

Abstract
Cytoplasmic localization of proline, glutamic acid, leucine-rich protein 1 (PELP1) is observed in ∼40% of women with invasive breast cancer. In mouse models, PELP1 overexpression in the mammary gland leads to premalignant lesions and eventually mammary tumors. In preliminary clinical studies, cytoplasmic localization of PELP1 was seen in 36% of women at high risk of developing breast cancer. Here, we investigated whether cytoplasmic PELP1 signaling promotes breast cancer initiation in models of immortalized human mammary epithelial cells (HMECs). Global gene expression analysis was performed on HMEC lines expressing vector control, PELP1-wt, or mutant PELP1 in which the nuclear localization sequence was altered, resulting in cytoplasmic localization of PELP1 (PELP1-cyto). Global gene expression analysis identified that PELP1-cyto expression in HMECs induced NF-κB signaling pathways. Western blotting analysis of PELP1-cyto HMECs showed up-regulation of inhibitor of κB kinase ϵ (IKKϵ) and increased phosphorylation of the NF-κB subunit RelB. To determine whether secreted factors produced by PELP1-cyto HMECs promote macrophage activation, THP-1 macrophages were treated with HMEC-conditioned medium (CM). PELP1-cyto CM induced changes in THP-1 gene expression as compared with control cell CM. Double conditioned medium (DCM) from the activated THP-1 cells was then applied to HMECs to determine whether paracrine signaling from PELP1-cyto-activated macrophages could in turn promote migration of HMECs. PELP1-cyto DCM induced robust HMEC migration, which was reduced in DCM from PELP1-cyto HMECs expressing IKKϵ shRNA. Our findings suggest that cytoplasmic localization of PELP1 up-regulates pro-tumorigenic IKKϵ and secreted inflammatory signals, which through paracrine macrophage activation regulates the migratory phenotype associated with breast cancer initiation.
Keywords: breast cancer, cell migration, cell signaling, inflammation, macrophage, NF-kB transcription factor
Introduction
Approximately 1.6 million breast biopsies are performed annually in the United States (1). Invasive breast cancer (IBC)2 is detected in 10–20% of biopsies, and surgical removal with or without chemotherapy and/or radiation is recommended for patients with IBC. In many biopsies negative for IBC, there is evidence of abnormal cells, including preinvasive lesions such as ductal carcinoma in situ (DCIS) or benign premalignant lesions such as atypical hyperplasia (AH). Both preinvasive and benign lesions are associated with an increased risk of developing IBC. Approximately 61,000 cases of non-invasive DCIS are diagnosed annually. Although only 20–30% of DCIS cases will progress to IBC, all patients are treated with surgery (with or without radiation). Of the 1.6 million biopsies performed annually, more than 1 million are found to be benign, and women with benign lesions such as hyperplasia and AH are classified as having benign breast disease (BBD) (2). BBD is stratified by histologic features and degree of cellular abnormality. BBD containing AH is considered a high risk lesion, resulting in four times the risk of developing IBC as compared with normal risk individuals (3). Despite an urgent clinical need to identify which women with DCIS or BBD will develop invasive disease, no molecular biomarkers have been identified to stratify women into those at high or low risk of developing IBC. Identification of such predictive molecular biomarkers would not only spare low risk women of unnecessary treatment but also lead to the development of novel targeted prevention strategies for high risk women.
Proline, glutamic acid, leucine-rich protein 1 (PELP1) is an emerging biomarker of breast cancer initiation and response to chemoprevention therapies. PELP1 is a large multidomain protein that contains 10 LXXLL motifs and several other motifs common to transcriptional regulators, but the overall protein structure is not homologous to other known proteins. Although PELP1 was first identified as an estrogen receptor (ER) co-activator (4), subsequent studies have found that PELP1 acts as a transcriptional regulator for many transcription factors and is associated with chromatin remodeling complexes (5–9). PELP1 has been shown to influence cancer cell biology through the regulation of proliferation; apoptosis and autophagy; migration, invasion, and metastasis; and endocrine resistance (5).
In addition to nuclear functions, PELP1 has been shown to regulate cytoplasmic signaling. PELP1 subcellular localization is primarily nuclear in normal breast tissue, but cytoplasmic localization is observed in ∼40% of IBC (10). Mutant PELP1 with an altered nuclear localization sequence results in a protein that is predominately cytoplasmic (PELP1-cyto) and leads to activation of cytoplasmic signaling in breast cancer cell line models (10). In mammary-specific transgenic mouse models, expression of wild-type PELP1 or PELP1-cyto induced mammary gland hyperplasia that was associated with increased Akt and Erk1/2 signaling (11, 12). Cytoplasmic PELP1 signaling has primarily been studied in breast cancer cell line models and in vivo mouse models (10, 11, 13). Recently, however, PELP1 localization was found to be altered in 4 of 11 (36%) atypical breast needle aspirate samples from women at high risk of developing breast cancer (14). These preclinical and preliminary clinical findings suggest that altered PELP1 localization may be an early event in breast cancer initiation.
In the present study, we examined whether signaling pathways, induced by cytoplasmic PELP1, promote breast cancer initiation in models of immortalized human mammary epithelial cells (HMECs). We found that PELP1-cyto expression in HMECs induced chemokine and cytokine gene expression and up-regulation of IKKϵ. In addition, PELP1-cyto-expressing HMECs activated macrophages, which then promoted mammary epithelial cell migration via paracrine signaling mechanisms. Macrophage activation was mediated in part through up-regulation of IKKϵ. These findings suggest that altered localization of PELP1 to the cytoplasm induces a cascade of pro-tumorigenic signaling that drives a migratory phenotype associated with breast cancer initiation.
Results
Cytoplasmic PELP1 Promotes Migration and Abnormal Acini Formation
We previously demonstrated that altered localization of PELP1 promotes HMEC survival in response to tamoxifen (14). To determine whether cytoplasmic PELP1 (PELP1-cyto) contributes to phenotypes associated with oncogenic signaling and breast cancer initiation, we first developed an additional HMEC model in MCF-10A cells to compare with our previously published HMEC-hTERT cell line model (14). These cell lines were chosen as models of spontaneously immortalized and hTERT-immortalized HMECs, respectively, that are susceptible to oncogene-induced transformation. Additionally, the MCF-10A model is useful for three-dimensional acini formation assays. As previously published for the HMEC-hTERT model (14), we established stable MCF-10A cell lines that express LXSN control or PELP1-cyto. Cells were selected for stable integration of PELP1 with G418. Clonal cell populations were screened for PELP1 localization by immunofluorescence (data not shown) and Western blotting of cytoplasmic and nuclear fractions. Clonal cell lines expressing PELP1-cyto (lanes C) showed increased PELP1 in the cytoplasm as compared with vector control (lanes V) cell lines (Fig. 1A). Western blotting for HDAC2 and MEK1 was performed as controls for protein loading and nuclear/cytoplasmic fractionation (Fig. 1A).
FIGURE 1.
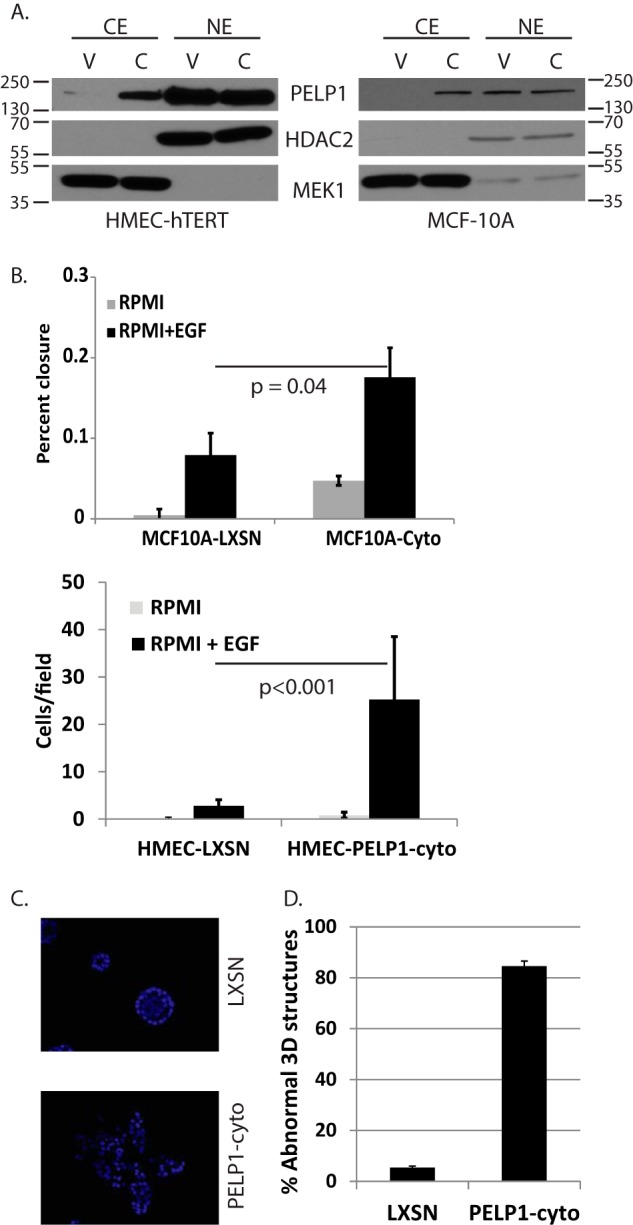
Cytoplasmic PELP1 alters migratory and three-dimensional growth phenotypes in mammary epithelial cell lines. A, MCF-10A and HMEC-hTERT lines expressing LXSN control (lanes V) or PELP1-cyto (lanes C) were examined by Western blotting of nuclear (NE) and cytoplasmic (CE) fractions to determine PELP1 localization. HDAC2 and MEK1 were used as nuclear and cytoplasmic fractionation and loading controls, respectively. B, scratch wound and Transwell migration assays for MCF-10A and HMEC-hTERT cells, respectively, in response to 20 ng/ml EGF. Each condition was performed in triplicate. The bars represent the means of triplicates with standard deviation. Student's t test was performed to determine the statistical significance between LXSN + EGF and PELP1-cyto + EGF conditions. C, immunofluorescent MCF-10A LXSN and PELP1-cyto three-dimensional cultures stained with DAPI. D, quantitation of multiacinar phenotype observed in MCF-10A cells expressing PELP1-cyto compared with MCF-10A LXSN control cells. The total numbers of structures (normal and abnormal) were counted from three random fields from three separate wells. The data are represented as the percentages of structures with an abnormal phenotype.
PELP1 has previously been shown to enhance the migratory potential of breast cancer cell lines (15–17). To determine the effect of altered PELP1 localization on epidermal growth factor (EGF)-induced migration of MCF10A and HMEC-hTERT, we tested MCF10A cells in scratch wound assays and HMEC-hTERT cells in Transwell migration assays (because the HMEC-hTERT cells do not form a compact sheet of cells compatible for the scratch wound assay). In the scratch wound assay, MCF-10A cells (LXSN and PELP1-cyto) grown to confluent monolayers were scratched, washed with PBS, and incubated in RPMI with or without 20 ng/ml EGF. Images were taken immediately after scratching and then again after a 16-h incubation. PELP1-cyto expression promoted a statistically significant 2-fold increase in EGF-induced migration of MCF-10A cells (p = 0.04; Fig. 1B). Of note, we consistently observed an increase in basal migration of MCF-10A PELP1-cyto cells independent of EGF. In the Transwell migration assay, serum-free RPMI supplemented with 20 ng/ml EGF was used as a chemoattractant in the bottom chamber; HMEC-hTERT cells (LXSN or PELP1-cyto) resuspended in RPMI were added to the upper chamber and incubated for 16 h. The cells that migrated through the Transwell were counted. PELP1-cyto expression enhanced EGF-induced migration of HMEC-hTERT cells almost 9-fold over control cells (p < 0.001; Fig. 1B).
Three-dimensional culture of MCF-10A cells on recombinant basement membrane results in formation of polarized acini structures that share features with the normal ductal structure of human breast tissue. This model and assay have been useful in examining the effects of oncogenes on the disruption of the epithelial architecture during breast cancer initiation (18). To determine whether altered localization of PELP1 disrupts MCF-10A three-dimensional acini formation, LXSN and PELP1-cyto cells were plated on recombinant basement membrane as previously described (18). After 2 weeks in culture, we found that the majority (over 80%) of PELP1-cyto three-dimensional structures displayed an abnormal multiacinar phenotype without a hollow lumen, whereas greater than 90% of the LXSN control cells generated spherical acini structures with a hollow lumen (Fig. 1, C and D).
PELP1-cyto Induces Changes in Global Gene Expression
To further elucidate the genes and pathways altered by PELP1-cyto expression in the HMEC-hTERT model, we performed global gene expression (GGE) analysis using Illumina bead chips. Hierarchical clustering of differentially expressed genes (>2-fold in any comparison with an adjusted p value of <1 × 10−11) showed that cells expressing PELP1-cyto had substantially altered gene programs, compared with HMECs expressing PELP1-wt or LXSN control (Fig. 2A). HMECs expressing PELP1-cyto had 58 genes up-regulated (>2-fold) as compared with control cells (Table 1). In contrast, HMECs expressing PELP1-wt had only three genes up-regulated as compared with control cells. In addition, HMECs expressing PELP1-cyto had 20 genes significantly down-regulated (>2-fold) as compared with control cells (Table 1), whereas HMECs expressing PELP1-wt had no genes significantly down-regulated (>2-fold).
FIGURE 2.

Cytoplasmic PELP1 signaling induces NF-κB and inflammatory signaling in mammary epithelial cell lines. A, heat map showing normalized expression values for differentially expressed transcripts (fold change >2 in at least one sample). RNA was isolated from biological duplicate cultures of HMEC-hTERT cell lines expressing LXSN, PELP1-wt, or PELP1-cyto (Cyto). Genes up-regulated or down-regulated are shown in red or blue, respectively. B, results of IPA showing pathways up-regulated (orange) or down-regulated (blue) based on genes regulated >2-fold between HMEC-hTERT PELP1-cyto and LXSN. IPA results are displayed by z-score. Each box represents a “disease of function” annotation within the indicated IPA category (i.e. cellular movement, etc.). C, GSEA plots from the MSigDB curated (C2) collection for two gene sets identified as up-regulated in PELP1-cyto cells as compared with LXSN indicating enrichment of NF-κB-dependent genes and inflammatory response. D, qRT-PCR to validate genes identified by GGE in HMEC-hTERT (left panel) and MCF-10A (right panel) cell lines. All conditions were performed in triplicate, and each bar represents the mean with standard deviation. Student's t test was performed to determine statistical significance between LXSN and PELP1-cyto conditions. *, p < 0.05.
TABLE 1.
Summary of genes found in GGE analysis
All 58 genes up-regulated and 20 down-regulated in HMEC-hTERT PELP1-cyto cells as compared with LXSN. Under IPA association, “I” indicates inflammatory/immune association, and “CM” indicates cellular movement. “qRT-PCR validated” refers to genes validated in HMEC-hTERT and MCF-10A PELP1-cyto cell lines. “IKKϵ regulated” refers to genes that were found downregulated in MCF-10A PELP1-cyto shIKKϵ cells as compared with PELP1-cyto shGFP cells. “ND” indicates not determined.
| IPA association | qRT-PCR validated | IKKϵ regulated | |
|---|---|---|---|
| Array genes up-regulated >2-fold (PELP1-cyto versus vector) | |||
| KRT81 | |||
| MYADM | |||
| PI3 | |||
| TGM2 | I, CM | ||
| C20orf100 | |||
| EPDR1 | |||
| IL13RA2 | CM | ||
| KYNU | |||
| LCP1 | I, CM | ||
| HSD17B2 | |||
| CTSH | I | ||
| TAGLN | CM | ||
| SLPI | I, CM | ||
| IGF2BP3 | |||
| IL1B | I, CM | YES | NO |
| L1TD1 | |||
| SAA1 | I, CM | YES | YES |
| TOX2 | |||
| CPNE1 | |||
| BAD | I | YES | |
| ACTG2 | |||
| B3GALNT1 | |||
| CYGB | |||
| CXCL1 | I, CM | YES | YES |
| CPNE1 | |||
| MYADM | |||
| MLPH | |||
| ARMCX1 | |||
| FAM129A | |||
| S100A9 | I, CM | YES | YES |
| PPARG | I, CM | ||
| C9orf169 | |||
| CSF3 | I, CM | YES | YES |
| FOXQ1 | |||
| FBN2 | |||
| IL8 | CM | YES | YES |
| C1orf85 | |||
| GCA | |||
| KRT34 | |||
| C1orf24 | |||
| ADA | I | ||
| CDC42EP5 | |||
| FUCA1 | |||
| PLD5 | |||
| CLIC3 | |||
| PTGS2 | I, CM | YES | ND |
| CES1 | |||
| TRPC4AP | |||
| G0S2 | |||
| SNCG | CM | ||
| IL1A | I, CM | YES | ND |
| MGMT | |||
| PTGES | I, CM | ||
| BCL2L1 | I | ||
| C20orf24 | |||
| MAP1B | CM | ||
| EDEM2 | |||
| ZGPAT | |||
| Array genes downregulated >2-fold (cytoplasm versus vector) | |||
| KRT15 | |||
| CXXC5 | |||
| GJB2 | I, CM | ||
| DOCK11 | |||
| ASNS | |||
| LGALS7 | YES | ND | |
| C14orf78 | |||
| MGC102966 | |||
| F12 | I | ||
| LOC400578 | |||
| TCTEX1D2 | |||
| NDRG1 | I, CM | ||
| FGFR3 | |||
| PRSS3 | |||
| LGALS7B | |||
| LOC100134134 | |||
| IGFL3 | |||
| AUTS2 | |||
| LEPREL1 | |||
| SYT7 | |||
We explored the importance of differentially regulated genes in HMECs expressing PELP1-cyto and vector control using Ingenuity Pathway Analysis (IPA). Pathways significantly regulated in PELP1-cyto cells (as compared with LXSN control cells) were cancer, cellular movement, tumor morphology, immune cell trafficking, and inflammatory response, among others (Fig. 2B). IPA “upstream analysis” also identified NF-κB and NF-κB-inducing cytokines as up-regulated in PELP1-cyto cells. We also employed Gene Set Enrichment Analysis (GSEA) to identify significantly regulated gene sets in PELP1-cyto cells as compared with control cells. Three gene set collections from the Molecular Signatures Database (MSigDB) were tested for enrichment: hallmark (H), curated (C2), and gene ontology (C5) (19, 20). We found multiple NF-κB transcription factor activation and inflammatory response gene sets significantly associated with PELP1-cyto cells, as well as expected oncogenic gene sets (supplemental Table S1). Representative GSEA plots for NF-κB and inflammation enriched gene sets are shown in Fig. 2C. We have validated a number of known NF-κB regulated genes that were identified from our GGE studies in both the HMEC-hTERT and MCF-10A models (Table 1). IL-8, CXCL1, and IL-1β—all known NF-κB-regulated genes involved in inflammation—are shown in Fig. 2D.
Up-regulation of IKKϵ in PELP1-cyto HMECs
Next we determined which components of the NF-κB signaling pathway were up-regulated or activated in PELP1-cyto HMECs. Western blotting of whole cell extracts (WCEs) and cytoplasmic or nuclear extracts from HMEC-hTERT and MCF-10A cells for NF-κB and IKK family members was performed. Of all the IKK and NF-κB family members tested, IKKϵ was the only family member significantly regulated by PELP1-cyto expression (Fig. 3). IKKϵ expression was increased in WCE collected from the PELP1-cyto-expressing HMEC-hTERT and MCF10A cells as compared with controls (Fig. 3A). Additionally, IKKϵ expression was increased in the cytoplasmic and nuclear extracts collected from the PELP1-cyto-expressing HMEC-hTERT and MCF10A cells as compared with control cells (Fig. 3B). Moreover, phosphorylation of RelB (p-RelB) at serine 522 was higher in cytoplasmic extracts from PELP1-cyto-expressing cells than from LXSN control cells (Fig. 3, A and B). Of note, modest increases in nuclear RelB and RelA/p65 and phosphorylation of RelA/p65 at serine 536 were observed in PELP1-cyto cells as compared with control cells (data not shown).
FIGURE 3.

IKKϵ is overexpressed in breast cancer and drives non-canonical NF-κB signaling in response to cytoplasmic PELP1 localization. A, representative Western blots from a minimum of three independent experiments. WCE from MCF-10A and HMEC-hTERT cells (LXSN (lanes V) and PELP1-cyto (lanes C)) were resolved by SDS-PAGE and probed with antibodies for PELP1, IKKϵ, and phospho-RelB (p-RelB). Actin was used as a loading control. B, cytoplasmic (CE) and nuclear extracts (NE) from MCF-10A and HMEC-hTERT cells were probed with antibodies to PELP1, IKKϵ, and p-RelB. HDAC2 and MEK1 were used as nuclear and cytoplasmic localization and loading controls, respectively. C, data from The Cancer Genome Atlas were analyzed for comparison of IKKϵ levels in normal breast tissue and invasive breast carcinoma. The data are represented in a whisker box plot (top whisker, 90%; top of box, 75%; line in box, median; bottom of box, 25%; bottom whisker, 10%).
IKKϵ amplification and overexpression and associated inflammatory gene expression in breast cancer has been described by others (21–26). Amplification of IKKϵ is associated with luminal breast cancer, whereas overexpression has been found in a subset of triple-negative breast cancer (21). Query of The Cancer Genome Atlas provisional breast tumor data set demonstrated that IKKϵ is significantly up-regulated in breast tumors as compared with normal breast tissue (Fig. 3C). Additionally, we found that there was a statistically significant (p = 0.032) tendency toward co-occurrent gene alterations for IKKϵ and PELP1 in this data set.
IKKϵ Knockdown Reduces Cytokine and Chemokine Gene Expression
To determine whether PELP1-cyto-induced IKKϵ expression is important for inflammatory gene expression, we knocked down IKKϵ in MCF-10A cells expressing LXSN control and PELP1-cyto as described under “Experimental Procedures.” Of the 5 shRNA constructs tested, only one resulted in significant down-regulation of IKKϵ levels. This pooled population was used for subsequent experiments. WCE, as well as cytoplasmic and nuclear extracts, were isolated from LXSN and PELP1-cyto MCF-10A cells expressing shGFP (control) or shIKKϵ. Western blotting revealed a loss of PELP1-cyto-induced p-RelB in shIKKϵ WCE and cytoplasmic extracts (Fig. 4A).
FIGURE 4.
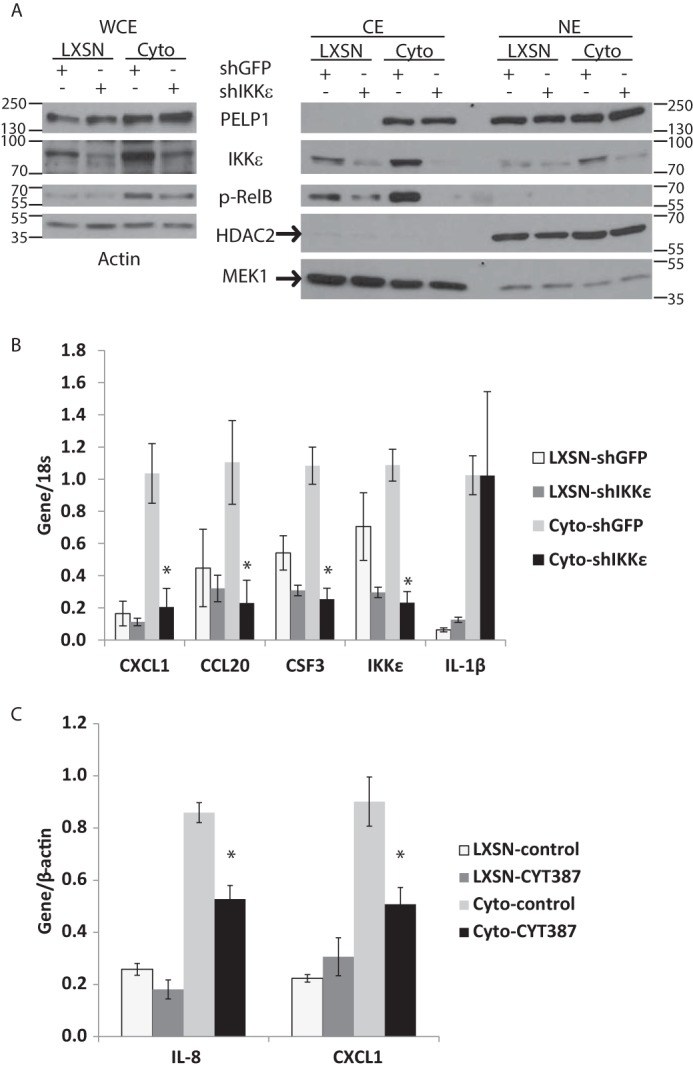
Knockdown of IKKϵ inhibits PELP1-cyto induced non-canonical NF-κB activation and inflammatory gene up-regulation. A, WCE (left panel) and cytoplasmic (CE) and nuclear (NE) extracts isolated from MCF-10A cells expressing LXSN or PELP1-cyto and either shGFP control or shIKKϵ. Lysates were examined by Western blotting for PELP1, IKKϵ, and phospho-RelB. Actin was used as the loading control for WCE, whereas HDAC2 and MEK1 were used as the nuclear and cytoplasmic fractionation and loading controls, respectively. The data are representative of at least three independent experiments. B, qRT-PCR for IKKϵ and inflammatory gene expression from MCF-10A cells (LXSN and PELP1-cyto) expressing shGFP or shIKKϵ. All conditions were performed in triplicate, and the data are represented as the means with standard deviation. Target gene expression values were normalized over their matched 18S values. Student's t test was performed to test for statistically significant differences in gene expression between PELP1-cyto shGFP and PELP1-cyto shIKKϵ samples. C, qRT-PCR gene expression from MCF-10A LXSN and PELP1-cyto cells treated with 5 μm CYT387 for 18 h. All conditions were performed in triplicate, and data are represented as the means with standard deviation. Student's t test was performed to test for statistically significant differences in gene expression between PELP1-cyto DMSO control-treated samples and PELP1-cyto CYT387-treated samples. In B and C, *, p < 0.05.
Next, we examined PELP1-cyto-induced gene expression in shGFP and shIKKϵ MCF-10A cell lines by qRT-PCR. We found that a number of inflammatory cytokines and chemokines up-regulated in PELP1-cyto MCF-10A cells were down-regulated by IKKϵ shRNA (Table 1). Shown in Fig. 4B are three genes identified in our GGE studies (CXCL1, CCL20, and CSF3). Additionally, qRT-PCR revealed slightly higher IKKϵ RNA levels in PELP1-cyto-expressing cells than in LXSN-shGFP controls; IKKϵ RNA levels were knocked down by IKKϵ shRNA in both the LXSN and PELP1-cyto MCF-10A cells (Fig. 3B). Not all genes up-regulated in PELP1-cyto-expressing cells were dependent on IKKϵ up-regulation. One example, IL-1β, is shown in Fig. 4B, which was consistently up-regulated in PELP1-cyto HMECs (both HMEC-hTERT and MCF10A) but never modulated by IKKϵ shRNA. We also treated cells with CYT387, a kinase inhibitor previously shown to inhibit IKKϵ-induced inflammatory gene expression (21). Treatment of HMEC-hTERT PELP1-cyto cells with 5 μm of CYT387 for 18 h resulted in statistically significant reduction in expression of IL-8 and CXCL1 as compared with HMEC-hTERT PELP1-cyto cells treated with DMSO control (Fig. 4C). Of note, CYT387 treatment did not have an effect on IL-8 or CXCL1 expression in HMEC-hTERT LXSN cells. These experiments suggest that increased expression of IKKϵ downstream of PELP1 facilitates inflammatory gene expression.
To determine whether other IKK family members are involved in PELP1-cyto-induced inflammatory gene regulation, we first examined IKKα, IKKβ, and TBK1 protein levels an localization by Western blotting cytoplasmic and nuclear extracts prepared from MCF-10A and HMEC-hTERT cells expressing either LXSN control or PELP1-cyto. As shown in Fig. 5A, no differences in the cytoplasmic or nuclear levels of IKKα, IKKβ, or TBK1 were observed in PELP1-cyto cells compared with LXSN control cells. To confirm that these IKK family members were not essential for IKKϵ-dependent regulation of inflammatory gene expression in PELP1-cyto expressing cells, we expressed shRNA to each of these genes in MCF-10A cells and then performed qRT-PCR for CXCL1, CCL20, and CSF3. In contrast to IKKϵ shRNA, shRNA to IKKα, IKKβ, and TBK1 did not inhibit PELP1-cyto-induced inflammatory gene expression in MCF10A cells. In fact, knockdown of IKKβ and TBK1 actually increased expression of CXCL1, CCL20, and CSF3 (Fig. 5B). Thus, IKKϵ appears to be the predominate IKK required for PELP-cyto-induced inflammatory gene expression.
FIGURE 5.
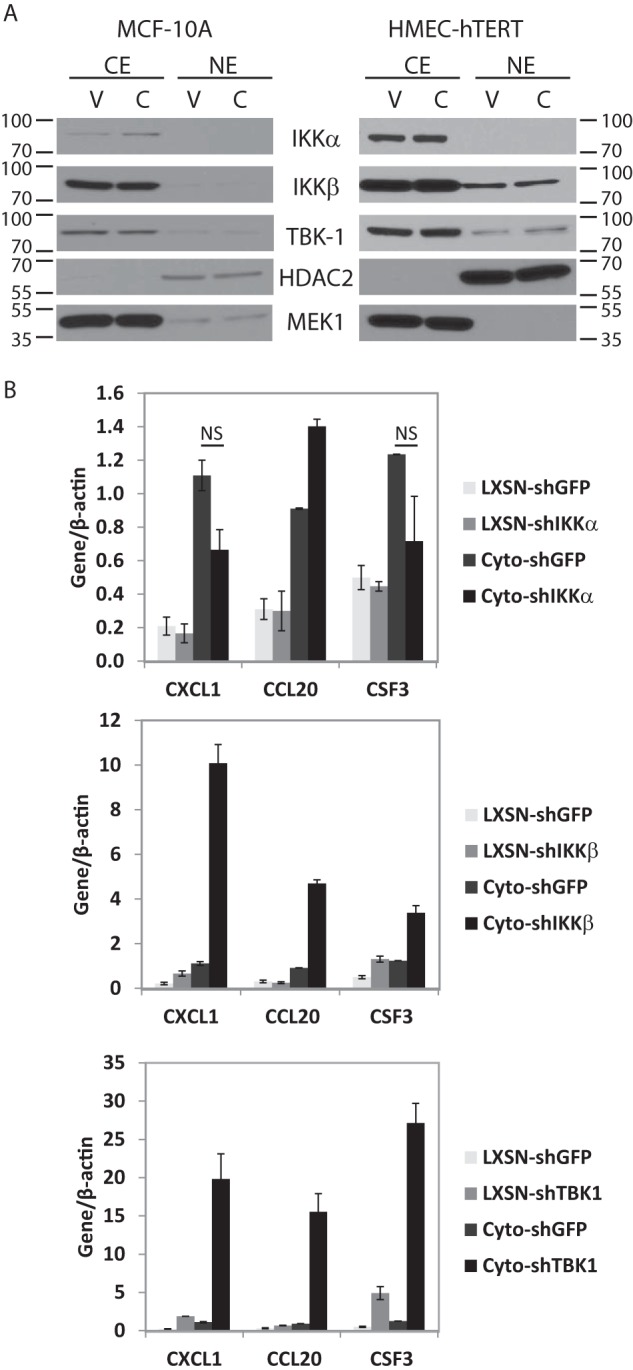
IKKα, IKKβ, and TBK1 do not regulate PELP1-cyto-induced inflammatory gene expression. A, MCF-10A and HMEC-hTERT lines expressing LXSN control (lanes V) or PELP1-cyto (lanes C) were examined by Western blotting of nuclear (NE) and cytoplasmic (CE) fractions to determine IKKα, IKKβ, and TBK1 expression levels and localization. HDAC2 and MEK1 were used as nuclear and cytoplasmic fractionation and loading controls, respectively. B, qRT-PCR for inflammatory gene expression from MCF-10A cells (LXSN and PELP1-cyto) expressing shGFP or shRNA targeting IKKα, IKKβ, or TBK1. Target gene expression values were normalized over their matched β-actin values. Student's t test was performed to test for statistically significant differences in gene expression between PELP1-cyto shGFP and PELP1-cyto shIKKα samples. NS, not significant.
Cytoplasmic PELP1 Promotes Macrophage Activation via IKKϵ
PELP1-cyto expression stimulated an increase in HMEC migration and three-dimensional acini formation (Fig. 1, B–D). We next tested whether IKKϵ expression is important for cytoplasmic PELP1-induced effects on cell migration and three-dimensional acini formation. Interestingly, IKKϵ shRNA did not significantly attenuate PELP1-cyto-induced EGF migration or abnormal acini formation (data not shown). However, because we found a significant increase in inflammatory chemokines and cytokines specifically in PELP1-cyto HMECs (Fig. 2D) and the known role of macrophages in breast tumor initiation and progression, we sought to determine whether paracrine signaling between PELP1-cyto HMECs and macrophages is a potential mechanism of PELP1-cyto-induced biology (27–30). We examined the effects of conditioned media (CM) from HMEC-hTERT and MCF-10A cells (LXSN and PELP1-cyto) on macrophage activation. The monocytic acute myeloid leukemia cell line, THP-1, was differentiated into macrophages with phorbol 12-myristate 13-acetate (PMA). THP-1 macrophages were then treated for 4 h with CM collected from HMECs expressing LXSN control or PELP1-cyto. CM from LXSN and PELP1-cyto cells induced expression of CCL20, IL-8, and IL-1β in differentiated THP-1 cells, but PELP1-cyto CM induced a more robust, statistically significant increase in expression of these genes (Fig. 6, A and B); this finding suggests HMECs expressing PELP1-cyto secrete paracrine factors (i.e. cytokines or chemokines) that promote macrophage activation.
FIGURE 6.

Cytoplasmic PELP1 localization in HMECs leads to activation and cross-talk with macrophages. A and B, representative qRT-PCR experiments from at least three independent experiments to measure gene expression from PMA-differentiated THP-1 cells that were incubated for 4 h in CM from HMEC-hTERT (A) or MCF-10A cells (B) expressing LXSN control or PELP1-cyto. The data are represented as the means with standard deviation of the target gene expression value normalized over the matched control gene 18S value (TBP-2 or 18S) of biological triplicates. C and D, representative experiments from at least three independent experiments for Transwell migration of HMEC-hTERT (C) or MCF10A (D) cell response to single-conditioned medium (CM) and DCM treatment for 18 h. All conditions were performed in triplicate, and the bars represent the average number of cells/well from four regions/Transwell with standard deviation. Student's t test was performed to determine the statistically significant differences in migration of HMECs exposed to LXSN and PELP1-cyto DCM. The p values are indicated on each graph. E, representative experiments from at least three independent experiments for Transwell migration of MCF-10A cell response to DCM treatment for 18 h. CM was taken from MCF-10A cells (LXSN and PELP1-cyto) expressing shGFP or shIKKϵ. MCF-10A cells were incubated with THP-1 cells to generate DCM. Student's t test was performed to test for statistically significant differences in migration of MCF-10A cells exposed to LXSN shGFP and PELP1-cyto shGFP DCM versus PELP1-cyto shGFP and PELP1-cyto shIKKϵ; the p values are indicated on each graph. F, model summarizing the data presented in Figs. 1–5. Cytoplasmic PELP1 expression, which has been shown to interact with growth factor receptors (GFR), induces an increase in IKKϵ protein expression (step 1), which leads to an increase in NF-κB-dependent gene expression (step 2). We propose that the secretion of these protein products from HMECs results in macrophage activation (step 3), which in turn secrete paracrine factors (step 4) that stimulates HMEC migration (step 5).
Macrophage activation influences the microenvironment to promote breast cancer initiation and progression through paracrine signaling. This includes stimulating new blood vessel growth, recruiting lymphocytes, and inducing migration of epithelial cells (31). Thus, we examined the effect of macrophage activation on HMEC migration. Differentiated THP-1 macrophages were incubated overnight in CM collected from HMECs expressing either LXSN or PELP1-cyto. This double conditioned media (DCM), first from LXSN or PELP1-cyto HMECs and then from THP-1 cells, was removed from THP-1 cells and used as the chemoattractant for HMEC-hTERT or MCF-10A cells in Transwell migration assays. DCM from PELP1-cyto cells induced a robust migratory effect as compared with LXSN DCM (Fig. 6, C and D). LXSN, PELP1-cyto, and THP-1 CM were used as controls, and very little migration was observed under these conditions.
Next, we determined whether the enhanced expression of IKKϵ in PELP1-cyto cells contributed to the migratory phenotype observed in response to PELP1-cyto DCM. DCM was generated from THP-1 cells incubated with CM from MCF-10A cells (LXSN or PELP1-cyto) expressing either shGFP or shIKKϵ. As expected, DCM from PELP-cyto/shGFP cells induced robust migration of MCF-10A cells as compared with DCM from LXSN/shGFP cells (p = 0.01). In contrast, MCF-10A cells exhibited a significant reduction in migration when exposed to DCM from PELP1-cyto/shIKKϵ cells as compared with DCM from PELP1-cyto/shGFP cells (Fig. 6E). Thus, increased expression of IKKϵ in PELP1-cyto HMECs contributes to macrophage activation that subsequently stimulates migration of HMECs through a loop of paracrine signaling. Interestingly, DCM from LXSN/shIKKϵ cells also displayed reduced migration as compared with LXSN/shGFP DCM, suggesting that IKKϵ expression is important for the migratory phenotype resulting from HMEC and macrophage paracrine cross-talk.
Discussion
Our study demonstrates a novel connection between cytoplasmic PELP1 signaling and breast cancer initiation phenotypes. We found that cytoplasmic PELP1 signaling in HMECs increased expression of inflammatory chemokines and cytokines via up-regulation of IKKϵ, leading to activation of macrophages. Interestingly, macrophage activation resulted in enhanced migration of HMECs. Thus, our data suggest that PELP1-cyto induced effects on the microenvironment may be an important mechanism of breast cancer initiation.
PELP1 Signaling and NF-κB Activation
IKK/NF-κB signaling is complex and context-dependent. Simplistically, canonical NF-κB activation involves cytokine-induced activation of the IKK complex containing IKKα/β/γ, phosphorylation and degradation of IκB proteins, and subsequent translocation of NF-κB homo- and heterodimers to the nucleus. Non-canonical NF-κB activation involves activation of IKKϵ, TBK1, IKKα, or NF-κB-inducing kinase and translocation of RelB/p52 or p50 heterodimers to the nucleus (32). Canonical and non-canonical NF-κB activation has a significant impact on cancer progression in a number of tumor types, including breast (21, 32–35). One prior report examined cytoplasmic PELP1 signaling on NF-κB activation and found that PELP1-cyto expression inhibited TNF-induced canonical NF-κB activation (13). We examined TNF-induced NF-κB activation and did not observe significant differences in IκBα or RelA/p65 phosphorylation in HMECs expressing PELP1-cyto as compared with control cells (data not shown), but these differences could be due to the use of the MCF-7 breast cancer cell line in the previous report and immortalized HMEC models in the present report. Interestingly, instead of canonical NF-κB activation, we observed up-regulation of the non-canonical IKK, IKKϵ. In our HMEC models, IKKϵ expression was required for expression of many, but not all, of the inflammatory cytokines and chemokines up-regulated by PELP1-cyto expression. IL-1β and other cytokines have been shown to induce IKKϵ expression (36), and co-expression of IL-1β and IKKϵ has been observed in immune-activated triple-negative breast cancer (21). Interestingly, IL-1β expression is increased 4–8-fold over control cells in our HMEC models. Knockdown of IKKϵ does not have an effect on IL-1β gene expression, suggesting that PELP1-cyto-induced expression of IL-1β may promote autocrine signaling that promotes IKKϵ expression, which then promotes expression of inflammatory cytokines and chemokines through activation of IKKϵ, NF-κB RelB, and/or RelA/p65 subunits. Of note, although we did observe modest increases in IKKϵ mRNA (Fig. 4B), increased IKKϵ protein expression is much greater than the relative increase in mRNA. This suggests that PELP1-cyto-induced IKKϵ up-regulation likely involves both transcriptional and post-transcriptional mechanisms.
Chronic NF-κB activation and its subsequent support of inflammatory signaling are known to promote tumor formation and aid in progression. Initially, NF-κB activation is necessary for immune system activation and destruction of transformed cells. However, this mechanism of clearance is typically not specific and potent enough to clear every malignant cell, which allows for subsequent adaptation and immune escape (37). This mechanism of tumor initiation may be exploited by cytoplasmic PELP1 signaling that drives sustained, non-canonical activation of NF-κB. Prior work from our lab demonstrating cytoplasmic PELP1 localization in asymptomatic, high risk women supports the idea that this may be an early event and driver of breast cancer initiation (14).
PELP1 Signaling, Breast Cancer Initiation, and Inflammatory Gene Expression
PELP1 dysregulation has been implicated in cellular transformation and tumorigenesis in breast cancer. Nuclear and cytoplasmic PELP1 signaling complexes have been shown to enhance cancer phenotypes both in vitro and in vivo. For example, in the nucleus PELP1 associates with chromatin remodeling complexes and regulates expression of genes involved in migration, invasion, and metastasis (17, 38, 39). In the cytoplasm PELP1 is associated with growth factor signaling pathways, such as the EGF receptor and promotes activation of Erk and Akt signaling pathways, which lead to tamoxifen resistance (10, 11). Fewer studies have been performed on the signaling functions of PELP1 in HMEC models, but Rajhans et al. (16) showed that PELP1 protein levels increased with increasing tumorigenicity in the MCF-10A model. Herein, we show that PELP1-cyto expression induces a multiacinar phenotype that is most similar to what has been observed with ErbB2 expression in MCF-10A cells (40). In a mouse model, mammary gland-specific PELP1 overexpression promotes hyperplasia and tumor formation of ER-positive carcinoma (12). A PELP1-cyto mammary gland specific mouse model has also been shown to induce hyperplasia and increase activation of Erk and Akt signaling (11). On the basis of the data presented here and our previously published work (14), we hypothesize that cytoplasmic PELP1 signaling is an oncogenic event. However, altered cellular localization cannot be tested using gene expression and alteration data available through cBioPortal or other genomic databases. Although PELP1-induced effects on proliferation are suspected to be the driving factor for hyperplasia and tumor formation in these models, effects on the tumor microenvironment have not been tested. Substantial work has demonstrated a strong link between chronic inflammation and carcinogenesis (41). PELP1 has recently been shown to induce expression of inflammatory genes in the brain that are critical for ER-mediated neuroprotection (42). Similarly, our work shows that cytoplasmic PELP1 drives inflammatory gene expression in HMECs. However, the inflammatory genes identified as regulated by PELP1 in the brain do not have significant overlap with the genes we have identified in HMECs; this is likely because the tissues and models are different (breast cancer initiation versus neuroprotection from global cerebral ischemia). Despite these differences in gene expression, our work and that of others indicates that PELP1 has the potential to regulate inflammatory processes in numerous tissues.
Macrophage Activation during Breast Cancer Initiation
During the last decade it has become increasingly appreciated that the tumor microenvironment plays an integral role in tumor initiation, progression, and metastasis (43). The tumor microenvironment contains adipocytes and immune, vascular, lymphatic, and fibroblast cells, as well as extracellular matrix. Tumor-associated macrophages (TAMs) have been found to promote breast cancer progression and metastasis in mouse models through paracrine signaling mechanisms (44–47), and the presence of TAMs in IBC is a poor prognostic indicator (29, 48). Importantly, TAMs have also been found associated with premalignant lesions (28) and the promotion of breast cancer initiation in mouse models (30). Macrophages have been characterized as M1 and M2, corresponding to pro- and anti-inflammatory functions within the wound healing environment, respectively. However, it has become increasingly clear that TAMs in the tumor microenvironment reside in a heterogeneous state between these two extreme phenotypes. For example, both pro-inflammatory cytokines (IL-1β and IL-6), which are considered M1, and anti-inflammatory cytokines (TGFβ and IL-10), which are considered M2, are pro-tumorigenic depending on the stage of tumor formation and progression. Herein, we show that conditioned media from PELP1-cyto HMECs can induce expression of CCL20, IL8, and IL-1β in THP-1 differentiated macrophages. Interestingly, chemokine (C-C motif) receptor 6, the receptor for CCL20, was recently shown to be important for mouse mammary tumor virus-polyoma middle T-induced tumor formation and the recruitment of pro-tumorigenic macrophages. CCL20 has also been shown to enhance migration and proliferation of breast epithelial cells (49, 50). The pro-inflammatory cytokine IL-1β has also been shown to play a role in breast cancer initiation and expression and has been linked to breast epithelial cell migration (51, 52). IL-8 is also known to enhance breast cancer cell survival and migration (53, 54). Thus, macrophage production of one or a combination of these cytokines/chemokines could be enhancing the HMEC migration we observed.
PELP1 or Inflammatory Markers as Biomarkers of Breast Cancer Initiation
In conclusion, the work presented here supports a model (Fig. 6F) in which altered localization of PELP1 to the cytoplasm results in expression of inflammatory cytokines, which promotes macrophage activation. Macrophage activation then results in production of paracrine factors that promote HMEC migration. Future studies are aimed at obtaining patient samples that contain BBD and examining PELP1 localization, IKKϵ expression, and inflammatory markers (such as the presence of macrophages or expression of CCL20, IL1β, or IL-8) to determine whether these markers could be developed as biomarkers of breast cancer initiation. These studies have the potential to directly impact clinical management of high risk women or women that have had breast biopsies positive for premalignant lesions, because these biomarkers could help guide early treatment decisions, such as surgical intervention and chemoprevention.
Experimental Procedures
Cell Lines and Reagents
MCF-10A and THP-1 cells were obtained from the American Type Culture Collection. HMEC-hTERT mammary epithelial cells were obtained from Lonza as primary cells and immortalized as previously described (55). MCF-10A cells were cultured as previously described (18). THP-1 cells were cultured in RPM1 1640 (Corning) supplemented with 10% fetal bovine serum and 0.05 mm 2-mercaptoethanol. HMEC-hTERT cells were cultured in HuMEC ready medium (1×) (Thermo Fisher Scientific).
Generation of PELP1 and IKKϵ Cell Lines
Retrovirus encoding an empty vector pLXSN or PELP1-cyto was generated as previously described (14). MCF-10A cells were transduced with control pLXSN or retrovirus encoding PELP1-cyto and were selected in 500 μg/ml of G418. Selected cells were plated as single cells to create clonal cell lines. Generation of HMEC-hTERT cell lines expressing PELP1 was described in Ref. 14. PELP1 expression and localization was confirmed using immunofluorescence and Western blotting.
We obtained five pLKO.1 shRNA constructs targeting IKKϵ from the RNAi Consortium. Lentivirus for all five shRNA was generated and tested in MCF-10A cells. Upon infection, the cells were selected in puromycin to generate a pooled population stably expressing the shRNA. IKKϵ mRNA transcript levels were measured by qRT-PCR to select the top knockdown construct. The target sequence achieving the greatest knockdown was 5′-GAGCATTGGAGTGACCTTGTA-3′. The results were verified by Western blotting for IKKϵ protein expression.
Western Blots
WCE were collected using supplemented RIPA buffer as previously described (14). Nuclear and cytoplasmic fractions were collected using the NE-PER nuclear protein extraction kit (Thermo Scientific). Lysates were quantitated, prepared, and resolved on an SDS-PAGE gel; transferred to polyvinylidene difluoride membrane; and processed for Western blotting as described in Ref. 14. The following antibodies were used: PELP1 (A300-180A-2; Bethyl Laboratories, Inc.), HDAC2, and p-RelB (sc-7899, lot no. E3014 and sc-101792, lot no. B2213; Santa Cruz Biotechnology, Inc.), MEK1 (07-641, lot no. 27102; EMD Millipore), and IKKϵ (D20G4, lot no. 2; Cell Signaling Technology).
Transwell Migration
HMEC-hTERT or MCF-10A cells in growth medium were trypsinized, washed once with PBS, resuspended in starvation medium, and counted. 750 μl of experimental medium was placed into the lower chamber of a 24-well plate. 5 × 104 cells in 350 μl were plated into the top of each 8-μm Transwell (Falcon) and placed into an incubator for 18 h. After 18 h, the top chamber was cleared of cells with a cotton-tipped applicator, washed in PBS, fixed in 4% paraformaldehyde, and stained with hematoxylin. Transwells were imaged at 10×, and four fields of migrated cells were counted/well. Each condition was performed in triplicate.
Scratch Wound Assay
In triplicate for each condition, 3 × 105 MCF-10A cells were plated into each well of a 12-well plate to achieve confluency the next day. At confluency, the cells were scratched with a pipette tip, rinsed with PBS to remove cell debris, placed into their experimental conditions, and imaged immediately (time = 0). The cells were returned to the incubator and imaged in the same location at 18 h. ImageJ was used to determine the percentage of scratch wound closure after 18 h had elapsed.
Three-dimensional Culture
MCF-10A cells expressing LXSN control or PELP1-cyto were grown in three-dimensional cultures as previously described by others (18). The medium was changed three times per week, and after 14 days the structures were fixed with 4% paraformaldehyde and stained with DAPI. Four fields/well and three wells/condition were imaged, and those that were not spherical and without a hollow lumen were considered abnormal.
Whole Genome Expression Analysis
RNA from hTERT-HMEC cells expressing control pLXSN or overexpression of PELP1-wt or PELP1-cyto was extracted in duplicate, independently processed, and hybridized to an Illumina bead chip HT-12v4 according to the manufacturer's protocols. Raw expression values were exported from BeadStudio software (Illumina) and imported into R software using the lumi package (56), in which values were log2-transformed and quantile-normalized. 27,382 probes (58%) were detected, and multiple probes that target the same gene were collapsed into a single value using the MaxMean algorithm in the R package gene filter. Differentially expressed genes were analyzed using the limma package using empirical Bayes. Group comparisons were reported with log2 fold change and the Benjamini and Hochberg (57) adjusted p value. Unsupervised hierarchical clustering of genes (scaled) was carried out using Euclidean distance and average linkage via the heat map function in the R package NMF (58).
IPA (Qiagen) was run with transformed and normalized expression data. IPA core analysis was completed with default settings, and a comparison analysis was completed with a fold change of ≥2.0, where p values were adjusted using the Benjamini and Hochberg method.
GSEA was performed with transformed and normalized expression data, and gene sets were derived from the MSigDB, version 5.1. Default settings were used, except genes were ranked by Diff_of_Classes and permutation type was gene_set. Enriched gene sets were considered significant with FDR < 0.05. GGE data has been submitted to the Gene Expression Omnibus (accession number GSE81447).
RNA Isolation, cDNA Synthesis, and qRT-PCR
RNA isolation was carried out using TriPure isolation reagent (Roche). cDNA synthesis and qRT-PCR were performed as previously described (14). Briefly, 2 × 105 cells were plated into 6-well plates and harvested after 24 h. In subsequent qRT-PCR, target gene expression was normalized over the expression of a housekeeping gene 18S, TBP-2, or β-actin. Primer sequences are included in supplemental Table S2.
Generation of Conditioned Media
Generation of CM and DCM was completed as follows. On day 1, THP-1 cells were plated at a density of 5 × 106 cells in growth medium with 20 ng/ml of PMA. HMECs were plated at 2 × 106 cells in a 100-mm dish. On day 2, the cells were starved in serum-free RPMI 1640. On day 3, CM was collected from the HMECs and THP-1 cell lines and centrifuged clarify the medium. CM was added to the dishes of THP-1 cells to generate DCM. On day 4, the DCM was collected from dishes of THP-1 cells and clarified by centrifugation.
Author Contributions
J. H. O. conceived and coordinated the study and wrote the paper. B. J. G. designed, performed, and analyzed the experiments present in all figures and contributed to the preparation of the manuscript. T. P. K. assisted in the design and analysis of the GGE data presented in Figs. 2 and 3. B. K. performed and analyzed experiments in Figs. 3 and 4. L. M. designed, performed, and analyzed experiments in Fig. 1. K. L. S. assisted in the design of experiments in Fig. 5. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Michael Franklin for critical reading and editing of the manuscript and Elizabeth Benner for technical assistance.
This work was supported in part by National Institutes of Health Grant K07CA131501, Institutional Research Grant 124166-IRG-58-001-52-IRG5 from the American Cancer Society, National Center for Advancing Translational Sciences of the National Institutes of Health Award UL1TR000114, and funds from the Masonic Cancer Center, University of Minnesota (to J. H. O.). J. H. O. received support from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1 and S2.
- IBC
- invasive breast cancer
- AH
- atypical hyperplasia
- BBD
- benign breast disease
- CCL20
- chemokine (C-C motif) ligand 20
- CM
- conditioned medium
- CXCL1
- chemokine (C-X-C motif) ligand 1
- DCIS
- ductal carcinoma in situ
- DCM
- double conditioned media
- ER
- estrogen receptor
- GGE
- global gene expression
- GSEA
- gene set enrichment analysis
- HDAC2
- histone deacetylase 2
- HMEC
- human mammary epithelial cell
- IKK
- inhibitor of κB kinase
- IPA
- Ingenuity Pathway Analysis
- MEK1
- mitogen-activated protein kinase kinase
- MSigDB
- Molecular Signatures Database
- PELP1
- proline, leucine, glutamic acid-rich protein 1
- PMA
- phorbol 12-myristate 13-acetate
- qRT-PCR
- quantitative real time PCR
- RPMI
- Roswell Park Memorial Institute
- TAM
- tumor-associated macrophage
- TBK1
- TANK-binding kinase 1
- WCE
- whole cell extract.
References
- 1. Davidson N. E., and Rimm D. L. (2015) Expertise vs evidence in assessment of breast biopsies: an atypical science. JAMA 313, 1109–1110 [DOI] [PubMed] [Google Scholar]
- 2. Hartmann L. C., Degnim A. C., and Dupont W. D. (2015) Atypical hyperplasia of the breast. N. Engl. J. Med. 372, 1271–1272 [DOI] [PubMed] [Google Scholar]
- 3. Degnim A. C., Visscher D. W., Berman H. K., Frost M. H., Sellers T. A., Vierkant R. A., Maloney S. D., Pankratz V. S., de Groen P. C., Lingle W. L., Ghosh K., Penheiter L., Tlsty T., Melton L. J. 3rd, Reynolds C. A., et al. (2007) Stratification of breast cancer risk in women with atypia: a Mayo cohort study. J. Clin. Oncol. 25, 2671–2677 [DOI] [PubMed] [Google Scholar]
- 4. Vadlamudi R. K., Wang R. A., Mazumdar A., Kim Y., Shin J., Sahin A., and Kumar R. (2001) Molecular cloning and characterization of PELP1, a novel human coregulator of estrogen receptor α. J. Biol. Chem. 276, 38272–38279 [DOI] [PubMed] [Google Scholar]
- 5. Girard B. J., Daniel A. R., Lange C. A., and Ostrander J. H. (2014) PELP1: a review of PELP1 interactions, signaling, and biology. Mol. Cell. Endocrinol. 382, 642–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cortez V., Mann M., Tekmal S., Suzuki T., Miyata N., Rodriguez-Aguayo C., Lopez-Berestein G., Sood A. K., and Vadlamudi R. K. (2012) Targeting the PELP1-KDM1 axis as a potential therapeutic strategy for breast cancer. Breast Cancer Res. 14, R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fanis P., Gillemans N., Aghajanirefah A., Pourfarzad F., Demmers J., Esteghamat F., Vadlamudi R. K., Grosveld F., Philipsen S., and van Dijk T. B. (2012) Five friends of methylated chromatin target of protein-arginine-methyltransferase[prmt]-1 (chtop), a complex linking arginine methylation to desumoylation. Mol. Cell. Proteomics 11, 1263–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mann M., Cortez V., and Vadlamudi R. (2013) PELP1 oncogenic functions involve CARM1 regulation. Carcinogenesis 34, 1468–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nair S. S., Mishra S. K., Yang Z., Balasenthil S., Kumar R., and Vadlamudi R. K. (2004) Potential role of a novel transcriptional coactivator PELP1 in histone H1 displacement in cancer cells. Cancer Res. 64, 6416–6423 [DOI] [PubMed] [Google Scholar]
- 10. Vadlamudi R. K., Manavathi B., Balasenthil S., Nair S. S., Yang Z., Sahin A. A., and Kumar R. (2005) Functional implications of altered subcellular localization of PELP1 in breast cancer cells. Cancer Res. 65, 7724–7732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumar R., Zhang H., Holm C., Vadlamudi R. K., Landberg G., and Rayala S. K. (2009) Extranuclear coactivator signaling confers insensitivity to tamoxifen. Clin. Cancer Res. 15, 4123–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cortez V., Samayoa C., Zamora A., Martinez L., Tekmal R. R., and Vadlamudi R. K. (2014) PELP1 Overexpression in the mouse mammary gland results in the development of hyperplasia and carcinoma. Cancer Res. 74, 7395–7405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rayala S. K., Mascarenhas J., Vadlamudi R. K., and Kumar R. (2006) Altered localization of a coactivator sensitizes breast cancer cells to tumor necrosis factor-induced apoptosis. Mol. Cancer Ther. 5, 230–237 [DOI] [PubMed] [Google Scholar]
- 14. Girard B. J., Regan Anderson T. M., Welch S. L., Nicely J., Seewaldt V. L., and Ostrander J. H. (2015) Cytoplasmic PELP1 and ERRgamma protect human mammary epithelial cells from Tam-induced cell death. PLoS One 10, e0121206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chakravarty D., Nair S. S., Santhamma B., Nair B. C., Wang L., Bandyopadhyay A., Agyin J. K., Brann D., Sun L. Z., Yeh I. T., Lee F. Y., Tekmal R. R., Kumar R., and Vadlamudi R. K. (2010) Extranuclear functions of ER impact invasive migration and metastasis by breast cancer cells. Cancer Res. 70, 4092–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rajhans R., Nair S., Holden A. H., Kumar R., Tekmal R. R., and Vadlamudi R. K. (2007) Oncogenic potential of the nuclear receptor coregulator proline-, glutamic acid-, leucine-rich protein 1/modulator of the nongenomic actions of the estrogen receptor. Cancer Res. 67, 5505–5512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roy S., Chakravarty D., Cortez V., De Mukhopadhyay K., Bandyopadhyay A., Ahn J. M., Raj G. V., Tekmal R. R., Sun L., and Vadlamudi R. K. (2012) Significance of PELP1 in ER-negative breast cancer metastasis. Mol. Cancer Res. 10, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Debnath J., Muthuswamy S. K., and Brugge J. S. (2003) Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268 [DOI] [PubMed] [Google Scholar]
- 19. Mootha V. K., Lindgren C. M., Eriksson K. F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., Houstis N., Daly M. J., Patterson N., Mesirov J. P., Golub T. R., et al. (2003) PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 [DOI] [PubMed] [Google Scholar]
- 20. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barbie T. U., Alexe G., Aref A. R., Li S., Zhu Z., Zhang X., Imamura Y., Thai T. C., Huang Y., Bowden M., Herndon J., Cohoon T. J., Fleming T., Tamayo P., Mesirov J. P., et al. (2014) Targeting an IKBKE cytokine network impairs triple-negative breast cancer growth. J. Clin. Invest. 124, 5411–5423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boehm J. S., Zhao J. J., Yao J., Kim S. Y., Firestein R., Dunn I. F., Sjostrom S. K., Garraway L. A., Weremowicz S., Richardson A. L., Greulich H., Stewart C. J., Mulvey L. A., Shen R. R., Ambrogio L., et al. (2007) Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 129, 1065–1079 [DOI] [PubMed] [Google Scholar]
- 23. Adli M., and Baldwin A. S. (2006) IKKι/IKKϵ controls constitutive, cancer cell-associated NF-κB activity via regulation of Ser-536 p65/RelA phosphorylation. J. Biol. Chem. 281, 26976–26984 [DOI] [PubMed] [Google Scholar]
- 24. Eddy S. F., Guo S., Demicco E. G., Romieu-Mourez R., Landesman-Bollag E., Seldin D. C., and Sonenshein G. E. (2005) Inducible IκB kinase/IκB kinase ϵ expression is induced by CK2 and promotes aberrant nuclear factor-κB activation in breast cancer cells. Cancer Res. 65, 11375–11383 [DOI] [PubMed] [Google Scholar]
- 25. Hutti J. E., Shen R. R., Abbott D. W., Zhou A. Y., Sprott K. M., Asara J. M., Hahn W. C., and Cantley L. C. (2009) Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKϵ promotes cell transformation. Mol. Cell 34, 461–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shen R. R., Zhou A. Y., Kim E., Lim E., Habelhah H., and Hahn W. C. (2012) IκB kinase ϵ phosphorylates TRAF2 to promote mammary epithelial cell transformation. Mol. Cell. Biol. 32, 4756–4768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bohrer L. R., and Schwertfeger K. L. (2012) Macrophages promote fibroblast growth factor receptor-driven tumor cell migration and invasion in a CXCR2-dependent manner. Mol. Cancer Res. 10, 1294–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hussein M. R., and Hassan H. I. (2006) Analysis of the mononuclear inflammatory cell infiltrate in the normal breast, benign proliferative breast disease, in situ and infiltrating ductal breast carcinomas: preliminary observations. J. Clin. Pathol. 59, 972–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Medrek C., Pontén F., Jirström K., and Leandersson K. (2012) The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 12, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schwertfeger K. L., Xian W., Kaplan A. M., Burnett S. H., Cohen D. A., and Rosen J. M. (2006) A critical role for the inflammatory response in a mouse model of preneoplastic progression. Cancer Res. 66, 5676–5685 [DOI] [PubMed] [Google Scholar]
- 31. Mantovani A., Schioppa T., Porta C., Allavena P., and Sica A. (2006) Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 25, 315–322 [DOI] [PubMed] [Google Scholar]
- 32. Baldwin A. S. (2012) Regulation of cell death and autophagy by IKK and NF-κB: critical mechanisms in immune function and cancer. Immunol. Rev. 246, 327–345 [DOI] [PubMed] [Google Scholar]
- 33. Shen R. R., and Hahn W. C. (2011) Emerging roles for the non-canonical IKKs in cancer. Oncogene 30, 631–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kendellen M. F., Bradford J. W., Lawrence C. L., Clark K. S., and Baldwin A. S. (2014) Canonical and non-canonical NF-κB signaling promotes breast cancer tumor-initiating cells. Oncogene 33, 1297–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang Z., Liu J. C., Chung P. E., Egan S. E., and Zacksenhaus E. (2014) Targeting HER2+ breast cancer: the TBK1/IKKϵ axis. Oncoscience 1, 180–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shimada T., Kawai T., Takeda K., Matsumoto M., Inoue J., Tatsumi Y., Kanamaru A., and Akira S. (1999) IKKι, a novel lipopolysaccharide-inducible kinase that is related to IκB kinases. Int. Immunol. 11, 1357–1362 [DOI] [PubMed] [Google Scholar]
- 37. Hoesel B., and Schmid J. A. (2013) The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 12, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chakravarty D., Roy S. S., Babu C. R., Dandamudi R., Curiel T. J., Vivas-Mejia P., Lopez-Berestein G., Sood A. K., and Vadlamudi R. K. (2011) Therapeutic targeting of PELP1 prevents ovarian cancer growth and metastasis. Clin. Cancer Res. 17, 2250–2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roy S. S., Gonugunta V. K., Bandyopadhyay A., Rao M. K., Goodall G. J., Sun L. Z., Tekmal R. R., and Vadlamudi R. K. (2014) Significance of PELP1/HDAC2/miR-200 regulatory network in EMT and metastasis of breast cancer. Oncogene 33, 3707–3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muthuswamy S. K., Li D., Lelievre S., Bissell M. J., and Brugge J. S. (2001) ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol. 3, 785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coussens L. M., and Werb Z. (2002) Inflammation and cancer. Nature 420, 860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sareddy G. R., Zhang Q., Wang R., Scott E., Zou Y., O'Connor J. C., Chen Y., Dong Y., Vadlamudi R. K., and Brann D. (2015) Proline-, glutamic acid-, and leucine-rich protein 1 mediates estrogen rapid signaling and neuroprotection in the brain. Proc. Natl. Acad. Sci. U.S.A. 112, E6673–E6682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barcellos-Hoff M. H., Lyden D., and Wang T. C. (2013) The evolution of the cancer niche during multistage carcinogenesis. Nat. Rev. Cancer 13, 511–518 [DOI] [PubMed] [Google Scholar]
- 44. Lin E. Y., Li J. F., Bricard G., Wang W., Deng Y., Sellers R., Porcelli S. A., and Pollard J. W. (2007) Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol. Oncol. 1, 288–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin E. Y., Li J. F., Gnatovskiy L., Deng Y., Zhu L., Grzesik D. A., Qian H., Xue X. N., and Pollard J. W. (2006) Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 66, 11238–11246 [DOI] [PubMed] [Google Scholar]
- 46. Wyckoff J., Wang W., Lin E. Y., Wang Y., Pixley F., Stanley E. R., Graf T., Pollard J. W., Segall J., and Condeelis J. (2004) A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 64, 7022–7029 [DOI] [PubMed] [Google Scholar]
- 47. Lin E. Y., Nguyen A. V., Russell R. G., and Pollard J. W. (2001) Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 193, 727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leek R. D., Lewis C. E., Whitehouse R., Greenall M., Clarke J., and Harris A. L. (1996) Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 56, 4625–4629 [PubMed] [Google Scholar]
- 49. Marsigliante S., Vetrugno C., and Muscella A. (2013) CCL20 induces migration and proliferation on breast epithelial cells. J. Cell. Physiol. 228, 1873–1883 [DOI] [PubMed] [Google Scholar]
- 50. Marsigliante S., Vetrugno C., and Muscella A. (2016) Paracrine CCL20 loop induces epithelial-mesenchymal transition in breast epithelial cells. Mol. Carcinog. 55, 1175–1186 [DOI] [PubMed] [Google Scholar]
- 51. Pantschenko A. G., Pushkar I., Anderson K. H., Wang Y., Miller L. J., Kurtzman S. H., Barrows G., and Kreutzer D. L. (2003) The interleukin-1 family of cytokines and receptors in human breast cancer: implications for tumor progression. Int. J. Oncol. 23, 269–284 [PubMed] [Google Scholar]
- 52. Goldberg J. E., and Schwertfeger K. L. (2010) Proinflammatory cytokines in breast cancer: mechanisms of action and potential targets for therapeutics. Current Drug Targets 11, 1133–1146 [DOI] [PubMed] [Google Scholar]
- 53. Hartman Z. C., Poage G. M., den Hollander P., Tsimelzon A., Hill J., Panupinthu N., Zhang Y., Mazumdar A., Hilsenbeck S. G., Mills G. B., and Brown P. H. (2013) Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 73, 3470–3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mohamed M. M., El-Ghonaimy E. A., Nouh M. A., Schneider R. J., Sloane B. F., and El-Shinawi M. (2014) Cytokines secreted by macrophages isolated from tumor microenvironment of inflammatory breast cancer patients possess chemotactic properties. Int. J. Biochem. Cell Biol. 46, 138–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ostrander J. H., McMahon C. M., Lem S., Millon S. R., Brown J. Q., Seewaldt V. L., and Ramanujam N. (2010) Optical redox ratio differentiates breast cancer cell lines based on estrogen receptor status. Cancer Res. 70, 4759–4766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Benjamini Y., and Hochberg Y. (1995) Controlling the false descovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 [Google Scholar]
- 58. Gaujoux R., and Seoighe C. (2010) A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.