

Abstract
The Akt protein, a serine/threonine kinase, plays important roles in cell survival, apoptosis, and oncogenes. Akt is translocated to the plasma membrane for activation. Akt-membrane binding is mediated by direct interactions between its pleckstrin homology domain (PHD) and phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3). It has been shown that Akt activation in breast cancer cells is modulated by calmodulin (CaM). However, the molecular mechanism of the interplay between CaM and membrane binding is not established. Here, we employed nuclear magnetic resonance (NMR) and biochemical and biophysical techniques to characterize how PI(3,4,5)P3, CaM, and membrane mimetics (nanodisc) bind to Akt(PHD). We show that PI(3,4,5)P3 binding to Akt(PHD) displaces the C-terminal lobe of CaM but not the weakly binding N-terminal lobe. However, binding of a PI(3,4,5)P3-embedded membrane nanodisc to Akt(PHD) with a 103-fold tighter affinity than PI(3,4,5)P3 is able to completely displace CaM. We also show that Akt(PHD) binds to both layers of the nanodisc, indicating proper incorporation of PI(3,4,5)P3 on the nanodisc surface. No detectable binding has been observed between Akt(PHD) and PI(3,4,5)P3-free nanodiscs, demonstrating that PI(3,4,5)P3 is required for membrane binding, CaM displacement, and Akt activation. Using pancreatic cancer cells, we demonstrate that inhibition of Akt-CaM binding attenuated Akt activation. Our findings support a model by which CaM binds to Akt to facilitate its translocation to the membrane. Elucidation of the molecular details of the interplay between membrane and CaM binding to Akt may help in the development of potential targets to control the pathophysiological processes of cell survival.
Keywords: breast cancer; calmodulin (CaM); isothermal titration calorimetry (ITC); nuclear magnetic resonance (NMR); pancreatic cancer; plasma membrane; membrane nanodisc; phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3); pleckstrin homology domain (PHD)
Introduction
Growth and survival signals initiated at the cell surface by receptor-ligand interactions often involve activation of Akt, a serine/threonine kinase, which is critical in regulating cell survival, apoptosis, and oncogenes (1–3). There are three members of the Akt family, termed Akt1, Akt2, and Akt3. These isoforms are products of distinct genes but are closely related and highly conserved. However, functional isoform-specific differences concerning proliferation, apoptosis, and migration in human cancers have been identified (4–7). Akt is made up of three conserved domains, consisting of an N-terminal pleckstrin homology domain (PHD),3 a central catalytic kinase domain (KD), and a C-terminal regulatory homology domain (HM) (8, 9). Components of the Akt activation pathway are worthy anti-cancer targets because constitutive activation of Akt is a common tumorigenic event (1, 3, 10, 11). The role of Akt in signaling was first identified when it was shown to be a direct downstream effect of phosphatidylinositol 3-kinase (PI3K) (12). PI3K is a lipid kinase upstream of Akt that has been implicated in numerous cellular functions, including cell survival growth (13), proliferation (14), and differentiation (15). Akt activation is initiated upon growth factor stimulation of PI3K, which generates 3′-phosphorylated phosphoinositides such as phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3) and phosphatidylinositol 3,4-bisphosphate (PI(3,4)P2) at the plasma membrane (PM) (11, 12, 16–18). These lipids, localized in the inner leaflet of the PM, serve as docking sites for Akt, which gets targeted to the membrane via its PHD (1, 11, 19). The PHD of Akt is formed by two antiparallel sheets, a C-terminal amphipathic helix and three variable loops (VL1, VL2, and VL3). These structural elements are recognized as global features for PHDs in hundreds of proteins in mammals and bacteria (20–22). The interaction between Akt(PHD) and PI(3,4,5)P3 is considered a hallmark in the activation pathways and is required to initiate a cascade of events, including phosphorylation and downstream activation of effector proteins. The PM-anchored Akt becomes fully activated upon phosphorylation of residues Thr-308 in the KD and Ser-473 in the HM through phosphoinositide-dependent kinase 1 (PDK1) and the mammalian target of rapamycin complex 2 (mTORC2), respectively (18).
Recent studies demonstrated that membrane targeting and subsequent activation of Akt are also modulated by phosphatidylserine (PS) (23). It has been proposed that PS interacts with specific residues in the PHD and HM of Akt to promote binding to PI(3,4,5)P3. The synergy between PS and PI(3,4,5)P3 is thought to induce Akt inter-domain conformational changes required for phosphorylation of Thr-308 and Ser-473 (23). Mutagenesis studies revealed that disruption of Akt-PS interaction impairs Akt signaling and increases susceptibility to cell death, suggesting that PS is critical for Akt activation and cell survival particularly in conditions with limited PI(3,4,5)P3 availability (23). Taken together, these studies imply that a synergy between membrane lipids is perhaps needed for Akt localization and activation.
In addition to binding to lipids, an emerging role for Akt(PHD) as a multifunctional domain that can also bind to proteins is now being discovered. Studies have shown that in cells under resting conditions, the PHD interacts with its KD in a closed conformation (24–26). In that stage, Thr-308 is blocked and is inaccessible to PDK1 (26). This closed conformation is only disrupted when the PHD binds to PI(3,4,5)P3 (24, 25). This disruption is required for subsequent exposure and phosphorylation of residue Thr-308 in the KD. Mutations in the PHD that disrupt PHD-KD interaction result in constitutive Akt activation (27). Furthermore, the PHD has been shown to bind to calmodulin (CaM) to promote Akt1 (will be called Akt throughout this paper) activation in many breast cancer cells (2, 28). CaM is a ubiquitous calcium-binding protein expressed in all eukaryotic cells (29–34). Upon epidermal growth factor (EGF) stimulation of these cells, CaM co-localizes with Akt at the PM to enable membrane anchoring and activation (2). Perturbations of this targeting mechanism by a CaM antagonist inhibited Akt-CaM translocation to the PM and led to apoptotic cell death in tumorigenic mammary carcinoma cells (2, 35). These studies are consistent with the findings that CaM is overexpressed in breast tumors and breast cancer cells lines (36, 37).
Until recently, the molecular mechanisms governing the Akt(PHD)-CaM interaction were not well characterized at the biochemical, biophysical, and structural levels. Attempting to understand the mechanism by which CaM and lipids contribute into Akt activation, we have initially characterized CaM interaction with Akt(PHD) (38). We have demonstrated that CaM forms a tight complex with the Akt(PHD) with a Kd of 100 nm (38). The CaM-binding interface on Akt(PHD) was mapped to two loops adjacent to the PI(3,4,5)P3-binding site, representing a novel CaM-binding motif and suggesting a synergistic relationship between CaM and PI(3,4,5)P3 for Akt activation (38). This proposed role of CaM is consistent with the recent findings that CaM enhances PI(3,4,5)P3 binding to the PHD of a homologous protein from the Tec kinase family called interleukin-2 inducible tyrosine kinase (Itk) and vice versa (39). Conversely, these findings are inconsistent with other studies that indicated that CaM competes with PI(3,4,5)3 for Akt(PHD) thereby decreasing the affinity of PI(3,4,5)P3 to Akt(PHD) (28). Taken together, we strongly believe that further investigation is required to clarify the interplay between CaM and PM lipids in Akt activation. To understand these mechanisms, it is essential to characterize at the molecular level how CaM and lipids bind to Akt(PHD).
Here, we employed NMR, biophysical, biochemical, and in vivo tools to understand the interplay between CaM and membrane interactions with Akt. We utilized dibutanoyl-phosphatidylinositol 3,4,5-trisphosphate ((diC4-PI(3,4,5)P3), a soluble analog of PI(3,4,5)P3, with truncated acyl chains. We show the following: (i) diC4-PI(3,4,5)P3 induces major changes in Akt(PHD); (ii) the glycerol backbone and acyl chains of PI(3,4,5)P3 may contribute to the interaction; (iii) diC4-PI(3,4,5)P3 binds to Akt(PHD) with a Kd of 13 μm; and (iv) CaM does not enhance binding of PI(3,4,5)P3 to Akt(PHD). By using PI(3,4,5)P3- and PS-incorporated nanodisc membrane model, we also show that Akt(PHD) binds tightly to membrane with a Kd of 19 nm, which is able to displace CaM from Akt(PHD). Finally, we demonstrate that inhibition of Akt-CaM binding attenuated Akt activation in pancreatic cancer cells. Elucidation of the mechanism by which Akt interacts with both CaM and lipids will help in the understanding of the activation mechanism, which may provide insight for new potential targets to control the pathophysiological processes of cell survival.
Results
It is established that membrane binding of Akt is regulated by specific interactions between PI(3,4,5)P3 and the PHD. However, the molecular mechanisms governing Akt(PHD)-membrane interactions and the interplay between membrane lipids and CaM are not well characterized. This is the first study aimed at characterization of the interplay of CaM and lipid/membrane binding to Akt(PHD).
Characterization of PI(3,4,5)P3 Binding to Akt(PHD) by NMR Spectroscopy
Previous NMR studies of Akt(PHD) have been conducted with inositol 1,4,5-trisphosphate (IP3), the polar head of phosphatidylinositol 4,5-bisphosphate, or inositol 1,3,4,5-tetrakisphosphate (IP4), the polar head of PI(3,4,5)P3 (40). In another study, the structure of the Akt(PHD) in complex with IP4 was solved by X-ray crystallography (41). IP3 and IP4 both lack the glycerol backbone and acyl chains that can potentially participate in protein-lipid interactions (42–45). In addition, previous studies have shown that IP4 and PI(3,4,5)P3 can play different roles in Akt activation (46) and that IP4 binds to the PHD of protein kinases with a lesser affinity than soluble PI(3,4,5)P3 analogs with truncated acyl chains (39, 47). Therefore, it is important to determine how IP4 and diC4-PI(3,4,5)P3 interact with Akt(PHD) and whether there are differences in binding affinity.
NMR chemical shifts are very sensitive to variations in the local molecular environment. Small changes in the 1H and 15N resonances obtained by collecting HSQC spectra on protein·protein and protein·lipid complexes can be used to map the binding interface. These experiments not only allow for identification of residues involved in the interaction and/or accompanying conformational changes, but they also provide an effective method for examining the stability and folding of the protein and assessing the strength of the interaction. We initially assessed how diC4-PI(3,4,5)P3 binds to Akt(PHD) using NMR chemical shift perturbations (CSPs) as detected in the 1H-15N HSQC spectra. Because of the propensity of Akt(PHD) to self-associate and form aggregates in solution at high protein concentrations (39, 40, 48, 49), which often complicates the NMR studies, we conducted the HSQC NMR experiments at low protein concentrations (60 μm). Titration of diC4-PI(3,4,5)P3 into a 15N-labeled Akt(PHD) resulted in significant CSPs of numerous signals (Fig. 1). Chemical shift changes ceased at 4:1 diC4-PI(3,4,5)P3/PHD. Interestingly, as indicated by the chemical shift changes a subset of signals are in fast exchange on the NMR scale between the free and bound forms of Akt(PHD) (e.g. Leu-12, Phe-27, Gly-33, and Gly-37), whereas numerous others are in the intermediate-to-slow exchange (e.g. His-13, Lys-14, Arg-15, Gly-16, Trp-22, Leu-28, Lys-23, Lys-25, Ala-58, and Gln-59). CSPs have been analyzed by plotting the normalized chemical shift changes versus residue number (Fig. 2A). The most pronounced CSPs (Δδ >0.1 ppm) were observed for signals corresponding to His-13, Lys-14, Arg-15, Gly-16, Glu-17, Trp-22, Lys-23, Lys-25, Ile-36, Thr-38, Arg-41, Asp-46, Gln-47, Ser-53, Ser-56, Ala-58, Gln-59, Thr-105, Val-106, and Asp-108. To identify the interaction interface of Akt(PHD), the CSPs were mapped on the structure of Akt(PHD). As shown in Fig. 2B, the significantly perturbed residues form a well defined interaction interface (red). The PI(3,4,5)P3 site is composed of a basic patch formed by β1/β2 strands and the loop that connects them (residues 13–23 and 53). This region defines a binding site that is similar to the previously published Akt(PHD) structure with IP4 (41).
FIGURE 1.

Two-dimensional 1H-15N HSQC spectra obtained on a 15N-labeled Akt(PHD) at 60 μm as titrated with diC4-PI(3,4,5)P3 (lipid/Akt(PHD), 0:1 (black); 0. 5:1 (red); 1:1 (green); 2:1 (blue); and 4:1 (orange)). 1H-15N resonances with the most substantial chemical shift changes are highlighted in magenta. Signals labeled in blue are folded in the spectrum by 20 ppm. Notice that only the side chain H-ϵ1 signal of Trp-22 is slightly perturbed upon lipid binding.
FIGURE 2.
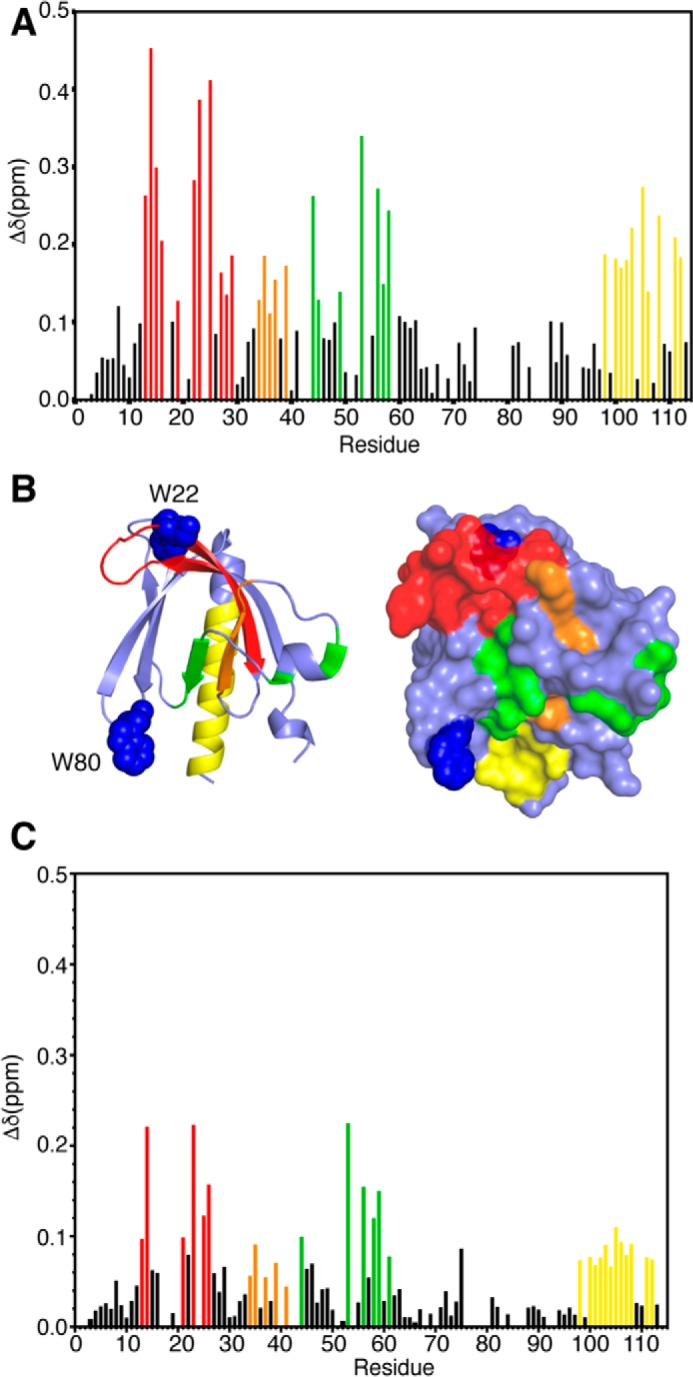
A, histogram of normalized 1H-15N chemical shift changes versus residue number calculated from the HSQC spectra for Akt(PHD) upon titration with diC4-PI(3,4,5)P3. B, Structure of the Akt(PHD) protein (Protein Data Bank code 1H10) highlighting residues that exhibited substantial (>0.1 ppm) chemical shift changes upon diC4-PI(3,4,5)P3 binding (red, residues 13–29; orange, residues 34–39; green, residues 49–58; yellow, 98–114). Tryptophan residues 22 and 80 are shown as blue spheres. C, histogram of normalized 1H-15N chemical shift changes versus residue number calculated from the HSQC spectra for Akt(PHD) upon titration with IP4.
Next, we conducted similar NMR studies with IP4. Titration of IP4 into a 15N-labeled Akt(PHD) led to significant CSPs for a number of signals. Titrations were conducted to 8:1 IP4/PHD. Further addition of IP4 led to sample precipitation. Overall, the chemical shift changes are similar to those caused by binding of diC4-PI(3,4,5)P3 (Fig. 2). The chemical shift changes in the HSQC spectra (data not shown) indicate a fast exchange, on the NMR scale, between free and bound forms. Some differences, however, were observed. For example, a subset of signals corresponding to some residues such as Arg-15, Trp-22, and Leu-28 exhibited more significant chemical shift changes upon binding of diC4-PI(3,4,5)P3 than that of IP4. NMR chemical shift changes under a fast exchange regime allowed for determination of the dissociation constant (Kd) of IP4 from Akt(PHD). Non-linear least squares fits of the titration data afforded a Kd of 202 ± 46 μm (data not shown). In contrast, determination of the Kd value from the NMR titration data of diC4-PI(3,4,5)P3 is more complicated because both fast and slow-to-intermediate exchange regimes are observed. Therefore, we resorted to ITC methods to measure the Kd and other thermodynamic parameters.
We obtained ITC data upon titration of diC4-PI(3,4,5)P3 into Akt(PHD). ITC provides values of Kd, stoichiometry of binding (n), enthalpy change (ΔH0), and the entropic terms (TΔS0). As shown in Fig. 3, binding of diC4-PI(3,4,5)P3 to Akt(PHD) is exothermic as indicated by the sign of the enthalpy. The binding data were fit into a one-site binding model and yielded the following thermodynamic parameters: Kd = 13 μm; n = 0.92 ± 0.06; ΔH0 = −2.7 kcal/mol; and ΔS0 = 13.0 cal/mol/degree. diC4-PI(3,4,5)P3 binding to Akt(PHD) is also stabilized by hydrophobic factors as indicated by the value of TΔS0 (Table 1). Collectively, our ITC data show that diC4-PI(3,4,5)P3 binds to Akt(PHD) in a 1:1 model and that both ionic and possible hydrophobic interactions contribute to the formation of the Akt(PHD)·PI(3,4,5)P3 complex. Interestingly, the affinity of diC4-PI(3,4,5)P3 binding to Akt(PHD) is 15-fold tighter than that observed for IP4, suggesting that the acyl chains and/or glycerol group contribute to the overall lipid binding. This result is also consistent with the NMR data.
FIGURE 3.
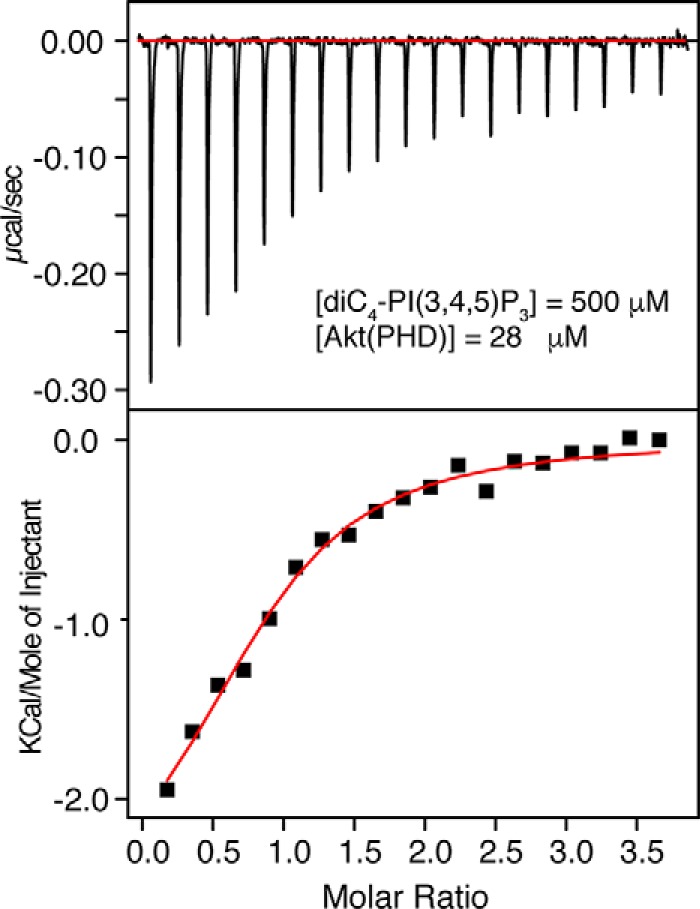
ITC data for binding of diC4-PI(3,4,5)P3 to Akt(PHD). Data were best fit to a one-site model with a Kd value of 13 μm.
TABLE 1.
Thermodynamic parameters for Akt(PHD) and Akt(PHD)-CaM binding to diC4-PI(3,4,5)P3 and nanodisc
| Lipid/membrane | Kd | ΔH | −TΔSa | ΔG | n |
|---|---|---|---|---|---|
| nm | kcal/mol | kcal/mol | kcal/mol | ||
| Akt(PHD) | |||||
| diC4-PI(3,4,5)P3 | 13,000 | −2.7 ± 0.3 | −3.8 ± 0.2 | −5.83 | 0.92 ± 0.06 |
| Nanodisc | 19 | −5.5 ± 0.2 | −5.0 ± 0.5 | −10.5 | 0.57 ± 0.02 |
| Akt(PHD)-CaM | |||||
| diC4-PI(3,4,5)P3 | 8000 | −3.1 ± 0.2 | −3.8 ± 0.3 | −6.90 | 1.0 ± 0.08 |
| Nanodisc | 200 | −4.9 ± 0.1 | −4.1 ± 0.2 | −9.01 | 0.63 ± 0.01 |
a Data were collected at 20 or 30 °C. Experiments were collected in triplicates.
Binding of PI(3,4,5)P3 and CaM to Akt(PHD), Competitive or Cooperative?
Akt(PHD) interacts with CaM to mediate membrane translocation and activation. Confocal microscopy studies have shown that upon EGF stimulation, CaM co-localizes with Akt(PHD) at the PM (2). We have recently demonstrated that Akt(PHD) binds tightly to CaM with a Kd of 100 nm and a 1:1 binding mode (38). Using NMR CSPs, we mapped the interaction region to two loops on Akt(PHD) and both lobes of CaM, establishing a novel CaM-binding motif. We found that the C-terminal lobe of CaM (CaM-C) recognizes the loop connecting β1 and β2 strands, whereas the N-terminal lobe (CaM-N) bind to the loop connecting β6 and β7 strands (38). Interestingly, this region is adjacent to the diC4-PI(3,4,5)P3-binding site described above. When compared with the HSQC spectra of the Akt(PHD)·CaM complex (38), many of the residues that are significantly affected by CaM binding are different from those affected by diC4-PI(3,4,5)P3 binding. For example, the side chain (ϵ1) 1H-15N resonances corresponding to Trp-80 and Trp-22 exhibited significant chemical shift changes upon binding of CaM but not diC4-PI(3,4,5)P3 (Fig. 1A). Residues that are involved in lipid binding such as Lys-14, Arg-15, Arg-23, Arg-25, and Asn-53 exhibit significant chemical shift changes when titrated with diC4-PI(3,4,5)P3 but not with CaM (Figs. 1 and 2). The aim here was to assess whether binding of CaM and diC4-PI(3,4,5)P3 to Akt(PHD) is competitive or cooperative and whether both CaM and diC4-PI(3,4,5)P3 are able to bind simultaneously to Akt(PHD).
To do so, we conducted 2D HSQC NMR experiments on a 15N-labeled Akt(PHD) sample upon binding to unlabeled CaM and diC4-PI(3,4,5)P3 (Fig. 4). First, unlabeled CaM was added to 15N-labeled samples of Akt(PHD) at 1:1 Akt(PHD)/CaM ratio followed by acquisition of 2D 1H-15N HSQC data. As expected, substantial chemical shift changes were observed for most of the 1H-15N resonances, indicating direct binding. Second, diC4-PI(3,4,5)P3 was titrated to the Akt(PHD)·CaM sample followed by acquisition of 1H-15N HSQC data (Fig. 4A). At 0.5:1:1 diC4-PI(3,4,5)P3/Akt(PHD)/CaM, signal intensity decreased for several signals (data not shown). Further addition of diC4-PI(3,4,5)P3 led to the appearance of several new peaks. Remarkably, at 1:1:1 diC4-PI(3,4,5)P3/Akt(PHD)/CaM, two separate resonances (doubling) for most of the signals (green) are observed (Fig. 4). Because the NMR spectra were quite complex to analyze, we picked a few signals corresponding to residues within the lipid-binding site (e.g. Lys-14, Arg-15, Arg-23, and Leu-28) to highlight these spectral changes (Fig. 4B). The chemical shifts of the doubling signals correspond to either CaM- or diC4-PI(3,4,5)P3-bound forms of Akt(PHD). This result indicates that peak doubling of these residues represents an equilibrium exchange of two conformational states, with one state representing the Akt(PHD)·diC4-PI(3,4,5)P3 complex and the other state representing the Akt(PHD)·CaM complex. Upon further addition of diC4-PI(3,4,5)P3 (1:1:2 CaM/PHD/lipid), signals representing the CaM/PHD disappeared, and those corresponding to the Akt(PHD)·diC4·PI(3,4,5)P3 complex became more intense, suggesting that diC4-PI(3,4,5)P3 binding led to displacement of CaM from Akt(PHD). No spectral changes were observed upon further addition of diC4-PI(3,4,5)P3.
FIGURE 4.

A, overlay of 2D 1H-15N HSQC spectra obtained for a 15N-labeled Akt(PHD) sample (60 μm) in the free state (black) and when in complex with Ca2+/CaM (red). diC4-PI(3,4,5)P3 was then added at a 1:1:1 (green) and a 2:1:1 ratio (orange) (diC4-PI(3,4,5)P3/Akt(PHD)/CaM) followed by acquisition of 2D 1H-15N HSQC. Spectra of selected regions showing signals of residues affected by either binding of lipid (B) or CaM (B) are shown. Red and magenta lines indicate the positions of signals of CaM- or lipid-bound states, respectively. diC4-PI(3,4,5)P3 binding to Akt(PHD) displaces the C-terminal lobe of CaM as signals of residues in this region (β1/β2) shift to the lipid bound form (B). However, lipid binding is not able to displace the distant N-terminal lobe of CaM (C).
The above result was unexpected because CaM affinity to Akt(PHD) is ∼103 times higher than that of PI(3,4,5)P3. One possible model to explain the above observation is that CaM binding to Akt(PHD) may induce conformational changes in Akt(PHD) to enhance lipid binding. To test this hypothesis, we have obtained ITC data upon titration of diC4-PI(3,4,5)P3 into the pre-formed Akt(PHD)·CaM complex under the same buffer conditions (Fig. 5). The binding data were fit into a one-site binding model and yielded the following thermodynamic parameters: Kd = 8 μm; n = 1.04 ± 0.08; ΔH0= −3.05 kcal; and ΔS0 = 12.9 cal/mol/degree. Interestingly, as indicated by the Kd values, the affinity of lipid binding to the Akt(PHD)·CaM complex is only slightly higher than that of Akt(PHD), suggesting that CaM does not significantly enhance lipid affinity to Akt(PHD).
FIGURE 5.
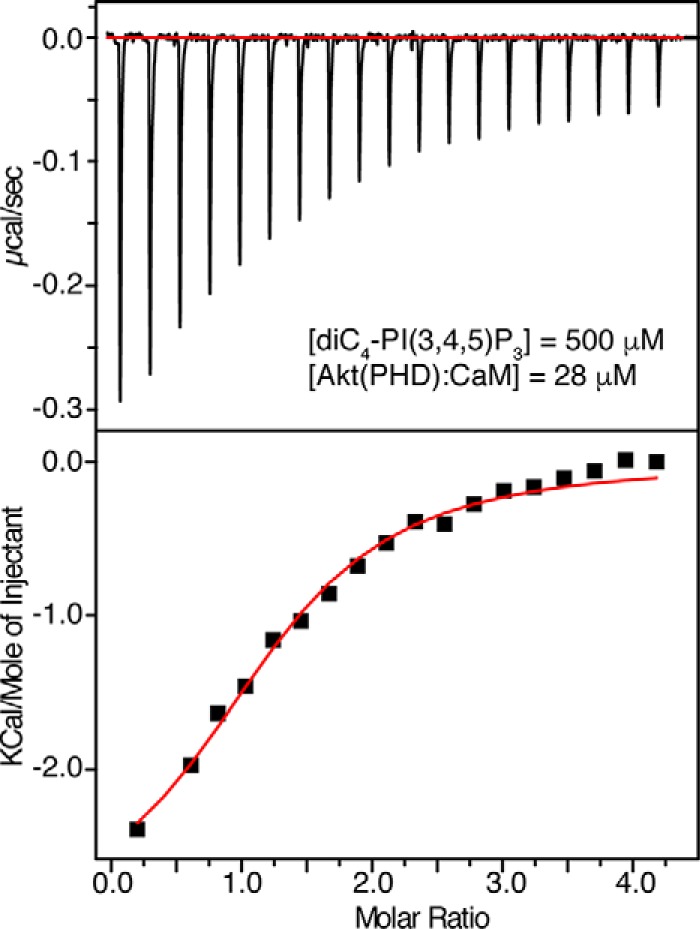
ITC data for binding of diC4-PI(3,4,5)P3 to Akt(PHD)·CaM complex. Data were best fit to a one-site model with a Kd value of 8 μm. The binding affinity is very similar to that obtained for diC4-PI(3,4,5)P3 titration to free Akt(PHD) in Fig. 2, indicating that CaM has minimal effect on lipid binding.
Another possible scenario for the above observation is that the lipid only displaces CaM-C, which is close to the lipid-binding site, but not CaM-N, which is far removed from it. To examine whether this is the case, we picked a few other signals corresponding to Akt(PHD) residues Thr-81, Thr-82, Ile-84, and Gln-113 that are distant from the PI(3,4,5)P3-binding site but are located in the β6 and β7 loop that binds to CaM-N (38). As shown in Fig. 4C, addition of diC4-PI(3,4,5)P3 did not affect these signals, suggesting that the β6 and β7 loop is still bound to CaM-N. Altogether, NMR data suggest that diC4-PI(3,4,5)P3 partially displaces CaM by only displacing the CaM-C lobe.
To determine whether CaM-N is still bound to Akt(PHD) upon lipid titration, we collected the 2D HSQC data on a 15N-labeled CaM-N sample as a function of added Akt(PHD) and diC4-PI(3,4,5)P3 (Fig. 6). As expected, addition of unlabeled Akt(PHD) to CaM-N led to modest chemical shift changes in the HSQC spectra, because CaM-N binds much weaker (112 μm) to Akt(PHD) than full CaM (38). Next, diC4-PI(3,4,5)P3 was titrated into the complex to saturation (Fig. 6). The 1H and 15N signals did not shift to positions similar to those observed for free CaM-N upon addition of diC4-PI(3,4,5)P3, indicating that lipid does not displace CaM-N from Akt(PHD). In a control experiment, no changes were observed upon diC4-PI(3,4,5)P3 titration into CaM. Based on these findings, we conclude that the ternary complex formation is not cooperative and that diC4-PI(3,4,5)P3 is able to compete with CaM, or at least displace the CaM-C lobe adjacent to the lipid-binding site.
FIGURE 6.
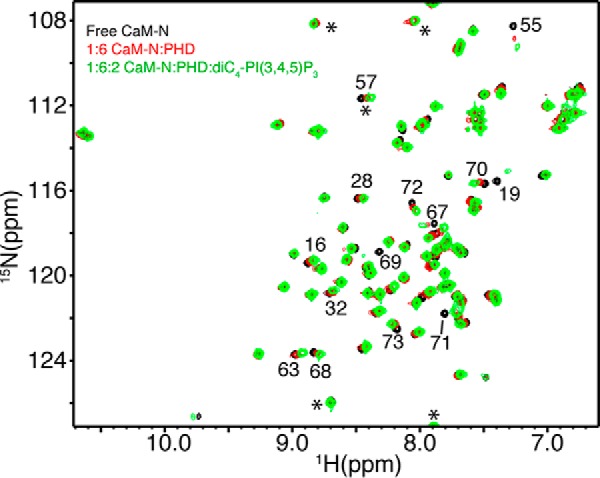
Two-dimensional 1H-15N HSQC spectra obtained on a 15N-labeled CaM-N at 60 μm as titrated with Akt(PHD). Spectra in black and red were obtained for CaM-N in the free state and in complex with excess Akt(PHD) to ensure saturation (1:6 ratio). The spectrum in green was obtained for the CaM-N·Akt(PHD) complex after diC4-PI(3,4,5)P3 was added at 2:1. The 1H-15N signals of CaM-N do not revert back to the positions observed for free CaM-N, indicating that Akt(PHD) is still bound to CaM-N upon titration of diC4-PI(3,4,5)P3. Signals denoted by asterisks are folded in the spectrum by 20 ppm.
Membrane Nanodisc Interactions with Akt(PHD)
The results above confirm that diC4-PI(3,4,5)P3 can potently compete with CaM to bind to Akt(PHD). However, to better understand the mechanism of Akt-membrane in the context of a biologically relevant system, we conducted these competition experiments with membrane nanodiscs with biologically relevant lipid composition. In these experiments, we also take into account the recent findings showing that PS enhances Akt(PHD)-membrane binding (23). Therefore, we wanted to assess the molecular mechanism of Akt(PHD) and Akt(PHD)·CaM binding to the membrane mimetic model containing PI(3,4,5)P3 and PS with native acyl chains. Phosphoinositides containing detergent micelles and liposomes have been previously used to study Akt-membrane interactions (23). However, these systems can have some drawbacks because detergents are not homogeneous and may affect protein stability and folding. Liposomes, however, are large with diameters on the order of microns and contain thousands to millions of phospholipids, which may not allow us to precisely characterize their binding to Akt(PHD). Therefore, we have utilized stable and homogeneous membrane nanodiscs, which have been used successfully in structural and biophysical studies (50–55).
Binding of nanodiscs to Akt(PHD) and Akt(PHD)·CaM has been examined by a gel filtration assay, ITC, and cobalt affinity pulldown assay. Two membrane nanodisc models with or without DPPI(3,4,5)P3 have been prepared as described previously (51, 56). All nanodiscs were run on a size exclusion column (Superdex 200) to ensure homogeneity. As shown in Fig. 7A, a single elution peak of 1:78:20:2 MSP1D1/POPC/POPS/DPPI(3,4,5)P3 nanodisc was observed at 45.5 ml. A sample mixture containing equimolar amounts of MSP1D1/POPC/POPS/DPPI(3,4,5)P3 nanodisc and Akt(PHD) at 70 μm was prepared and loaded onto a gel filtration column (Superdex 200). The nanodisc·Akt(PHD) complex yielded a peak at 45.2 ml and a broad peak at ∼85 ml (Fig. 7A). SDS-PAGE analysis of peak fractions indicate complex formation between Akt(PHD) and nanodisc (Fig. 7B). These results demonstrate direct binding of Akt(PHD) to nanodisc. SDS-PAGE data revealed that the protein eluting at ∼85 ml is unbound Akt(PHD). To test whether Akt(PHD) binds to nanodisc in the absence of DPPI(3,4,5)P3, a mixture of equimolar amounts of Akt(PHD) and nanodisc made of MSP1D1/POPC/POPS were mixed and run on size exclusion chromatography. Two peaks indicating nanodisc and Akt(PHD) were observed (data not shown). Analysis of the fractions via SDS-PAGE showed no evidence of Akt(PHD)-nanodisc binding. Our data unequivocally validate our approach and demonstrate that DPPI(3,4,5)P3 is required for Akt(PHD) binding to membranes.
FIGURE 7.
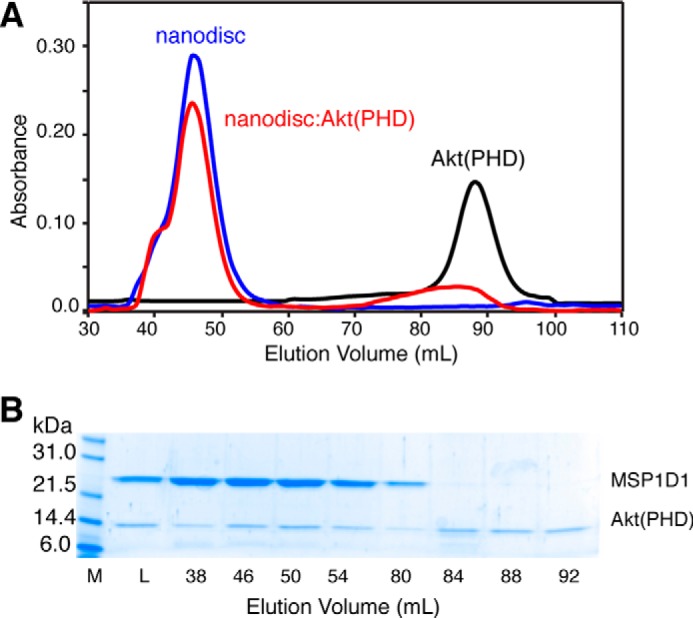
A, elution profiles of MSP1D1/POPC/POPS/DPPI(3,4,5)P3 nanodisc, Akt(PHD), and their mixture at 1:1 on a HiLoad Superdex 200 column. The elution volume of nanodisc is consistent with formation of a homogeneous nanodisc. B, fractions were analyzed by SDS-PAGE. Notice that Akt(PHD) elutes with nanodisc. Unbound Akt(PHD) is also detected. M, marker; L, load.
Next, we aimed at obtaining quantitative data on nanodisc binding to Akt(PHD), including binding affinity and stoichiometry. We resorted to ITC methods as it provides such information and other thermodynamic properties. A sample of MSP1D1/POPC/POPS/DPPI(3,4,5)P3 nanodisc at 60 μm was titrated into Akt(PHD) at 10 μm at identical buffer conditions (Fig. 8A). This experiment was collected in triplicate and yielded similar values. Data were best fit to one-site binding model and yielded a Kd value of 19 nm, an affinity that is ∼103 tighter than that obtained for diC4-PI(3,4,5)P3 titration. As indicated by the ΔH0 value, the interaction is exothermic and enthalpically driven (Fig. 8A and Table 1). This result is striking and suggests that Akt(PHD) interaction with the membrane is governed by multiple forces. Of particular note, the stoichiometry of binding (n) value is 0.57, which suggests that two molecules of Akt(PHD) bind to one nanodisc. The n value indicating the binding stoichiometry suggests that two Akt(PHD) molecules bind to one nanodisc, perhaps one molecule per membrane layer. ITC experiments conducted in the absence of DPPI(3,4,5)P3 showed no detectable binding (Fig. 8A), again demonstrating that PI(3,4,5)P3 is indispensable for Akt(PHD) binding to membranes.
FIGURE 8.
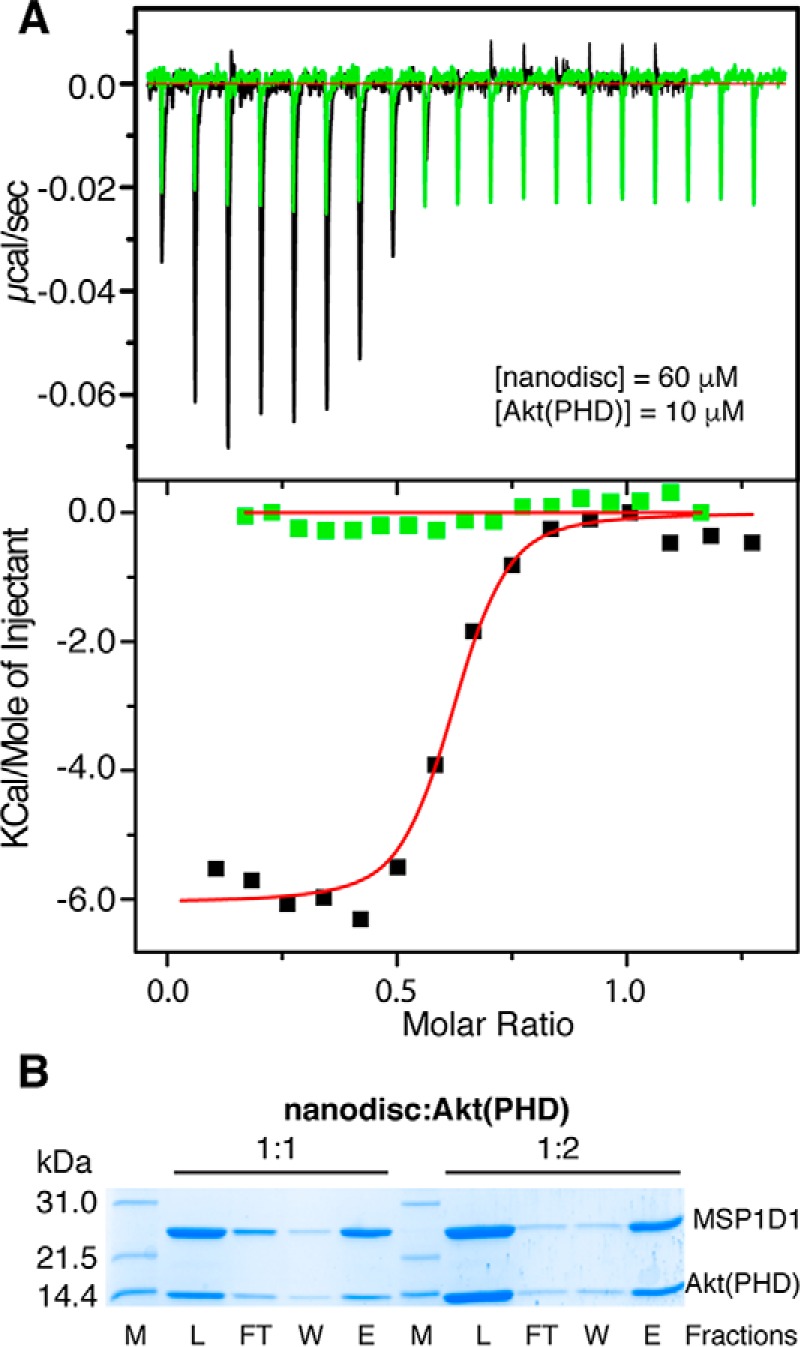
A, ITC data obtained for titration of MSP1D1/POPC/POPS/DPPI(3,4,5)P3 nanodisc at 60 μm into 10 μm Akt(PHD) (black). Data fitting afforded a Kd value of 19 nm. However, no binding is detected upon titration of MSP1D1/POPC/POPS nanodisc at 210 μm into 18 μm Akt(PHD) (green), indicating that DPPI(3,4,5)P3 is indispensable for binding. B, SDS-PAGE analysis of cobalt affinity pulldown of Akt(PHD) with nanodisc. Notice that the band representing Akt(PHD) is half of the intensity of MSP1D1 at 1:1 but is equally intense at 1:2 (nanodisc/Akt(PHD)), indicating that two molecules of Akt(PHD) bind to nanodisc. Protein marker, load, flow-through, wash, and elution fractions are denoted as M, L, FT, W, and E, respectively.
To further assess the stoichiometry of Akt(PHD)-nanodisc binding, we employed a cobalt-affinity pulldown assay. A DPPI(3,4,5)P3-nanodisc was immobilized onto a cobalt resin via a His6 tag fused to the N terminus of the MSP1D1 protein. It is important to note that a nanodisc contains two copies of the MSP1D1 protein. Akt(PHD) was added to the immobilized nanodisc at a 1:1 or 1:2 ratio of nanodisc/Akt(PHD). Fractions of the flow-through, washes, and elutions were analyzed via SDS-PAGE. As shown in Fig. 8B, two bands representing nanodisc and Akt(PHD) are observed. As expected, at 1:1 nanodisc/Akt(PHD) the MSP1D1 band representing the nanodisc is more intense than the Akt(PHD) because two molecules of MSP1D1 wrap around the phospholipid bilayer. However, at 1:2 nanodisc/Akt(PHD) the intensity of the MSP1D1 protein is very similar to that of the Akt(PHD) band indicating similar protein ratios. Consistent with the ITC data, the cobalt pulldown data indicate that two molecules of Akt(PHD) are bound to the nanodisc.
Does Membrane Nanodisc Displace CaM?
To elucidate the molecular mechanism of Akt activation, it is essential to understand the interplay of CaM and membrane binding to Akt(PHD). The NMR experiments performed with the diC4-PI(3,4,5)P3 lipid show that the lipid is able to partially displace CaM by competing with the C-terminal lobe. However, we wanted to examine this competition by using a nanodisc model, which is more biologically relevant. One aim was to test whether both CaM and nanodisc could simultaneously bind to Akt(PHD) and to determine whether CaM enhances or inhibits lipid binding to Akt(PHD). To do so, nanodisc was added to a pre-formed Akt(PHD)·CaM complex at 1:1:1 stoichiometry and loaded onto the size exclusion column (Superdex 200). As shown in the elution profile in Fig. 9A, two peaks at 45.5 and 62.1 ml were observed. SDS-PAGE analysis demonstrates that two different complexes were formed, a Akt(PHD)·nanodisc complex at 45.5 ml (fractions 38–54) and Akt(PHD)·CaM complex at 62.1 (fractions 60–70) (Fig. 9, A and B). No CaM protein is detected in the Akt(PHD)-nanodisc fractions (38–54), indicating that CaM is not associated with the Akt(PHD)·nanodisc complex.
FIGURE 9.

A, elution profiles of Akt(PHD)·CaM, MSPI/POPC/POPS/DPPI(3,4,5)P3, and their mixtures loaded on the size exclusion column (Superdex 200). B, fractions were analyzed via SDS-PAGE. Notice that no CaM is detected in the first peak eluting at 45.2 ml, indicating that membrane binding to Akt(PHD) displaced the CaM protein. C, SDS-PAGE of cobalt affinity pulldown of the Akt(PHD)·CaM complex with nanodisc. Again, no CaM is detected in the elution fraction. Protein marker, flow-through, wash, and elution fractions are denoted as M, FT, W, and E, respectively. D, ITC data obtained for titration of MSP1D1/POPC/POPS/DPPI(3,4,5)P3 nanodisc at 210 μm into 25 μm Akt(PHD)·CaM complex. Data fitting afforded an apparent Kd value of 200 nm. E, SDS-PAGE of nickel affinity pulldown assay of Akt(PHD)·nanodisc complex in the presence of CaM. Akt(PHD)·nanodisc complex was prepared at 2:1 stoichiometry (lane 1). CaM was then added at 2:2:1 (CaM/PHD·nanodisc) (lane 2). Lanes 3, 4–6, and 7 indicate flow-through, washes 1–3, and elution fractions. As shown, all Akt(PHD) is still captured by the nanodisc as CaM is not able to pull it down. L, load.
To confirm this result, we recapitulated this finding using a co-affinity pulldown assay. Solution fractions of the Akt(PHD)·CaM complex were added onto a nanodisc immobilized on a cobalt resin at 1:1 stoichiometry. SDS-PAGE data show that nanodisc binding displaces CaM (Fig. 9C). However, consistent with our gel filtration data, we observe a fraction of the Akt(PHD) protein in the flow-through fractions with CaM (Fig. 9C) suggesting that both CaM and PI(3,4,5)P3 may have a similar affinity for Akt(PHD). ITC data show that nanodiscs bind to Akt(PHD) 10-fold tighter than CaM (18 versus 195 nm at 150 mm NaCl). Therefore, we wanted to examine whether CaM binding to Akt(PHD) enhanced or decreased affinity of binding to nanodisc. We obtained ITC data upon titration of nanodisc into the Akt(PHD)·CaM complex (Fig. 9D). Data fit best to a one-site model and provided an apparent Kd of 200 nm, which suggests that CaM may moderately decrease lipid binding to Akt(PHD). However, the ITC data can also be complicated by the fact the heat generated in the cell is a combined effect of formation of two different complexes, nanodisc·Akt(PHD) and CaM·Akt(PHD).
Next, we sought to find out whether CaM is able to capture Akt(PHD) when prebound to nanodisc. This experiment is important because it may help in understanding at the cellular level whether CaM can capture Akt when bound to the PM. To do so, we designed a cobalt pulldown completion assay in which a preformed nanodisc containing 2% PI(3,4,5)P3 was first bound to Akt(PHD). CaM was then added to the complex. The resulting complex was bound to cobalt resin, washed, and eluted, and fractions were analyzed by SDS-PAGE. As shown in Fig. 9E, the CaM protein was washed out, whereas Akt(PHD) remained exclusively captured by nanodisc. This result indicates that CaM is not able to bind and pull down Akt(PHD) when it is bound with membrane. In summary, our results demonstrate that Akt(PHD) has a higher affinity to membranes containing PI(3,4,5)P3 and that CaM is displaced and cannot bind simultaneously, a result that has potential biological implications on Akt activation.
CaM Inhibition Attenuates Akt Activation in Pancreatic Cancer Cells
To evaluate the functional role of CaM in Akt activation in cancer cells, we determined the effect of CaM inhibition on the activation of Akt by EGF, a growth factor that has been shown to activate Akt in breast cancer cells (2, 28). We found that EGF induced Akt phosphorylation at both Ser-473 and Thr-308 in pancreatic cancer MiaPaCa-2 cells (Fig. 10A). Inhibition of CaM by using trifluoperazine (TFP) markedly attenuated EGF-induced Akt phosphorylation and activation. Using immunofluorescent staining and confocal microscopy, we further confirmed that EGF induced phosphorylation and activation of Akt. Furthermore, we show that CaM co-localizes with Akt on the membrane in MiaPaCa-2 cells (Fig. 10B). Immunoprecipitation analysis further shows a direct interaction of CaM and Akt upon EGF stimulation (Fig. 10C), which is inhibited by TFP. Taken together, these results demonstrate that CaM·Akt binding is important for regulating Akt activation in pancreatic cancer cells.
FIGURE 10.

CaM inhibition attenuates Akt activation. Pancreatic cancer cells, MiaPaCa-2, were exposed to serum-free media with or without TFP (10 μm) for 30 min and subsequently exposed to EGF (20 ng/ml) with or without TFP. A, Western blotting analysis of the phosphorylation of Akt at Ser-473 and Thr-308 in response to EGF for up to 15 min. B, confocal imaging analysis of EGF-induced Akt phosphorylation (pAkt, red) and co-localization of pAKT (red) with CaM (green) at 5 min. Enlarged image shows co-localization of pAkt with CaM. Scale bar, 10 μm. C, interaction of CaM and Akt, assessed by immunoprecipitation (IP) with CaM antibody followed by Western blotting analysis of CaM and Akt with specific antibodies. Representative results of at least three independent experiments are shown.
Discussion
Akt plays a key role in many cellular processes, including cell survival, apoptosis, and oncogenesis. Growth and survival signals initiated at the cell surface often involve activation of Akt, which is required for Akt to directly phosphorylate its downstream cellular targets such as the BH3-only protein BAD, glycogen synthase kinase 3, and tuberous sclerosis complex 2 (1–3, 11, 57). The PHD, a conserved structural fold found in organisms ranging from mammals to bacteria (20–22), binds specifically to PI(3,4,5)P3 on the inner leaflet of the PM (1, 11, 19). PS has also been shown to enhance Akt(PHD) binding to membrane (23). The mechanisms of Akt translocation to the PM for phosphorylation and subsequent activation of its effector proteins are critical steps in the activation pathway. Understanding these mechanisms became even more complex after the discovery of a role of CaM in mediating Akt-membrane translocation and activation in breast cancer cells (2, 35). Disruption of the Akt-CaM interaction using a CaM antagonist blocked translocation to the membrane and led to apoptotic cell death in tumorigenic mammary carcinoma cells (2, 35).
To elucidate the mechanism of Akt activation, it is essential to characterize the molecular requirements for these interactions. In a recent report (38), we have characterized the interactions between CaM and Akt(PHD) by NMR, biochemical, and biophysical methods. We have shown that CaM binds tightly to Akt(PHD) (Kd = 100 nm). The interaction between CaM and Akt(PHD) is enthalpically driven and dependent on salt concentration, suggesting that electrostatic interactions contribute to the formation of the complex. By using NMR CSPs, we mapped the CaM-binding interface in Akt(PHD) to two loops, of which one of them (connecting β1/β2 strands) is adjacent to the PI(3,4,5)P3-binding site. We have also shown that Akt(PHD) engages both CaM lobes simultaneously.
In this study, we employed a battery of methods and techniques to elucidate the interplay between CaM and membrane interactions with Akt(PHD). Structural characterization of protein·lipid complexes is often performed with either the polar head of membrane lipids or other soluble analogs bearing truncated acyl chains. Previous NMR and X-ray crystallography studies of Akt(PHD) have used IP3 and IP4, respectively (40, 41). Previous studies have shown that IP4 and PI(3,4,5)P3 can play different roles in Akt activation (46) and that IP4 binds to the PHD of protein kinases with a lesser affinity than soluble PI(3,4,5)P3 analogs with truncated acyl chains (39, 47). Herein, we show that although both bind to the same site, diC4-PI(3,4,5)P3 binds to Akt(PHD) 15-fold tighter than IP4, which suggests that the acyl chains and glycerol group contribute to binding. Indeed, several additional residues adjacent to the lipid-binding site exhibited chemical shift changes suggesting a direct contact with the acyl chains and glycerol group.
To be able to understand the role of CaM in Akt activation, it is important to assess whether CaM and membrane binding to Akt(PHD) is competitive or cooperative. Our NMR competition assay demonstrates that the ternary complex formation is not cooperative and that diC4-PI(3,4,5)P3 competes with CaM to bind to Akt(PHD). The finding that diC4-PI(3,4,5)P3 displaces only the C-terminal lobe of CaM was interesting because CaM binds tighter than the lipid. Therefore, it was necessary to establish a competition assay with membranes containing biologically relevant lipid composition, including PI(3,4,5)P3 and PS. By employing a nanodisc binding assay, we demonstrate that Akt(PHD) binds very tightly to the membrane with a Kd of 19 nm. This interaction is exothermic and enthalpically driven, suggesting that multiple forces govern Akt(PHD)-membrane interaction. We provide evidence that CaM is fully displaced upon Akt(PHD) binding to nanodisc.
Our findings have biological implications on Akt activation because we provide the first mechanistic evidence for the interplay of both CaM and membrane binding to Akt(PHD). Furthermore, we demonstrate that inhibition of CaM·Akt binding attenuated Akt activation in pancreatic cancer cells, which is also associated with disruption of membrane co-localization of CaM and Akt. Our results are consistent with previous observations in breast cancer cells that a CaM antagonist prevented translocation of both Akt and CaM to the PM (2). These results suggest that CaM binds to Akt(PHD) in the cytoplasm and mediates its transport to the PM. Studies have shown that under no stimulus in the cytoplasm, the PHD interacts with the KD in a closed formation (24, 25). The Akt(PHD)-KD interface was mapped to β1/β2 strands and VL1 and VL3 of Akt(PHD) with the PI(3,4,5)P3 binding pocket partially blocked (26). Interestingly, we mapped the CaM binding domain to the VL1 and VL3 loops of Akt(PHD) (38). We propose a model (Fig. 11) by which CaM binding to Akt(PHD) disrupts its interaction with the KD, leading to enhancement of membrane targeting and localization because studies have shown that mutations that disrupt the Akt(PHD)-KD leads to constitutive Akt activation (27). Indeed, this role is consistent with the finding that membrane binding to Akt(PHD) disrupts the Akt(PHD)-KD interaction (24, 25). Confocal microscopy studies have shown that upon EGF stimulation, CaM co-localizes with Akt(PHD) at the PM suggesting that CaM may play a role in shuttling of Akt to the membrane.
FIGURE 11.
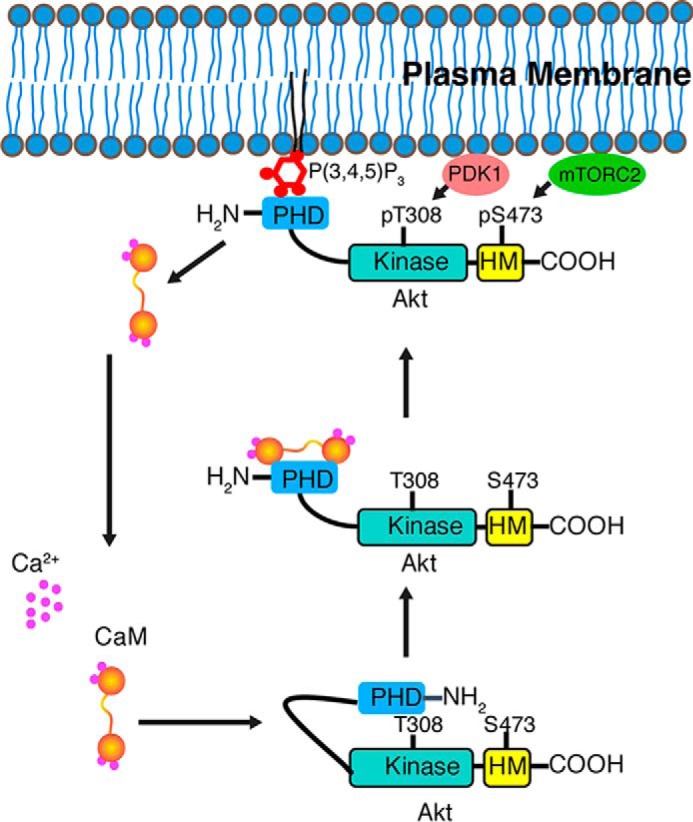
Proposed model highlighting the role of CaM in Akt activation. Based on our data, we propose that upon EGF stimulation CaM interacts with the PHD of Akt and mediates Akt translocation to the PM for phosphorylation and activation. CaM is then released upon binding of Akt to the PM. These results are consistent with previous studies suggesting that CaM may be regulating Akt activity by modulating its subcellular location.
Interestingly, our findings are not in agreement with previous studies which indicated that CaM competes with PI(3,4,5)3 for Akt(PHD) thereby decreasing the affinity of PI(3,4,5)P3 to Akt(PHD) (28). These studies have been performed by using a pulldown assay, phosphoinositide membrane strip, and GST-tagged Akt(PHD) protein. However, our studies were conducted with native and untagged Akt(PHD) protein and a physiologically relevant membrane mimetic containing PS, which has been shown to enhance membrane binding (23). It is possible that these differences in experimental design can adversely affect the results. In another report (39), it has been shown that CaM act in a positive feedback loop to potentiate Ca2+ signaling downstream of the Tec kinase family member Itk. Similar to our findings, the CaM-binding site was mapped to two loops adjacent to the lipid binding pocket within the Itk(PHD). However, in a drastic difference Itk(PHD) bound synergistically to CaM and PI(3,4,5)P3 (and not IP4) such that binding to CaM enhanced the binding to PI(3,4,5)P3 and vice versa. Altogether, it appears that the interplay between CaM and phosphoinositide is key to activation of Akt and analogous PHD-containing proteins such as Itk. Elucidation of the interplay between membrane lipids and CaM-mediated Akt activation in cancer cells will advance our understanding of the mechanisms governing Akt activation in general and may contribute to the development of novel anti-cancer drugs that disrupt these interactions.
Experimental Procedures
Sample Preparation
Expression and purification of the CaM protein were performed as described (58). CaM samples were stored in a buffer containing 50 mm HEPES or Tris (pH 7.0), 150 mm NaCl, and 1 mm CaCl2. The CaM-N domain (residues 1–80) was expressed and purified as described (59). Because CaM-N has zero extinction coefficient, protein concentration was measured using the bicinchoninic acid assay (BCA; Thermo Scientific). A plasmid encoding for the Akt-PHD protein fused to a small ubiquitin-like modifier tag was constructed as described (38). The Akt protein sequence is 98% identical to that of human Akt1 (Swiss-Prot code P31749). Expression and purification of Akt(PHD) were conducted as described (38). The Akt(PHD) protein was stored in 50 mm Tris or HEPES (pH 7.0), 150 mm NaCl, and 1 mm CaCl2. diC4-PI(3,4,5)P3 and IP4 were used as received from Echelon Biosciences (Salt Lake City, UT). Stock solutions of IP4 and diC4-PI(3,4,5)P3 at 5–10 mm were prepared for NMR titrations.
Membrane Nanodiscs
A nanodisc membrane model is a non-covalent assembly of phospholipids and a genetically engineered membrane scaffold protein (MSP) (51–55, 60). Phospholipids (∼120–160 molecules total) associate as a bilayer domain with two molecules of MSP wrapping around the edges of the discoidal structure in a belt-like configuration. Nanodiscs have been successfully utilized to study membrane proteins and protein-membrane interactions (51–55, 60). MSP1D1 nanodisc with a diameter of 98 Å was prepared as described (51, 56). POPC and POPS (Avanti Polar Lipids, Alabaster, AL) were stored as stock chloroform solutions. After evaporation, POPC and POPS (at a molar ratio of 78:20) were resuspended in a buffer containing 10 mm Tris (pH 7.4), 100 mm NaCl, 100 mm sodium cholate, and 0.01% sodium azide. Several freeze-thaw cycles were used to properly redissolve the phospholipids in the buffer. 2% DPPI(3,4,5)P3 (Cayman Chemicals, Ann Arbor, MI) dissolved in H2O was added to the phospholipids and mixed with MSP1D1 to give a final ratio of 1:78:20:2 MSP1D1/POPC/POPS/DPPI(3,4,5)P3. A nanodisc free of DPPI(3,4,5)P3 was also made with 1:80:20 MSP1D1/POPC/POPS. Sodium cholate was removed from the nanodiscs using Bio-BeadsTM (Bio-Rad). Nanodiscs were further purified by gel filtration on a Superdex 200 10/300 GL column (GE Healthcare).
Gel Filtration Assay
The mobility of Akt(PHD) in complex with either CaM, nanodisc, or both CaM and nanodisc was analyzed by a gel filtration assay. Briefly, 1.0 ml of ∼70 μm sample solutions of Akt(PHD), Akt(PHD)·CaM, and nanodisc (1:1:1) complexes were loaded onto a Superdex 200 10/300 column in a buffer containing 50 mm HEPES (pH 7.0), 150 mm NaCl, and 1 mm CaCl2. Protein fractions were analyzed by SDS-PAGE.
Cobalt Affinity Pulldown Assay
200 μl of His6-tagged nanodisc at 50 μm was mixed with 200 μl of cobalt resin slurry and rocked for 30 min at 4 °C. 100 μl of Akt(PHD) or Akt(PHD)·CaM samples at 50 μm were then added to the resin and washed three times with a buffer containing 50 mm Tris (pH 7.0), 150 mm NaCl, 1 mm CaCl2, and 50 mm imidazole. Proteins were eluted with a buffer containing 300 mm imidazole. Fractions of the flow-through, wash, and elution were analyzed via SDS-PAGE. In a competition experiment, 80 μl of nanodisc at 94 μm was mixed with 175 Akt(PHD) at 86 μm and then added to 200 μl of nickel resin slurry and rocked for 30 min at 4 °C. Resin was washed three times with a buffer containing 50 mm Tris (pH 7.0), 150 mm NaCl, 1 mm CaCl2, and 50 mm imidazole. 165 μl of CaM at 90 μm was then added to the resin and washed three times. Proteins were eluted with a buffer containing 300 mm imidazole. Fractions were analyzed via SDS-PAGE.
ITC
Thermodynamic parameters of phospholipid and nanodisc binding to the Akt(PHD) and Akt(PHD)·CaM complex were determined using an Auto-ITC200 microcalorimeter (Malvern Instruments). ITC experiments were conducted on protein samples in a buffer containing 50 mm HEPES (pH 7.0), 150 mm NaCl, and 1 mm CaCl2. diC4-PI(3,4,5)P3 at 500 μm was titrated into 28 μm Akt(PHD) or Akt(PHD)·CaM complex under the same buffer conditions. An MSP1D1/POPC/POPS.DPPI(3,4,5)P3 nanodisc at 60 or 210 μm was titrated into 10 μm Akt(PHD) or 25 μm Akt(PHD)·CaM complex, respectively. An MSP1D1/POPC/POPS nanodisc (without DPPI(3,4,5)P3) at 210 μm was titrated into 18 μm Akt(PHD). Heat of reaction was measured at 20 or 30 °C for 16 or 19 injections. Heat of dilution was measured by titrating diC4-PI(3,4,5)P3 or nanodisc into buffer and subtracting it from the heat of binding. Data analysis was performed using the Microcal Origin package (version 8.1). Binding curves were analyzed, and Kd values were determined by nonlinear least square fitting of the baseline-corrected data. The formula used to fit the data as one binding site is as follows: ΔQ(i) = Q(i) + (dVi/Vo)((Q(i) + Q(i − 1))/2) − Q(i − 1), where ΔQ(i) is the heat released at the ith injection; Q(i) is the total heat content of the solution; dVi is the injection volume, and Vo is the total volume.
NMR Spectroscopy
NMR data were collected at 25 °C on a Bruker Avance II (700 MHz 1H) spectrometer equipped with a cryogenic triple-resonance probe, processed with NMRPIPE (61), and analyzed with NMRVIEW (62) or CPPN analysis (63). NMR titration samples were prepared in buffer containing 50 mm Tris-d11 (pH 7.0), 150 mm NaCl, and 1 mm CaCl2. NMR chemical shifts for CaM are reported elsewhere (64, 65). Proton, carbon, and nitrogen signals for Akt(PHD) and Akt(PHD)·CaM complex were reported previously (38).
Cell Culture, Antibodies, and Reagents
The human pancreatic cancer cell line MiaPaCa-2 was purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were grown in Dulbecco's modified Eagle's media (DMEM) (Corning) and supplemented with penicillin (5 units/ml), streptomycin (5 μg/ml), and 10% heat-inactivated FBS. Antibodies were commercially available. Anti-Akt, phospho-Akt (Ser-473), and phospho-Akt (Thr-308) antibodies were from Cell Signaling (Danvers, MA). CaM and anti-GAPDH antibodies were from Millipore (Billerica, MA) and Fitzgerald (Concord, MA), respectively. TFP and EGF were used as purchased (Sigma).
Western Blotting Analysis
Proteins were extracted, quantified with a BCA protein assay kit (Thermo Scientific, Waltham, MA), separated by SDS-PAGE, and transferred to Immobilon P membranes (Millipore, Billerica, MA) as described (66). Membranes were blocked in 5% nonfat milk and incubated with primary antibodies overnight at 4 °C. Horseradish peroxidase-conjugated secondary antibodies in the blocking buffer were incubated for 1 h at room temperature. Signals were detected using Immobilon Western chemiluminescent horseradish peroxidase substrate detection kit (Millipore).
Immunoprecipitation
Immunoprecipitation was performed as described (66). Protein extracts (1000 μg) were incubated with 1 μg of anti-CaM antibody for 1 h and subsequently incubated with 50 μl of 1:1 slurry of protein G-agarose beads overnight at 4 °C. Beads were washed, and 20 μl of 2× Laemmli sample buffer was added to the beads followed by heating at 95 °C for 5 min and chilling on ice. After brief centrifugation, proteins in the supernatant were analyzed by Western blotting with CaM and Akt antibodies.
Immunofluorescence Staining
The protocol for immunofluorescence staining is described elsewhere (67). MiaPaCa-2 cells were seeded on to 18× 18-mm glass coverslips in a 6-well plate and exposed to EGF with or without TFP for 5 min. Cells were washed with ice-cold PBS and fixed with 4% (w/v) paraformaldehyde, blocked with 1% BSA, and incubated with primary antibody, a mouse monoclonal anti-CaM, and a rabbit polyclonal anti-phospho-Akt(Thr-308) antibody (10 μg/ml) at room temperature for 1 h and then secondary antibody, Alexa Fluor 488 anti-mouse IgG, or Alexa Fluor 594 anti-rabbit IgG (10 μg/ml) for 30 min. Microscopic images were taken at ×100 magnification using a confocal microscope (Nikon A1R, Nikon Instruments Inc., Melville, NY).
Author Contributions
C. A., R. H. G., F. X., Y. S., Y. C., and J. S. S. designed and executed the experiments and analyzed the data. C. A., Y. C., and J. S. S. compiled the figures and wrote the manuscript.
Acknowledgments
We thank the Comprehensive Cancer Center, University of Alabama at Birmingham, for supporting the High-Field NMR facility and the X-ray core facility that houses the Auto-ITC200 (acquired through National Institutes of Health Grant 1S10RR026478).
This work was supported in part by the Comprehensive Cancer Center, University of Alabama at Birmingham (funded by National Institutes of Health NCI Grant P30 CA013148), pilot grant (to J. S. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PHD
- pleckstrin homology domain
- CaM
- calmodulin
- PI(3,4,5)P3
- phosphatidylinositol 3,4,5-trisphosphate
- diC4-PI(3,4,5)P3
- dibutanoylphosphatidylinositol 3,4,5-trisphosphate
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- IP4
- inositol 1,3,4,5-tetrakisphosphate
- MSP
- membrane scaffold protein
- HSQC
- heteronuclear single quantum coherence
- ITC
- isothermal titration calorimetry
- PM
- plasma membrane
- KD
- kinase domain
- HM
- homology domain
- DPPI(3,4,5)P3
- 1,2-dipalmitoylphosphatidylinositol 3,4,5-trisphosphate
- TFP
- trifluoperazine
- PS
- phosphatidylserine
- IP3
- inositol 1,4,5-trisphosphate
- CSP
- chemical shift perturbation.
References
- 1. Altomare D. A., and Testa J. R. (2005) Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464 [DOI] [PubMed] [Google Scholar]
- 2. Coticchia C. M., Revankar C. M., Deb T. B., Dickson R. B., and Johnson M. D. (2009) Calmodulin modulates Akt activity in human breast cancer cell lines. Breast Cancer Res. Treat. 115, 545–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pérez-Tenorio G., Stål O., and Southeast Sweden Breast Cancer Group (2002) Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br. J. Cancer 86, 540–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cristiano B. E., Chan J. C., Hannan K. M., Lundie N. A., Marmy-Conus N. J., Campbell I. G., Phillips W. A., Robbie M., Hannan R. D., and Pearson R. B. (2006) A specific role for AKT3 in the genesis of ovarian cancer through modulation of G2-M phase transition. Cancer Res. 66, 11718–11725 [DOI] [PubMed] [Google Scholar]
- 5. Héron-Milhavet L., Franckhauser C., Rana V., Berthenet C., Fisher D., Hemmings B. A., Fernandez A., and Lamb N. J. (2006) Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol. Cell. Biol. 26, 8267–8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Irie H. Y., Pearline R. V., Grueneberg D., Hsia M., Ravichandran P., Kothari N., Natesan S., and Brugge J. S. (2005) Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J. Cell Biol. 171, 1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yun S. J., Tucker D. F., Kim E. K., Kim M. S., Do K. H., Ha J. M., Lee S. Y., Yun J., Kim C. D., Birnbaum M. J., and Bae S. S. (2009) Differential regulation of Akt/protein kinase B isoforms during cell cycle progression. FEBS Lett. 583, 685–690 [DOI] [PubMed] [Google Scholar]
- 8. Liao Y., and Hung M. C. (2010) Physiological regulation of Akt activity and stability. Am. J. Transl. Res. 2, 19–42 [PMC free article] [PubMed] [Google Scholar]
- 9. Hanada M., Feng J., and Hemmings B. A. (2004) Structure, regulation and function of PKB/AKT–a major therapeutic target. Biochim. Biophys. Acta 1697, 3–16 [DOI] [PubMed] [Google Scholar]
- 10. Carpten J. D., Faber A. L., Horn C., Donoho G. P., Briggs S. L., Robbins C. M., Hostetter G., Boguslawski S., Moses T. Y., Savage S., Uhlik M., Lin A., Du J., Qian Y. W., Zeckner D. J., et al. (2007) A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444 [DOI] [PubMed] [Google Scholar]
- 11. Dillon R. L., White D. E., and Muller W. J. (2007) The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene 26, 1338–1345 [DOI] [PubMed] [Google Scholar]
- 12. Franke T. F., Yang S. I., Chan T. O., Datta K., Kazlauskas A., Morrison D. K., Kaplan D. R., and Tsichlis P. N. (1995) The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81, 727–736 [DOI] [PubMed] [Google Scholar]
- 13. Ahmed N. N., Grimes H. L., Bellacosa A., Chan T. O., and Tsichlis P. N. (1997) Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc. Natl. Acad. Sci. U.S.A. 94, 3627–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Valius M., and Kazlauskas A. (1993) Phospholipase C-gamma 1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor's mitogenic signal. Cell 73, 321–334 [DOI] [PubMed] [Google Scholar]
- 15. Kaliman P., Viñals F., Testar X., Palacín M., and Zorzano A. (1996) Phosphatidylinositol 3-kinase inhibitors block differentiation of skeletal muscle cells. J. Biol. Chem. 271, 19146–19151 [DOI] [PubMed] [Google Scholar]
- 16. Yap T. A., Garrett M. D., Walton M. I., Raynaud F., de Bono J. S., and Workman P. (2008) Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr. Opin. Pharmacol. 8, 393–412 [DOI] [PubMed] [Google Scholar]
- 17. Miao B., and Degterev A. (2011) Targeting phosphatidylinositol 3-kinase signaling with novel phosphatidylinositol 3,4,5-triphosphate antagonists. Autophagy 7, 650–651 [DOI] [PubMed] [Google Scholar]
- 18. Franke T. F. (2008) Intracellular signaling by Akt: bound to be specific. Sci. Signal. 1, pe29. [DOI] [PubMed] [Google Scholar]
- 19. Cooray S. (2004) The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 85, 1065–1076 [DOI] [PubMed] [Google Scholar]
- 20. Lemmon M. A. (2007) Pleckstrin homology (PH) domains and phosphoinositides. Biochem. Soc. Symp. 74, 81–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Q., Bateman A., Finn R. D., Abdubek P., Astakhova T., Axelrod H. L., Bakolitsa C., Carlton D., Chen C., Chiu H. J., Chiu M., Clayton T., Das D., Deller M. C., Duan L., et al. , (2010) Bacterial pleckstrin homology domains: a prokaryotic origin for the PH domain. J. Mol. Biol. 396, 31–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu J. W., Mendrola J. M., Audhya A., Singh S., Keleti D., DeWald D. B., Murray D., Emr S. D., and Lemmon M. A. (2004) Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol. Cell 13, 677–688 [DOI] [PubMed] [Google Scholar]
- 23. Huang B. X., Akbar M., Kevala K., and Kim H. Y. (2011) Phosphatidylserine is a critical modulator for Akt activation. J. Cell Biol. 192, 979–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Calleja V., Laguerre M., Parker P. J., and Larijani B. (2009) Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 7, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Calleja V., Alcor D., Laguerre M., Park J., Vojnovic B., Hemmings B. A., Downward J., Parker P. J., and Larijani B. (2007) Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 5, e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu W. I., Voegtli W. C., Sturgis H. L., Dizon F. P., Vigers G. P., and Brandhuber B. J. (2010) Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 5, e12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parikh C., Janakiraman V., Wu W. I., Foo C. K., Kljavin N. M., Chaudhuri S., Stawiski E., Lee B., Lin J., Li H., Lorenzo M. N., Yuan W., Guillory J., Jackson M., Rondon J., et al. , (2012) Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc. Natl. Acad. Sci. U.S.A. 109, 19368–19373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dong B., Valencia C. A., and Liu R. (2007) Ca2+/calmodulin directly interacts with the pleckstrin homology domain of AKT1. J. Biol. Chem. 282, 25131–25140 [DOI] [PubMed] [Google Scholar]
- 29. Chin D., and Means A. R. (2000) Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 10, 322–328 [DOI] [PubMed] [Google Scholar]
- 30. Hoeflich K. P., and Ikura M. (2002) Calmodulin in action: diversity in target recognition and activation mechanisms. Cell 108, 739–742 [DOI] [PubMed] [Google Scholar]
- 31. Ishida H., and Vogel H. J. (2006) Protein-peptide interaction studies demonstrate the versatility of calmodulin target protein binding. Protein Pept. Lett. 13, 455–465 [DOI] [PubMed] [Google Scholar]
- 32. Osawa M., Tokumitsu H., Swindells M. B., Kurihara H., Orita M., Shibanuma T., Furuya T., and Ikura M. (1999) A novel target recognition revealed by calmodulin in complex with Ca2+-calmodulin-dependent kinase kinase. Nat. Struct. Biol. 6, 819–824 [DOI] [PubMed] [Google Scholar]
- 33. Vetter S. W., and Leclerc E. (2003) Novel aspects of calmodulin target recognition and activation. Eur. J. Biochem. 270, 404–414 [DOI] [PubMed] [Google Scholar]
- 34. Yamniuk A. P., and Vogel H. J. (2004) Calmodulin's flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol. Biotechnol. 27, 33–57 [DOI] [PubMed] [Google Scholar]
- 35. Deb T. B., Coticchia C. M., and Dickson R. B. (2004) Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J. Biol. Chem. 279, 38903–38911 [DOI] [PubMed] [Google Scholar]
- 36. Desai K. V., Xiao N., Wang W., Gangi L., Greene J., Powell J. I., Dickson R., Furth P., Hunter K., Kucherlapati R., Simon R., Liu E. T., and Green J. E. (2002) Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proc. Natl. Acad. Sci. U.S.A. 99, 6967–6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krishnaraju K., Murugesan K., Vij U., Kapur B. M., and Farooq A. (1991) Calmodulin levels in oestrogen receptor positive and negative human breast tumours. Br. J. Cancer 63, 346–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Agamasu C., Ghanam R. H., and Saad J. S. (2015) Structural and biophysical characterization of the interactions between calmodulin and the pleckstrin homology domain of Akt. J. Biol. Chem. 290, 27403–27413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang X., Boyken S. E., Hu J., Xu X., Rimer R. P., Shea M. A., Shaw A. S., Andreotti A. H., and Huang Y. H. (2014) Calmodulin and PI(3,4,5)P(3) cooperatively bind to the Itk pleckstrin homology domain to promote efficient calcium signaling and IL-17A production. Sci. Signal. 7, ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Auguin D., Barthe P., Augé-Sénégas M. T., Stern M. H., Noguchi M., and Roumestand C. (2004) Solution structure and backbone dynamics of the pleckstrin homology domain of the human protein kinase B (PKB/Akt). Interaction with inositol phosphates. J. Biomol. NMR 28, 137–155 [DOI] [PubMed] [Google Scholar]
- 41. Thomas C. C., Deak M., Alessi D. R., and van Aalten D. M. (2002) High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr. Biol. 12, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 42. Vlach J., and Saad J. S. (2013) Trio engagement via plasma membrane phospholipids and the myristoyl moiety governs HIV-1 matrix binding to bilayers. Proc. Natl. Acad. Sci. U.S.A. 110, 3525–3530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saad J. S., Miller J., Tai J., Kim A., Ghanam R. H., and Summers M. F. (2006) Structural basis for targeting HIV-1 Gag to virus assembly sites on the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 103, 11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saad J. S., Ablan S. D., Ghanam R. H., Kim A., Andrews K., Nagashima K., Soheilian F., Freed E. O., and Summers M. F. (2008) Structure of the myristoylated HIV-2 MA protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J. Mol. Biol. 382, 434–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gorbatyuk V. Y., Nosworthy N. J., Robson S. A., Bains N. P., Maciejewski M. W., Dos Remedios C. G., and King G. F. (2006) Mapping the phosphoinositide-binding site on chick cofilin explains how PIP2 regulates the cofilin-actin interaction. Mol. Cell 24, 511–522 [DOI] [PubMed] [Google Scholar]
- 46. Sauer K., Park E., Siegemund S., French A. R., Wahle J. A., Sternberg L., Rigaud S., Jonsson A. H., Yokoyama W. M., and Huang Y. H. (2013) Inositol tetrakisphosphate limits NK cell effector functions by controlling PI3K signaling. Blood 121, 286–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frech M., Andjelkovic M., Ingley E., Reddy K. K., Falck J. R., and Hemmings B. A. (1997) High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/protein kinase B and their influence on kinase activity. J. Biol. Chem. 272, 8474–8481 [DOI] [PubMed] [Google Scholar]
- 48. Meuillet E. J. (2011) Novel inhibitors of AKT: assessment of a different approach targeting the pleckstrin homology domain. Curr. Med. Chem. 18, 2727–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fushman D., Cahill S., and Cowburn D. (1997) The main-chain dynamics of the dynamin pleckstrin homology (PH) domain in solution: analysis of 15N relaxation with monomer/dimer equilibration. J. Mol. Biol. 266, 173–194 [DOI] [PubMed] [Google Scholar]
- 50. Bayburt T. H., and Sligar S. G. (2003) Self-assembly of single integral membrane proteins into soluble nanoscale phospholipid bilayers. Protein Sci. 12, 2476–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bayburt T. H., and Sligar S. G. (2010) Membrane protein assembly into nanodiscs. FEBS Lett. 584, 1721–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Borch J., and Hamann T. (2009) The nanodisc: a novel tool for membrane protein studies. Biol. Chem. 390, 805–814 [DOI] [PubMed] [Google Scholar]
- 53. Hagn F., Etzkorn M., Raschle T., and Wagner G. (2013) Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 135, 1919–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kobashigawa Y., Harada K., Yoshida N., Ogura K., and Inagaki F. (2011) Phosphoinositide-incorporated lipid-protein nanodiscs: a tool for studying protein-lipid interactions. Anal. Biochem. 410, 77–83 [DOI] [PubMed] [Google Scholar]
- 55. Yokogawa M., Kobashigawa Y., Yoshida N., Ogura K., Harada K., and Inagaki F. (2012) NMR analyses of the interaction between the FYVE domain of early endosome antigen 1 (EEA1) and phosphoinositide embedded in a lipid bilayer. J. Biol. Chem. 287, 34936–34945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bayburt T. H., Grinkova Y. V., and Sligar S. G. (2002) Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2, 853–856 [Google Scholar]
- 57. Manning B. D., and Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Samal A. B., Ghanam R. H., Fernandez T. F., Monroe E. B., and Saad J. S. (2011) NMR, biophysical and biochemical studies reveal the minimal calmodulin-binding domain of the HIV-1 matrix protein. J. Biol. Chem. 286, 33533–33543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fernandez T. F., Samal A. B., Bedwell G. J., Chen Y., and Saad J. S. (2013) Structural and biophysical characterization of the interactions between the death domain of Fas receptor and calmodulin. J. Biol. Chem. 288, 21898–21908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ritchie T. K., Grinkova Y. V., Bayburt T. H., Denisov I. G., Zolnerciks J. K., Atkins W. M., and Sligar S. G. (2009) Chapter 11–reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 62. Johnson B. A., and Blevins R. A. (1994) NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 63. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 64. Ghanam R. H., Fernandez T. F., Fledderman E. L., and Saad J. S. (2010) Binding of calmodulin to the HIV-1 matrix protein triggers myristate exposure. J. Biol. Chem. 285, 41911–41920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ikura M., Kay L. E., and Bax A. (1990) A novel approach for sequential assignment of 1H, 13C, and 15N spectra of larger proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. application to calmodulin. Biochemistry 29, 4659–4667 [DOI] [PubMed] [Google Scholar]
- 66. Yuan K., Sun Y., Zhou T., McDonald J., and Chen Y. (2013) PARP-1 regulates resistance of pancreatic cancer to TRAIL therapy. Clin. Cancer Res. 19, 4750–4759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mao X., Debenedittis P., Sun Y., Chen J., Yuan K., Jiao K., and Chen Y. (2012) Vascular smooth muscle cell Smad4 gene is important for mouse vascular development. Arterioscler. Thromb. Vasc. Biol. 32, 2171–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]