9661), Ki67 (ab16667, Abcam), cyclin D1 (ab16663, Abcam), tubulin (T5168, Sigma-Aldrich), anti-FLAG (F3165, Sigma-Aldrich), anti-HA (3F10 and 12CA5, Roche Applied Science), anti-Myc (sc-40, Santa Cruz Biotechnology), biotin-conjugated anti-rabbit IgGs (E0432, DakoCytomation), HRP-conjugated streptavidin (P0397, DakoCytomation), and HRP-conjugated anti-rabbit IgGs (GE Healthcare). Unless otherwise indicated, antibodies were purchased from Cell Signaling Technology.
Cell Culture
CT26, HepG2, and human embryonic kidney (HEK) 293T cells were maintained in DMEM containing 10% fetal calf serum (FCS). Cells were stimulated with 15d-PGJ2 (10 μm), 1,2-NQ (50 μm), or tBHQ (100 μm) in the absence or presence of SP600125 (20 μm), SB230580 (20 μm), U0126 (20 μm), LY294002 (20 μm), or MG132 (10 μm) for the indicated times. 1,2-NQ was freshly dissolved in DMSO and used for each experiment.
Mice
Il11ra1−/− mice were provided by L. Robb and described previously (23), and they were back-crossed to C57BL/6 mice for at least 7 generations. C57BL/6 mice were purchased from Japan-SLC. All experiments were performed according to the guidelines approved by the Institutional Animal Experiment Committee of Juntendo University School of Medicine and Toho University School of Medicine.
Western Blotting
Cells were lysed in a radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 25 mm β-glycerophosphate, 1 mm sodium orthovanadate, 1 mm sodium fluoride, 1 mm PMSF, 1 μg/ml aprotinin, and 1 μg/ml leupeptin). After centrifugation, cell lysates were subjected to SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore). The membranes were immunoblotted with the indicated antibodies. The membranes were developed with Super Signal West Dura extended duration substrate (Thermo Scientific) and analyzed by a LAS4000 (GE Healthcare) or Amersham Biosciences imager (GE Healthcare). In some experiments, blots were quantified using ImageJ version 1.49 (National Institutes of Health).
Quantitative PCR (qPCR) Assays
Total RNAs were extracted from CT26 and HepG2 cells or the cecum from mice of the indicated genotype. cDNAs were synthesized with the ReverTra Ace qPCR RT kit (Toyobo). qPCR analysis was performed with the 7500 Fast Real-Time PCR detection system with the SYBR Green method of the target genes together with an endogenous control, murine Hprt or human GAPDH, with 7500 SDS software (Applied Biosystems). The following primers were used in this study: murine Hmox1, 5′-GTCAAGCACAGGGTGACAGA-3′ and 5′-ATCACCTGCAGCTCCTCAAA-3′; human HMOX1, 5′-AAGATTGCCCAGAAAGCCCTGGAC-3′ and 5′-AACTGTCGCCACCAGAAAGCTGAG-3′; murine Il11, 5′-CTGCACAGATGAGAGACAAATTCC-3′ and 5′-GAAGCTGCAAAGATCCCAATG-3′; human IL11, 5′-GTGGCCAGATACAGCTGTCGC-3′ and 5′-GGTAGGACAGTAGGTCCGCTC-3′; murine Fosl1, 5′-TAAGGCGCGAGCGGAACAAG-3′ and 5′-TCGCTGCAGCCCAGATTTCTC-3′; human FOSL1, 5′-TCGCTGCAGCCCAGATTTCTC-3′ and 5′-CTGCTGCTGTCGATGCTTG-3′; human NRF2 (NFE2L2), 5′-TGCCCCTGGAAGTGTCAAACA-3′ and 5′-CAACAGGGAGGTTAATGATTT-3′; murine Hprt, 5′-AACAAAGTCTGGCCTGTATCC AA-3′ and 5′-GCAGTACAGCCCCAAAATGG-3′; human GAPDH, 5′-AGCCACATCGCTCAGACAC-3′ and 5′-GCCCAATACGACCAAATCC-3′.
ELISA
CT26 cells and HepG2 cells were stimulated with 1,2-NQ (50 μm) for 18 h. Concentrations of murine IL-11 and human IL-11 in the culture supernatants were determined by ELISA according to the manufacturer's instructions (R&D Systems).
Knockdown of Target Genes by siRNAs
CT26 or HepG2 cells were transfected with the indicated siRNAs using Lipofectamine 2000 (Invitrogen) at a final concentration of 50 nm. At 24 or 36 h after transfection, the medium was changed to 1% FCS-containing DMEM, and then cells were stimulated with 1,2-NQ for the indicated times. Control (Gfp) siRNA (D-001300-01) and ON-TARGETplus SMARTpool siRNAs targeting human FOSL1 (L-004341) and murine Jund (L-054829) and Stealth RNAiTM siRNA negative control and Med GC and Stealth siRNAs targeting human NRF2 (NFE2L2) (HSS181506 and HSS107130) and murine Junb (MSS247328 and MSS247330) were purchased from Thermo Fisher Scientific. The efficiency of siRNAs was analyzed by immunoblotting with the indicated antibodies using cell lysates or qPCR analysis.
EMSA
EMSA was performed as described previously (24). Briefly, nuclear extracts were prepared from unstimulated or 1,2-NQ-stimulated HepG2 cells in the absence or presence of U0126. The two AP-1-containing oligonucleotides (5′-AGGGAGGGTGAGTCAGGATGTGTCAGGCC-3′ and 5′-AGGGCGGCCTGACACATCCTGACTCACCCT-3′) were labeled with T4 polynucleotide kinase in the presence of [γ-32P]ATP (PerkinElmer Life Sciences). Then nuclear extracts were incubated with the labeled oligonucleotides. The composition of the AP-1 complex was examined by supershift analysis with the indicated antibodies.
Luciferase Assay
The reporter vectors containing the murine Il11 gene promoter upstream of luciferase, including pGL3-Il11 (−1.3 kb) (−1094/+134), pGL3-Il11 (−1.3 kb, M1), pGL3-Il11 (−1.3 kb, M2), and pGL3-Il11 (−1.3 kb, M1M2), were described previously (12). M1 and M2 indicate the vectors harboring mutations of 5′ AP-1 and 3′ AP-1 sites, respectively. pGL3-Il11 (−0.8 kb) (−605/+134) and pGL3-Il11 (−0.8 kb, M1M2) (−605/+134) were generated by subcloning the PCR-amplified products containing the indicated fragments using pGL3-Il11 (−1.3 kb) (−1094/+134) and pGL3-Il11 (−1.3 kb, M1M2) as templates into pGL3-basic vector (Promega). The following primers were used in this study: pGL3-Il11 (−0.8 kb), 5′-GGGGGGCTCGAGGTGTATGTACCATCACTCTG-3′ and5′-CGGGTCGACACAGGCCAGGGGTTCCCCAGGGCA-3′. An expression vector for Myc-tagged NRF2 was generated by subcloning of the RT-PCR product of murine Nrf2 into pcDNA3-myc vector using the following primer: murine Nrf2, 5′-GCGCAATTGATGATGATGGACTTGGAGTTGCC-3′ and5′-GCGCTCGAGCTAGTTTTTCTTTGTATCTG-3′.
HepG2 cells were transfected with the indicated reporter vector along with pRL-TK vector (Promega) using Lipofectamine 2000. At 24 h after transfection, fresh growth medium was added to the transfection reaction, cells were stimulated with 1,2-NQ for 18 h, and luciferase activities were measured using the Pica Gene Dual-Luciferase kit (Toyo Ink) on a Luminometer (Berthold).
ChIP Assay
The ChIP assay was performed as described previously with slight modification (25). Briefly, HepG2 cells were unstimulated or stimulated with the indicated concentrations of 1,2-NQ for 2 h. Cells were fixed with 1% formalin for 30 min and then harvested and lysed with a ChIP assay buffer. After brief sonication by using a Bioruptor II (BM Equipment), the lysates were immunoprecipitated with control Ig or anti-NRF2 antibody. After extensive washing, immunoprecipitated DNA fragments were released and subjected to qPCR. The primers to amplify the promoter regions of IL11 (−177/−9) and NQO1 (−429/−302) and an unrelated region of the NQO1 (+8068/+8163) gene were as follows: IL11 (−162/−80), 5′-GAGCGCGGCGGCGTGAGCCCT-3′ and 5′-GACACATCCTGACTCACCCT-3′; NQO1 (−429/−302), 5′-CATGTCTCCCCAGGACTCTC-3′ and 5′-TTTTAGCCTTGGCACGAAAT-3′; NQO1 (+8068/+8163), 5′-CGTGTGTGCTTTGTGTGTGT-3′ and 5′-GCCTCCTTCATGGCATAGTT-3′. The amounts of a target DNA in immunoprecipitates with control rabbit Ig (Thermo Fisher Scientific) or anti-NRF2 antibody were quantified by qPCR using 7500 SDS software (Applied Biosystems). In brief, the ratios of the amounts of a target DNA fragment in each immunoprecipitate to those in the DNAs before immunoprecipitation (input DNA) were calculated from each cycle threshold value.
CT26 cells were stimulated with H2O2 or 1,2-NQ for 2 h and subjected to ChIP assays as described above except for using control Ig or anti-Fra-1 antibody for immunoprecipitation. The primers to amplify the promoter regions of the murine Il11 (−257/−206) gene were as follows: Il11 (−257/−206), 5′-CGGCTCGTCTGAATGGAAAA-3′ and 5′-TGACACATCCTGACTCACCC-3′.
Transient Transfection and Co-immunoprecipitation
HEK293T cells were transfected with the indicated expression vectors by polyethyleneimine “Max” (Polysciences). Expression vectors for FLAG-tagged Fra-1, HA-tagged MEKK1ΔN (constitutive active mutant), and HA-tagged ERK2 were described previously (12, 26). After an 18-h transfection, cells were lysed in Buffer A (50 mm Tris-HCl, pH 8.0, 250 mm NaCl, 0.5% Nonidet P-40, 25 mm β-glycerophosphate, 1 mm sodium orthovanadate, 1 mm sodium fluoride, 1 mm PMSF, 1 μg/ml aprotinin, and 1 μg/ml leupeptin). After immunoprecipitation with control or anti-FLAG antibodies, immunoprecipitates were subjected to SDS-PAGE, and co-immunoprecipitated proteins were analyzed by immunoblotting with anti-Myc, anti-HA, or anti-FLAG antibodies. Expression of each transfected protein in the lysates was verified by immunoblotting with the indicated antibodies.
For transient expression of NRF2, HepG2 cells were transfected with Myc-tagged NRF2 as described above. After an 18-h transfection, cells were lysed in a radioimmune precipitation assay buffer, and cell lysates were subjected to SDS-PAGE analysis.
Induction of Peritonitis
8–10-week-old WT, Il11ra1+/−, and Il11ra1−/− mice were injected intraperitoneally with 1,2-NQ (2.5 or 5.0 mg/kg) that was freshly prepared by using a Bioruptor II (BM Equipment) just before administration. Mice were sacrificed 1 or 3 days after injection.
Histological and Immunohistochemical Analyses
Small intestine, cecum, and colon were fixed in 10% formalin and embedded in paraffin blocks. Paraffin-embedded intestinal sections were used for H&E staining. Paraffin-embedded sections were stained with anti-Ki67, anti-cyclin D1, and anti-cleaved caspase 3 (CC3) antibodies and visualized with biotin-conjugated donkey anti-rabbit IgG antibody and streptavidin-conjugated HRP. Pictures were obtained with an all-in-one microscope (BZ-X700, Keyence) and analyzed with Axio version 3.0 (Zeiss).
Statistical Analyses
Statistical analysis was performed by unpaired Student's t test. In some experiments that involved more than two conditions, Tukey's analysis of variance test was performed. p values of <0.05 were considered to be significant.
Author Contributions
T. N., Y. K., and H. N. designed the research; T. N., Y. D., R. M., S. Y., and Y. K. performed the research; Y. S., K. O., and Y. K. contributed to new reagents/analytical tools; T. N., Y. K., and H. N. analyzed the data; T. N. and H. N. wrote the paper.
Supplementary Material
Acknowledgments
We thank L. Robb for providing Il11ra1−/− mice. We thank Y. Tabe, M. Nishida, H. Yagita, and H. Motohashi for helpful advice. We also thank T. Ikegami and T. Ikeda (Division of Molecular and Biochemical Research, Biomedical Research Center, Juntendo University Graduate School of Medicine) for technical support.
This work was supported in part by Grants-in-Aid for Scientific Research (B) 24390100 (to H. N.) and Scientific Research (C) 26460397 (to T. N.) and Grant-in-Aid for Young Scientists (B) 24790328 (to T. N.), and Challenging Exploratory Research 25670167 (to H. N.) from the Japan Society for the Promotion of Science (JSPS). This work was also supported by Grant-in-Aid for Scientific Research on Innovative Areas 26110003 (to H. N.) from the MEXT (Ministry of Education, Culture, Sports, Science, and Technology), Japan, the Fund for the Advancement of Science in commemoration of Toho University's 60th anniversary (to T. N.), and Project Research Grant of Toho University School of Medicine 28-3 (to T. N.). This work was also supported by research grants from the NOVARTIS Foundation for the Promotion of Science (to H. N.), the Naito Science Foundation (to H. N.), the Uehara Science Foundation (to H. N.), and the Takeda Science Foundation (to H. N.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. 1–4.
- 15d-PGJ2
- 15d-prostaglandin J2
- tBHQ
- tert-butylhydroxyquinone
- 1,2-NQ
- 1,2-Naphthoquinone
- EGFR
- EGF receptor
- PTP1B
- protein-tyrosine phosphatase 1B
- IEC
- intestinal epithelial cell
- HEK
- human embryonic kidney
- qPCR
- quantitative PCR.
References
- 1. Rudolph T. K., and Freeman B. A. (2009) Transduction of redox signaling by electrophile-protein reactions. Sci. Signal. 2, re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uchida K., and Shibata T. (2008) 15-Deoxy-Δ(12,14)-prostaglandin J2: an electrophilic trigger of cellular responses. Chem. Res. Toxicol. 21, 138–144 [DOI] [PubMed] [Google Scholar]
- 3. Fujii S., Sawa T., Nishida M., Ihara H., Ida T., Motohashi H., and Akaike T. (2016) Redox signaling regulated by an electrophilic cyclic nucleotide and reactive cysteine persulfides. Arch. Biochem. Biophys. 595, 140–146 [DOI] [PubMed] [Google Scholar]
- 4. Kansanen E., Jyrkkänen H. K., and Levonen A. L. (2012) Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Biol. Med. 52, 973–982 [DOI] [PubMed] [Google Scholar]
- 5. Marnett L. J., Riggins J. N., and West J. D. (2003) Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J. Clin. Invest. 111, 583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumagai Y., Shinkai Y., Miura T., and Cho A. K. (2012) The chemical biology of naphthoquinones and its environmental implications. Annu. Rev. Pharmacol. Toxicol. 52, 221–247 [DOI] [PubMed] [Google Scholar]
- 7. Iwamoto N., Sumi D., Ishii T., Uchida K., Cho A. K., Froines J. R., and Kumagai Y. (2007) Chemical knockdown of protein-tyrosine phosphatase 1B by 1,2-naphthoquinone through covalent modification causes persistent transactivation of epidermal growth factor receptor. J. Biol. Chem. 282, 33396–33404 [DOI] [PubMed] [Google Scholar]
- 8. Motohashi H., and Yamamoto M. (2004) Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 10, 549–557 [DOI] [PubMed] [Google Scholar]
- 9. Nguyen T., Sherratt P. J., and Pickett C. B. (2003) Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 43, 233–260 [DOI] [PubMed] [Google Scholar]
- 10. Miura T., Kakehashi H., Shinkai Y., Egara Y., Hirose R., Cho A. K., and Kumagai Y. (2011) GSH-mediated S-transarylation of a quinone glyceraldehyde-3-phosphate dehydrogenase conjugate. Chem. Res. Toxicol. 24, 1836–1844 [DOI] [PubMed] [Google Scholar]
- 11. Putoczki T. L., and Ernst M. (2015) IL-11 signaling as a therapeutic target for cancer. Immunotherapy 7, 441–453 [DOI] [PubMed] [Google Scholar]
- 12. Nishina T., Komazawa-Sakon S., Yanaka S., Piao X., Zheng D.-M., Piao J.-H., Kojima Y., Yamashina S., Sano E., Putoczki T., Doi T., Ueno T., Ezaki J., Ushio H., Ernst M., et al. (2012) Interleukin-11 links oxidative stress and compensatory proliferation. Sci. Signal. 5, ra5. [DOI] [PubMed] [Google Scholar]
- 13. Obana M., Maeda M., Takeda K., Hayama A., Mohri T., Yamashita T., Nakaoka Y., Komuro I., Takeda K., Matsumiya G., Azuma J., and Fujio Y. (2010) Therapeutic activation of signal transducer and activator of transcription 3 by interleukin-11 ameliorates cardiac fibrosis after myocardial infarction. Circulation 121, 684–691 [DOI] [PubMed] [Google Scholar]
- 14. Beei C., Iwamoto N., Inaba T., Shinkai Y., and Kumagai Y. (2013) Activation of EGFR/MEK/ERK/AP-1 signaling mediated by 1,2-naphthoquinone, an atmospheric electrophile, in human pulmonary A549 cells. J. Toxicol. Sci. 38, 793–797 [DOI] [PubMed] [Google Scholar]
- 15. Eferl R., and Wagner E. F. (2003) AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868 [DOI] [PubMed] [Google Scholar]
- 16. Friling R. S., Bergelson S., and Daniel V. (1992) Two adjacent AP-1-like binding sites form the electrophile-responsive element of the murine glutathione S-transferase Ya subunit gene. Proc. Natl. Acad. Sci. U.S.A. 89, 668–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malhotra D., Portales-Casamar E., Singh A., Srivastava S., Arenillas D., Happel C., Shyr C., Wakabayashi N., Kensler T. W., Wasserman W. W., and Biswal S. (2010) Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chio I. I., Jafarnejad S. M., Ponz-Sarvise M., Park Y., Rivera K., Palm W., Wilson J., Sangar V., Hao Y., Öhlund D., Wright K., Filippini D., Lee E. J., Da Silva B., Schoepfer C., et al. (2016) NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 166, 963–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wages P. A., Lavrich K. S., Zhang Z., Cheng W. Y., Corteselli E., Gold A., Bromberg P., Simmons S. O., and Samet J. M. (2015) Protein sulfenylation: a novel readout of environmental oxidant stress. Chem. Res. Toxicol. 28, 2411–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Menegon S., Columbano A., and Giordano S. (2016) The dual roles of NRF2 in cancer. Trends Mol. Med. 22, 578–593 [DOI] [PubMed] [Google Scholar]
- 21. Lörchner H., Pöling J., Gajawada P., Hou Y., Polyakova V., Kostin S., Adrian-Segarra J. M., Boettger T., Wietelmann A., Warnecke H., Richter M., Kubin T., and Braun T. (2015) Myocardial healing requires Reg3β-dependent accumulation of macrophages in the ischemic heart. Nat. Med. 21, 353–362 [DOI] [PubMed] [Google Scholar]
- 22. Loonen L. M., Stolte E. H., Jaklofsky M. T., Meijerink M., Dekker J., van Baarlen P., and Wells J. M. (2014) REG3γ-deficient mice have altered mucus distribution and increased mucosal inflammatory responses to the microbiota and enteric pathogens in the ileum. Mucosal Immunol. 7, 939–947 [DOI] [PubMed] [Google Scholar]
- 23. Robb L., Li R., Hartley L., Nandurkar H. H., Koentgen F., and Begley C. G. (1998) Infertility in female mice lacking the receptor for interleukin 11 is due to a defective uterine response to implantation. Nat. Med. 4, 303–308 [DOI] [PubMed] [Google Scholar]
- 24. Nakano H., Oshima H., Chung W., Williams-Abbott L., Ware C. F., Yagita H., and Okumura K. (1996) TRAF5, an activator of NF-κB and putative signal transducer for the lymphotoxin-β receptor. J. Biol. Chem. 271, 14661–14664 [DOI] [PubMed] [Google Scholar]
- 25. Katsuoka F., Motohashi H., Engel J. D., and Yamamoto M. (2005) Nrf2 transcriptionally activates the mafG gene through an antioxidant response element. J. Biol. Chem. 280, 4483–4490 [DOI] [PubMed] [Google Scholar]
- 26. Nakajima A., Komazawa-Sakon S., Takekawa M., Sasazuki T., Yeh W. C., Yagita H., Okumura K., and Nakano H. (2006) An antiapoptotic protein, c-FLIPL, directly binds to MKK7 and inhibits the JNK pathway. EMBO J. 25, 5549–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.