

Abstract
The epithelial sodium channel (ENaC) is the limiting entry point for Na+ reabsorption in the distal kidney nephron and is regulated by numerous hormones, including the mineralocorticoid hormone aldosterone. Previously we identified ankyrin G (AnkG), a cytoskeletal protein involved in vesicular transport, as a novel aldosterone-induced protein that can alter Na+ transport in mouse cortical collecting duct cells. However, the mechanisms underlying AnkG regulation of Na+ transport were unknown. Here we report that AnkG expression directly regulates Na+ transport by altering ENaC activity in the apical membrane. Increasing AnkG expression increased ENaC activity while depleting AnkG reduced ENaC-mediated Na+ transport. These changes were due to a change in ENaC directly rather than through alterations to the Na+ driving force created by Na+/K+-ATPase. Using a constitutively open mutant of ENaC, we demonstrate that the augmentation of Na+ transport is caused predominantly by increasing the number of ENaCs at the surface. To determine the mechanism of AnkG action on ENaC surface number, changes in rates of internalization, recycling, and membrane delivery were investigated. AnkG did not alter ENaC delivery to the membrane from biosynthetic pathways or removal by endocytosis. However, AnkG did alter ENaC insertion from constitutive recycling pathways. These findings provide a mechanism to account for the role of AnkG in the regulation of Na+ transport in the distal kidney nephron.
Keywords: epithelial sodium channel (ENaC), kidney, membrane trafficking, sodium transport, vesicles, Ankyrin, apical transport, collecting duct, distal nephron
Introduction
The epithelial sodium channel (ENaC)2 in the distal nephron of the kidney plays a critical role in responding to hormonal cues that maintain blood pressure homeostasis (1–3). Following a decrease in blood volume or plasma sodium (Na+) concentration, a hormonal cascade results in aldosterone release to stimulate Na+ transport. Transcription and translation of ENaC and other aldosterone-sensitive proteins is increased in response to aldosterone, to increase Na+ reabsorption and maintain Na+ and volume homeostasis. Because ENaC is the rate-limiting step for Na+ reabsorption across the distal nephron, dysregulation of the channel has been linked to both hypo- and hypertension (2, 4, 5), indicating its significance in kidney physiology and Na+ homeostasis.
ENaC is a heterotrimeric channel composed of α-, β-, and γ-subunits, and its activity is determined by the number of channels (N) at the apical membrane and the open probability (Po) or activity of those channels. Numerous factors alter ENaC N and/or Po at the apical surface (6–9). ENaC residing in the apical plasma membrane is ubiquitinylated by the E3 ligase Nedd4-2 and internalized to reduce apical channel density. The channel can then either recycle to the apical membrane via constitutive vesicle recycling pathways or be targeted for degradation by the lysosome (10–12). Several studies have reported that cytoskeletal proteins have a dynamic role in controlling ENaC activity. Actin binds the C terminus of α-ENaC (13) and increases ENaC activity (7, 14–16), whereas disruption of the actin cytoskeleton eliminates ENaC activation by small G proteins (17). The cortical actin-associated protein cortactin reduces ENaC activity by reducing Po via a link to the Arp2/3 complex (9). ENaC co-immunoprecipitates with ankyrin G (AnkG) and α-spectrin (18), and the association with α-spectrin may assist apical membrane targeting (19). These studies highlight the importance of the cytoskeleton in Na+ transport regulation and link AnkG to ENaC.
AnkG is found in a variety of tissues, including the kidneys, lungs, heart, and brain (20–22). In neurons and cardiomyocytes, AnkG is essential for assembly of the axon initial segment (23–25) and the intercalated disk (26, 27), respectively, and elimination of AnkG results in mislocalized voltage-gated Na+ and K+ channels (25, 28, 29). In the lungs and kidney, AnkG plays a role in lateral membrane biogenesis (30–32) and has been shown to regulate endocytosis of E-cadherin (33). Electron microscopic images from the kidney have shown that AnkG is localized to both the apical and basolateral submembranous cytoskeleton (34), where it may associate with a wide range of transmembrane proteins. This diverse range of activities suggests that AnkG may have multiple distinct roles depending on where it is located and the proteins with which it interacts.
Although several studies have examined AnkG association with and regulation of voltage-gated channels (29, 35–37), the functional significance of AnkG interaction with ENaC has not been examined. We recently identified AnkG as a potential regulator of Na+ transport in mouse cortical collecting duct (mCCD) cells (38). AnkG expression is modified by aldosterone-sensitive microRNAs (38). Here, we examined the role of AnkG on ENaC apical delivery, recycling, and internalization to determine the mechanism underlying the increase by AnkG of Na+ transport in mCCD cells.
Results
AnkG Is Expressed at the Plasma Membrane Where It Alters Vectorial Na+ Transport
We previously showed that AnkG expression is altered by aldosterone via a change in microRNA expression (38). AnkG localization was determined by immunofluorescent confocal imaging. As shown in the z stack projection images in Fig. 1 (A and B), AnkG is localized at the plasma membrane, as well as some intracellular punctae. The cortical actin ring was used to establish mCCD cell polarity. To test whether a change in AnkG expression altered ENaC activity, AnkG was knocked down or overexpressed in mCCD cells, and ENaC-mediated Na+ transport determined by short circuit current (Isc) measurements in Ussing chambers. Reducing AnkG expression resulted in a 69 ± 2% (n = 15) reduction in the amiloride-sensitive Isc, whereas overexpressed AnkG increased ENaC currents by 67 ± 11% (Fig. 1, C and D). Alterations in AnkG expression were verified by Western blotting (see Fig. 3).
FIGURE 1.

AnkG expression in mCCD cells alters ENaC activity. A, confocal images of immunofluorescently labeled AnkG (green) in mCCD cells. B, magnified z stack images of mCCD cells with AnkG (green) and actin (red). The middle panel shows labeling of the cortical actin ring, and the right panel demonstrates AnkG at apical, lateral, and basal submembranous surfaces. C, representative short circuit current (Isc) recordings of ENaC activity with control (black), AnkG KD (light gray), and OE (dashed, dark gray). 10 μm amiloride is added at the end of the recording to determine the ENaC-specific contribution of the current recordings. D, summary of normalized Isc from several experiments. AnkG KD reduces ENaC current (0.31 ± 0.06 versus 1.00 ± 0.03, n ≥ 15, p < 0.001), and AnkG OE increases ENaC current (1.67 ± 0.11 versus 1.00 ± 0.06, n ≥ 12, p < 0.001).
FIGURE 3.

Altering AnkG does not impact whole cell ENaC expression but does alter surface channel number. A, representative Western blots showing total ENaC subunit expression with AnkG OE and KD relative to control. B, summary graph of normalized densitometry from Western blots analyzing the expression levels of AnkG and ENaC and the Na+/K+-ATPase expression. AnkG KD reduced total AnkG expression levels (0.35 ± 0.10 versus 1.00 ± 0.12, n = 5; p < 0.01). AnkG KD did not significantly affect the expression levels of any ENaC subunits or Na+/K+-ATPase in mCCD cells. AnkG OE also had no effect on ENaC subunits, Na+/K+-ATPase, or endogenous AnkG. C, summarized results of the effect of FRTs expressing WT or βS518K (locked) ENaC with AnkG. AnkG OE increases βS518K current (1.55 ± 0.11, n = 11) similarly to WT (1.45 ± 0.05, n = 45), demonstrating that AnkG OE altered ENaC N at the membrane surface.
Na+/K+-ATPase Localization and Transport Activity Is Unaffected by AnkG Expression
Previous studies have shown that all three ankyrin family members, AnkR, AnkB, and AnkG can bind to domains in Na+/K+-ATPase (44–48) and that AnkR expression is required for Na+/K+-ATPase targeting to the plasma membrane (49). Because the Na+/K+- ATPase is critical for establishing the electrochemical gradient that drives Na+ entry through ENaC, we wanted to verify that changes in vectorial Na+ transport observed in Fig. 1 were due to changes in ENaC activity at the apical surface rather than through indirect changes to the Na+/K+-ATPase localization or function. Using immunofluorescence labeling in polarized mCCDs, we determined that AnkG knockdown in mCCD cells did not impact basolateral targeting of the Na+/K+-ATPase (Fig. 2A). We observed a decrease in lateral height (6.2 ± 0.12 μm versus 5.4 ± 0.14 μm, n ≥39, p < 0.001) and an increase in cell width (7.9 ± 0.48 μm versus 9.5 ± 0.43 μm, n = 30, p < 0.05) with AnkG knockdown (Fig. 2B), which is consistent with previous studies of AnkG knockdown in epithelial cells (30, 31, 33). Consistent with an increase in cell width to compensate for decreased lateral height, capacitance measurements of apical and basolateral surface area confirmed that AnkG knockdown and overexpression did not significantly alter total membrane surface area in the mCCD cells (Fig. 2F).
FIGURE 2.

AnkG modifies Na+ transport by regulating ENaC, and not Na+/K+-ATPase. A, representative projection image of α-Na+/K+-ATPase (green) in control or mCCD cells with AnkG KD. White scale bars are 10 μm. B, lateral heights and cell widths were determined using line analysis. In control cells, the average lateral height of mCCDs grown on filters was 6.2 ± 0.1 μm (n = 50), and the average cell width was 7.9 ± 0.5 μm (n = 30). In contrast, cells with AnkG knockdown had significantly shorter lateral heights of 5.4 ± 0.1 (n = 39, p < 0.001), and significantly longer cell widths (9.4 ± 0.4, n = 30, p < 0.05). C, representative current (black) and capacitance (gray) traces of apical membrane permeabilization. The spike in current activity following permeabilization is the result of the rapid influx of Na+. After reaching a steady state, Na+/K+-ATPase current was determined by addition of ouabain (30 μm). D, representative current (black) and capacitance (gray) traces depicting basolateral permeabilization. ENaC current was determined by the addition of amiloride (10 μm). E, results of AnkG KD on isolated Na+/K+-ATPase and ENaC currents. AnkG KD did not alter Na+/K+-ATPase conductance; however, it significantly reduced ENaC current (0.57 ± 0.04, n = 5 versus 1.00 ± 0.14, n = 4; p < 0.05). F, summarized capacitance data. AnkG KD did not impact the apical or basolateral surface area of polarized mCCD epithelial monolayers.
To test the impact of AnkG knockdown on isolated Na+/K+-ATPase and ENaC currents, we selectively permeabilized the apical or basolateral membranes of polarized monolayers, respectively, in modified Ussing chambers to isolate each membrane domain. Using the ionophore nystatin, membrane permeablizations were carried out as described previously (42). To verify permeabilization of the apical membrane, 10 μm amiloride was applied to the apical surface to demonstrate that the amiloride-sensitive current was eliminated. Membrane capacitance measurements performed concurrently with Isc recordings verified apical permeabilization caused by the appearance of basolateral membrane capacitance. We observed a spike in Isc following apical permeabilization (Fig. 2C), the result of rapid influx of Na+ into cells from the higher extracellular concentrations. Within a minute, the pump activity reached a plateau, at which point ouabain was added to determine the steady state Na+/K+-ATPase activity. For basolateral permeabilizations, experiments were performed with a low Na+ concentration Ringer's solution in the basolateral chamber to provide a driving force for Na+ entry via ENaC when the Na+/K+-ATPase transport was eliminated as previously described (42). Basolateral permeabilization was verified by application of 30 μm ouabain, which did not significantly impact Na+ transport through ENaC (Fig. 2D). Na+/K+-ATPase activity and membrane surface area were unaffected by AnkG knockdown; however, ENaC activity was significantly reduced (Fig. 2E), suggesting the that changes in Na+ transport seen in Fig. 1 were due to changes in ENaC activity at the apical surface.
AnkG Does Not Alter ENaC Whole Cell Expression Levels, but Increases Surface Number
To test whether the increased AnkG altered endogenous whole cell ENaC expression, Western blots of cell lysates from mCCD cells in which AnkG was depleted or overexpressed were carried out. Knockdown of AnkG reduced total AnkG expression but did not alter expression of the ENaC subunits or the α-subunit of Na+/K+-ATPase. Likewise, AnkG overexpression did not change endogenous AnkG, ENaC, or Na+/K+-ATPase protein levels (Fig. 3, A and B).
ENaC current is determined by the number and the open probability (Po) of the channels at the plasma membrane. Because AnkG is linked to the cytoskeleton and actin binding to ENaC has been shown to alter channel Po, we next wanted to determine whether AnkG regulation of ENaC activity was via a change in Po. In these experiments we used Fisher rat thyroid (FRT) cells. Unlike mCCD cells, the FRT cells lack endogenous ENaC. Experiments utilizing ENaC mutations were all performed in FRT cells to exclude the contribution of endogenously expressed ENaC from WT channels that would be present in mCCD cells. FRT cells were transfected with wild type α,β,γ-ENaC or α,β(S518K), γ-ENaC. The β-ENaC mutant locks the channel in an open confirmation, resulting in a channel that has a Po close to 1 (50). If AnkG altered ENaC Po, preventing this regulation by using the “locked” mutant would eliminate the increase in ENaC-mediated Na+ transport observed with AnkG overexpression. Overexpression (OE) of AnkG in FRT cells increased total current of both WT (1.00 ± 0.03 versus 1.45 ± 0.05, n = 44, p < 0.001) and βS518K (1.00 ± 0.09 versus 1.55 ± 0.11, n = 11, p < 0.001) ENaC. The percentage of increase between WT AnkG OE and βS518K AnkG OE was not significantly different (p = 0.369), demonstrating that AnkG predominantly increases N to increase Na+ transport (Fig. 3C).
AnkG Does Not Affect Internalization Rates of ENaC
To alter the number of channels at the plasma membrane, AnkG could regulate the rate of surface delivery, apical recycling, cAMP-regulated recycling, and/or removal of ENaC from the membrane. As mentioned in the introduction, ubiquitylation and internalization of ENaC is a well established means of reducing the number of ENaCs at the plasma membrane (12, 51). We therefore tested whether AnkG overexpression reduced ENaC internalization rates. FRT cells expressing WT ENaC or a cleavage-resistant (CR) ENaC were co-transfected with GFP or AnkG and subjected to an electrophysiological pulse-chase experiment. The CR ENaC mutations prevent cleavage by endogenous proteases, which keeps them in an inactive state (electrically silent) at the surface membrane (52). CR channels can be activated by the addition of an exogenous protease like trypsin in the apical compartment (41, 53). This allowed us to activate a pool of channels at the membrane with a trypsin pulse (followed by trypsin washout) and observe the internalization of the active ENaCs as a measure of the reduction in current over time. After 30–60 min of rundown, we reapplied trypsin to determine whether inactive channels had been delivered to the membrane during the rundown period. A sample trace of one such experiment comparing WT ENaC and CR ENaC is shown in Fig. 4A, and a sample trace comparing CR ENaC control with CR ENaC AnkG OE is shown in Fig. 4B. Introduction of the CR mutations did not change the AnkG phenotype on ENaC, namely increased ENaC current with AnkG overexpression (Fig. 4C). After normalizing to the initial current following trypsin activation (pulse), we plotted the average internalization curves of multiple experiments (Fig. 4D). The Isc rundowns for WT ENaC were slower than those of the CR ENaC because contributions from the biosynthetic and recycling pools were not eliminated. However, whereas the apparent CR ENaC internalization rates were faster as expected, AnkG overexpression did not significantly alter Isc rundown. This suggests that AnkG was not altering the rate of ENaC endocytosis.
FIGURE 4.

AnkG overexpression does not impact ENaC internalization rates. A, representative Isc traces of WT and CR ENaC expressed in FRT cells following trypsin activation, washout, secondary trypsin activation, and amiloride block. CR ENaC is electrically silent until trypsin cleavage activates the pool of ENaC at the surface. Following removal of trypsin, the decay of current in the CR ENaC transfected FRTs gives the rate of internalization without contribution from channels being inserted into the membrane from biosynthetic pathway. B, representative Isc trace of CR ENaC with or without AnkG OE. C, summary of WT ENaC and CR ENaC Isc phenotypes. FRT cells heterologously expressing ENaC and a control plasmid, or AnkG. AnkG OE significantly increased WT ENaC current (1.45 ± 0.05 versus 1.00 ± 0.02, n = 45, p < 0.001) and also significantly increased the trypsin activated CR ENaC current (1.40 ± 0.16 versus 1.00 ± 0.04, n = 11, p < 0.05). D, average current decay curves of WT and CR ENaC with control or AnkG OE. WT ENaC decay rates are shown to demonstrate their slower rate caused by the contribution of channel insertion from the recycling and biosynthetic pathways. As expected, CR ENaC current decay is faster than WT ENaC; however, there is no significant change in the rate of removal with overexpression of AnkG in either CR or WT ENaC experiments.
cAMP-dependent ENaC Recycling Is Not Affected by AnkG Expression
Next we looked at the effect of AnkG on ENaC trafficking in response to cAMP stimulation and stimulus washout. We previously characterized an intracellular ENaC pool, which is acutely inserted into the membrane in response to activation of cAMP (40, 42). Using 5 μm forskolin to repeatedly mobilize these vesicles, we measured the rates of ENaC insertion and removal, as well as the relative percentage increase in current resulting from insertion of this pool of channels in control transfected and AnkG depleted mCCD cells. A representative trace is shown in Fig. 5A. After normalization to the initial current, we did not observe changes in either the percentages of increase or the rates of insertion following forskolin stimulation (Fig. 5B) or any changes in the percentage decrease or rates of removal with AnkG knockdown (Fig. 5C). These data suggest AnkG is not altering the ENaC recycling through the cAMP-regulated pool.
FIGURE 5.
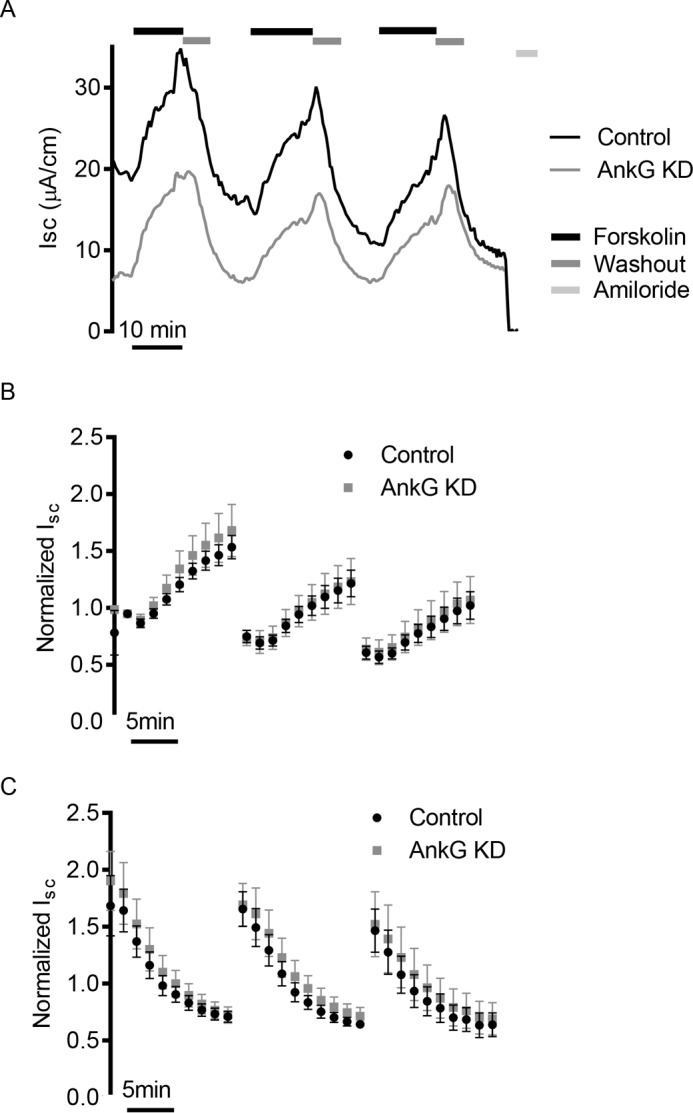
AnkG knockdown does not alter rates of delivery and internalization from the cAMP-regulated recycling pool. A, representative Isc traces of mCCD cells undergoing three rounds of 5 μm forskolin stimulation and washout. Forskolin increases intracellular levels of cAMP, which in turn results in the insertion of an intracellular pool of ENaC channels into the membrane. B, summarized results of rate and percentages of increase from initial current over time for mCCDs following forskolin stimulation. The data points are the averages of n = 5 experiments and are normalized to the initial current value of each experiment. There is no significant difference in the rate or relative percentage of channels inserted into the membrane. C, summarized results of ENaC internalization following removal of forskolin. The data are normalized to the initial current of each experiment. There is no significant difference in the rate of removal or percentage of channels internalized (n = 5).
AnkG Increases the Rate of ENaC Membrane Insertion
To test whether AnkG increased the rate of ENaC delivery to the apical membrane, we performed a variation of the electrophysiological pulse-chase described above using mCCD cells. First, we stimulated mCCD cells with forskolin to insert ENaC from the cAMP-regulated pool into the plasma membrane. We next irreversibly blocked all the channels at the surface using 25 μm phenamil (54). Following washout of the inhibitor, any recovery of the Isc represents newly inserted ENaC to the apical surface from a blocker-inaccessible intracellular sources, such as channels from the biosynthetic or non-cAMP-regulated recycling pathways. The increase in Isc was normalized to the initial (unblocked) value. A representative trace of the entire protocol is shown in Fig. 6A, and a trace of the normalized recovery following phenamil block is shown in Fig. 6B. After 30 min, the control traces had recovered to 15 ± 1.8% (n = 20) of the initial starting current, whereas cells with AnkG knockdown recovered to 8 ± 2.8% (n = 8, p < 0.05), and cells with AnkG overexpression had recovered 24 ± 2.5% (n = 9, p < 0.01) of the starting current (Fig. 6C). These data demonstrate that AnkG increases ENaC surface delivery. In addition, whereas the AnkG overexpressing cells recovered faster, the currents also trended to a plateau, indicating that a new steady state was being reached with surface and intracellular pools.
FIGURE 6.

AnkG alters ENaC activity by increasing the rate of surface delivery. A, representative Isc trace of mCCD cell current recovery after 5 μm forskolin stimulation and block by 25 μm phenamil. Phenamil irreversibly blocks all channels at the surface, and once the phenamil has been removed, the amiloride-sensitive current that appears over time is the result of channels being inserted from the biosynthetic and non-cAMP-regulated pathway. Pretreatment with forskolin inserts channels from the recycling pool into the surface, eliminating them as possible contributions to the recovery current. B, current recovery plotted as a percentage of preinhibited values over time. The rate and extent of recovery in AnkG OE cells is greater than control or AnkG KD mCCDs. C, percentage recovery summary demonstrated that after 30 min, significantly more channels were delivered to the surface with AnkG OE (24 ± 2.5%, n = 9; p < 0.01) and significantly fewer channels with AnkG KD (8 ± 2.8%, n = 8, p value<0.05) relative to control (15 ± 1.8%, n = 20). D, representative Isc trace of FRTs expressing CR ENaC in combination with Liddle's mutation with or without AnkG OE. Following trypsin activation and washout, electrically silent channels were being delivered to the plasma membrane during the current run down time course. After 30 min, the cells were restimulated with trypsin. The increase from the base current is indicative of the number of channels delivered to the surface during the 30-min time period. E, summarized Isc increase for the second trypsin pulse following the 30-min interval from n > 4 experiments like those in Figs. 4B and 6D, respectively. AnkG overexpression delivered 75% ± 16% more ENaC in 30 min than control cells (n = 4, p < 0.001) without the Liddle's mutation, and this increase was absent with the β-ENaC Liddle's mutant. F, absolute Isc values following the second trypsin pulse. AnkG OE did not significantly change the number of channels that are being delivered from the biosynthetic pathway because no significant increase in delivery of silent ENaC was observed with AnkG OE with the Liddle's mutation, as was the case in non-Liddle's ENaC.
To verify this accelerated surface delivery, we examined ENaC appearance in the FRT cell model with CR ENaC. As previously shown (Fig. 4, A and B), after the 30-min washout period following the initial trypsin pulse, the second trypsin treatment was reapplied to determine the number of inactive channels that had been delivered to the plasma membrane. AnkG overexpression increased the magnitude of the second trypsin pulse, suggesting an increase in the number of channels delivered to the membrane relative to WT within 30 min (1.41 ± 0.13 μA/cm2 versus 0.98 ± 0.0 8μA/cm2, n = 15, p < 0.01) These data confirmed that AnkG increased ENaC membrane delivery relative to controls but do not distinguish whether the increase in channel delivery was from channels being delivered from the biosynthetic pathway or from intracellular recycling pools. To dissect these two possibilities, we repeated the CR ENaC trypsin pulse-chase experiment with the addition of the Liddle's mutation, P616L β-ENaC (55). This mutation prevents ubiquitination and delays internalization of ENaC from the plasma membrane. These channels would therefore not contribute to a constitutive ENaC recycling pool, and the role of AnkG in altering delivery to the surface from the biosynthetic pathway could be investigated. Fig. 6D depicts a representative trace of the CR Liddle's ENaC trypsin pulse-chase experiment. The Liddle's mutation significantly increased ENaC activity as expected but not to the same extent as AnkG overexpression (Fig. 6E). Following the 30-min rundown, there was a limited response to the second trypsin pulse, and no significant difference in the number of channels delivered with control or AnkG OE Liddle's mutant ENaCs (1.00 ± 0.03 μA/cm2 versus 0.89 ± 0.04 μA/cm2, n = 8, p = 0.09) (Fig. 6F). These results demonstrate that the increases in current resulting from the second trypsin pulse in the CR ENaC experiments are predominantly from non-cAMP-regulated recycling channels rather than channels being delivered from the Golgi through the biosynthetic pathway. Taken together, these experiments indicate that AnkG increases the number of channels at the surface by facilitating the insertion of ENaC via the constitutive recycling pathway.
Discussion
Ankyrins are a family of intracellular proteins that link integral membrane proteins via ankyrin repeats to spectrin based cytoskeletal structures and may play a crucial role in trafficking proteins to specific domains within the plasma membrane (56). Although it has been known for some time that AnkG is highly expressed in the kidney (21, 22, 34, 44), the roles of this protein in kidney physiology and ion homeostasis remain largely uncharacterized. Previous studies have suggested that AnkR and AnkG may be essential for basolateral sorting of the Na+/K+-ATPase and ammonium (RhBG) transporters, respectively (44, 57). Work by San-Cristobal et al. (58) has shown that AnkG inhibits current through the potassium channel Kv1.1, which establishes a favorable driving force for Mg2+ entry into the distal tubules of mice.
A large cohort of proteins are involved in apical cargo transport, and a number of them have been described to interact with ENaC. Along this pathway, ENaC is transported by and interacts with a network of small GTPases, motor proteins, docking and fusion proteins, exocytic proteins, and the cytoskeleton itself. Small GTPases such as Rho and Rab family members have roles in reorganizing the cytoskeleton and regulating intracellular vesicle trafficking, respectively (17). Rho has been shown to activate ENaC by increasing membrane density (17). A number of Rabs, including Rab11 and Rab27, can also increase or decrease ENaC activity, potentially via control of vesicle formation, vesicle fusion, or association of vesicular cargo with motor proteins (11, 59, 60). AnkG has been shown to link cargo vesicles to kinesin to traffic along microtubules in neurons (61). Additionally, the ankyrin family member AnkB associates with dynactin to recruit the motor protein dynein to organelle membranes (62). It is possible that AnkG assists ENaC cargo vesicles to interact with the appropriate apical membrane targeting molecular motors to facilitate surface recycling, thereby increasing ENaC apical membrane density; however, this remains to be determined.
Our previous work shows that AnkG expression is regulated by aldosterone-sensitive miRNAs and that AnkG protein levels are up-regulated following aldosterone stimulation because of repression of aldosterone-regulated miRNAs (38). For AnkG to be a physiologically relevant in the aldosterone signaling cascade, increased AnkG protein expression should lead to an increase in Na+ transport. In this study, we demonstrate by varying AnkG expression that Na+ transport via ENaC is directly altered. By isolating Na+ transport at the apical or basolateral membrane, we confirmed that AnkG could increase Na+ transport via ENaC with no impact on Na+/K+-ATPase localization or function. This is surprising given the high expression of AnkG at the lateral surface (34, 44, 63) as well as the established ability of Na+/K+-ATPase to bind ankyrins (46–48). However, from these studies it is not clear which form of ankyrin Na+/K+-ATPase may be binding. A recent study by Stankewich et.al. (34) analyzed ankyrin expression and localization along the nephron using immunofluorescence and TEM, and they observed AnkG at both the apical and basolateral membranes. Additionally, TEM images of kidney tubules from this paper show AnkG closely associated with some but not all vesicles near the apical surface, whereas AnkG on the lateral side appears more tightly associated with the plasma membrane itself (34). The 190-kDa AnkG isoform was also identified as an apically associated protein in an apical membrane proteomics screen of cultured mouse cortical collecting duct (mpkCCD-clone 11) cells (64). It is possible therefore, that AnkG serves a structural role along the basolateral membrane, such as its anchoring effect on E-cadherin (33, 63), whereas near the apical surface, its association with vesicles plays a role in recycling.
An additional reported function of AnkG is its role in determining lateral cell height in lung and kidney epithelial cells. We could confirm these observations in the mCCD cells and demonstrated that AnkG knockdown decreased lateral height in mCCD cells similar to previously published findings (30, 31, 33). However, this height reduction appears to be compensated by an increase in cell width because we did not observe significantly altered apical or basolateral surface membrane area.
Given the essential role of Nedd4-2 ubiquitination-dependent endocytosis in regulating the number of ENaCs at the membrane and the role of AnkG in endocytosis, it was unexpected to determine that altering AnkG expression did not have a significant effect on ENaC internalization or cAMP-stimulated trafficking of ENaC. We therefore investigated apical surface delivery as a possible mechanism for AnkG action on ENaC. To isolate this step in the life cycle of ENaC, the contribution of channels at the surface and in the cAMP-stimulated recycling pool were eliminated by irreversibly blocking these surface channels with phenamil and recording the appearance of new channels at the apical membrane as an increase in current over time. These experiments established that AnkG was increasing apical delivery of non-cAMP-regulated recycling channels or channels being delivered from the biosynthetic pathway. Overexpression of AnkG appeared to re-establish a steady state of recycling ENaC faster than cells in which AnkG was depleted. We verified this increased delivery of ENaC using FRT cells and cleavage-resistant subunits. AnkG overexpression increased the number of channels delivered from the intracellular pool to the surface during that time period. By using a Liddle's mutant, which prevents normal endocytosis in combination with the CR ENaC, we were able to establish that AnkG is accelerating constitutive recycling rather than increasing delivery of channels from the biosynthetic pathway.
Taken together, these experiments demonstrate the role of a novel aldosterone-induced ENaC regulator, which may contribute to Na+ handling in the distal nephron. Uncovering AnkG as essential for ENaC surface delivery contributes to our general knowledge of ENaC trafficking, which has broader implications for blood volume homeostasis.
Experimental Procedures
Antibodies and Reagents
Chemical reagents were from Sigma-Aldrich or Fisher Science unless noted otherwise. Antibodies against ankyrin G were from Neuromab (immunofluorescence, clone N106/36, catalog no. 73-146, lot no. 437–5VA-5) and Santa Cruz (Western blot, catalog no. sc-28561, lot no. E2512). Polyclonal antibodies directed against the N terminus of α-ENaC were previously described and obtained from Genescript (11) or Stressmarq Biosciences (catalog no. SPC-403D, lot no. 130911). Polyclonal antibodies against β-ENaC and γ-ENaC were obtained from Stressmarq Biosciences (catalog no. SPC-404D, lot no. 150101, lot no. 2653 and SPC-405D lot no. PA-14299, respectively). Monoclonal antibodies against GFP and Na+/K+-ATPase were purchased from Santa Cruz (catalog no. sc-9996, lot no. D2009 and catalog no. sc-21712, lot no. D2213, respectively). Alexa Fluor 568-linked phalloidin was used to image F-actin in mCCDs (catalog no. A12380).
Cell Culture
Mouse cortical collecting duct (mCCDcl-11) cells (provided by Laurent Schild and Bernard Rossier, Université de Lausanne, Switzerland) were cultured in 75-cm flasks (passage 28–38) in 2% FBS supplemented medium as previously described (38, 39). The cells for transfection were seeded onto 6-well dishes and transiently transfected with Lipofectamine 2000 according to the manufacturer's instructions. For overexpression, the cells were transfected with 2 μg of pEGFP-C1 vector as a control (Clontech) or ankyrinG-190-GFP (Addgene, plasmid no. 31059). Transfection efficiency for the cDNA, assessed by GFP fluorescent signal, was consistently>30% of cells. For knockdown, cells were transfected with 25 nm of a commercially available control RNA scramble sequence (Integrated DNA Technologies, Coralville, IA) or dicer specific silencing RNA (DsiRNA) targeting AnkG, sequence 5′-ACACGUUAGAUAAUGUGAACCUGGU-3′ (Integrated DNA Technologies). The DsiRNA construct transfection efficiency was routinely >60%, as assessed by Cy3 transfection control DsiRNA and AnkG immunofluorescent staining (Integrated DNA Technologies). 24 h post-transfection, the cells were seeded onto 0.33 cm2 or 1 cm2 Costar Transwell filters and allowed to polarize for at least 48 h before performing experiments (40).
Fisher Rat Thyroid (FRT cells) from ATCC were grown in 5% FBS medium in 75-cm flasks as previously described (40). For transfection, the cells were seeded onto 6-well plates and transfected with Lipofectamine 2000 using 0.6 μg of α-, β-, or γ-ENaC (wild type or cleavage-resistant α (R202A/R205A/R208A/R231A) and γ (R143A/RKRK186QQQ)) (41) and Liddle's β (P616L) with 1 μg of pEGFP-C1 or AnkyrinG-190-GFP (ENaC constructs kindly provided by Thomas Kleyman, Renal Division, University of Pittsburgh School of Medicine). Following a 24-h transfection, the cells were seeded onto 0.33-cm2 Transwell filters and cultured for 24 h before performing electrophysiological experiments.
Immunoblotting
The cells were lysed in lysis buffer (0.4% sodium deoxycholate, 1% Nonidet P-40,10 mm Tris, 50 mm EGTA, pH 7.5) on ice for 15min before pelleting insoluble material at 12,000rpm for 10min. Protein concentrations for lysates were determined using Bio-Rad protein assay according to manufacturer's instruction, and 50 μg of sample was run as total lysate lanes. Total cell lysates were denatured with 2× SDS sample buffer with 200 mm DTT at 70 °C for 15 min, separated on 7.5% SDS-PAGE gels, and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were blocked with 5% skim milk power in TBST (10 mm Tris, 150 mm NaCl, 0.05% Tween 20, pH 7.5) and probed with appropriate primary antibodies overnight at 4 °C. After washing three times for 10 min each with TBST, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies in TBST with 5% milk for 1 h. The membranes were again washed three times for 10 min with TBST and then treated with chemiluminescent substrate to visualized proteins of interest.
Short Circuit Current Measurements
Cultured cells on filter supports were mounted in modified Ussing chambers (P2300; Physiological Instruments, San Diego, CA) bathed in 4 ml of Ringer's solution (120 mm NaCl, 25 mm NaHCO3, 3.3 mm KH2PO4, 0.8 mm K2HPO4, 1.2 mm MgCl2, 1.2 mm CaCl2, 10 mm glucose, pH 7.4) in each hemi-chamber and bubbled with 5% CO2 at 37 °C, as previously described (11, 38, 40, 42). Short circuit currents (Isc) were obtained with an automatic voltage-clamp (VCC MC8; Physiological Instruments). Transepithelial resistance was measured by applying a 2-mV bipolar pulse and calculated using Ohm's law. In some experiments, Isc and total membrane capacitance were measured simultaneously with an automated voltage clamp system (designed by W. Van Driesche, KU Leuven, Leuven, Belgium) as described previously (42, 43). Briefly, the equipment were comprised of two digital signal processing boards: one that recorded Isc and one that recorded epithelial capacitance (CT). The recordings were digitized using PowerLab (Ad Instruments, Colorado Springs, CO). For basolateral permeabilization experiments, the basolateral chamber contained at low Na+ Ringer's solution, in which 120 mm NaCl was replaced with N-methyl-d-glucamine chloride. To stimulate cAMP, 5 μm forskolin was added basolaterally. To determine ENaC-sensitive Isc, 10 μm amiloride or 25 μm phenamil was added apically, whereas the contribution of Na+/K+-ATPase was determined by addition of 30 μm ouabain, basolaterally. Electrically silent, uncleaved ENaC channels were activated by apical addition of 1 μm trypsin. When washing out phenamil or trypsin in the electrical pulse-chase assays, chambers were washed six times with a total of 25–30 ml Ringer's solution.
Immunofluorescent Labeling and Confocal Microscopy
All preparation steps were performed at 4 °C unless otherwise noted. Polarized cells on filters were washed three times with ice-cold PBS (0.1 mm CaCl2, 1 mm MgCl2) and then fixed with 4% paraformaldehyde for 45 min. After an additional three washes with PBS, the cells were permeabilized with 0.1% Triton X-100 and 0.1% Nonidet P-40 in PBS for 25 min. Following three washes with PBS, the cells were incubated overnight with primary antibody (1:50) in blocking buffer (10% nonfat milk in PBS) at 4 °C. The cells were washed three times with PBS and then incubated with Alexa-conjugated secondary antibodies in blocking buffer for 3 h at room temperature. The nuclei were stained with Hoescht (1:10,000) for 5 min. After a final PBS wash, the cells were mounted on coverslips with Fluoromount G (Southern Biotech, Birmingham, AL). The images were captured on a confocal Nikon A1 Plus microscope using a Plan Apo VC 60× Oil DIC objective with 1.4 numerical aperture controlled by Nikon Elements AR software (Nikon, Tokyo, Japan) at the University of Pittsburgh Center for Biologic Imaging. Lateral cell heights and cell widths were determined using line analysis and computed by Nikon Elements AR software.
Statistics and Curve Fitting
All data were analyzed using GraphPad Prism (GraphPad, Inc., La Jolla, CA). Data from multiple replicates were normalized to control values for each experiment. Unpaired Student's t tests were performed to evaluate statistical significance, with p values <0.05 considered significant. Curves were fit using non-linear regression analysis of data points averaged from multiple experiments. The rate of removal of channels from the surface (FRT cleavage-resistant ENaC and mCCD forskolin washout experiments) was modeled as y = (y0 − plateau)*e−Kx + plateau, where y0 is the y value at x = 0, the plateau is the steady state Isc, K is the rate constant expressed as min−1, and x is time in min. Channel membrane insertion curves (mCCD forskolin stimulation and phenamil recovery) are one-phase association curves, given as y = y0 + (plateau − y0)*(1 − e−Kx), where y0 is the initial y value, plateau is the steady state Isc, K is the rate constant expressed as min−1, and x is time in min. The data are presented as means ± S.E. In the graphs, the error bars depict the S.E., *, **, and *** indicate significant difference from control where p < 0.05, p < 0.01, or p < 0.001, respectively.
Author Contributions
C. A. K., R. S. E., L. K., X. L., and M. B. B. performed experiments, collected data, and analyzed results. C. A. K., R. S. E., and M. B. B. conceived the study and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
This project used the University of Pittsburgh Center for Biologic Imaging facility.
This work was supported by NIDDK, National Institutes of Health Grant DK102843 (to M. B. B.) and Predoctoral Grant PRE25680068 from the American Heart Association (to C. A. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ENaC
- epithelial sodium channel
- AnkG
- ankyrin G
- mCCD
- mouse cortical collecting duct
- KD
- knockdown
- OE
- overexpression
- CR
- cleavage-resistant
- FRT
- Fisher rat thyroid.
References
- 1. Rossier B. C. (2014) Epithelial sodium channel (ENaC) and the control of blood pressure. Curr. Opin. Pharmacol. 15, 33–46 [DOI] [PubMed] [Google Scholar]
- 2. Soundararajan R., Pearce D., Hughey R. P., and Kleyman T. R. (2010) Role of epithelial sodium channels and their regulators in hypertension. J. Biol. Chem. 285, 30363–30369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schild L. (2010) The epithelial sodium channel and the control of sodium balance. Biochim. Biophys. Acta 1802, 1159–1165 [DOI] [PubMed] [Google Scholar]
- 4. Schafer J. A. (2002) Abnormal regulation of ENaC: syndromes of salt retention and salt wasting by the collecting duct. Am. J. Physiol. Renal Physiol. 283, F221–F235 [DOI] [PubMed] [Google Scholar]
- 5. Luft F. C. (2003) Mendelian forms of human hypertension and mechanisms of disease. Clin. Med. Res. 1, 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morimoto T., Liu W., Woda C., Carattino M. D., Wei Y., Hughey R. P., Apodaca G., Satlin L. M., and Kleyman T. R. (2006) Mechanism underlying flow stimulation of sodium absorption in the mammalian collecting duct. Am. J. Physiol. Renal Physiol. 291, F663–F669 [DOI] [PubMed] [Google Scholar]
- 7. Reifenberger M. S., Yu L., Bao H. F., Duke B. J., Liu B. C., Ma H. P., Alli A. A., Eaton D. C., and Alli A. A. (2014) Cytochalasin E alters the cytoskeleton and decreases ENaC activity in Xenopus 2F3 cells. Am. J. Physiol. Renal Physiol. 307, F86–F95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mueller G. M., Maarouf A. B., Kinlough C. L., Sheng N., Kashlan O. B., Okumura S., Luthy S., Kleyman T. R., and Hughey R. P. (2010) Cys palmitoylation of the β subunit modulates gating of the epithelial sodium channel. J. Biol. Chem. 285, 30453–30462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ilatovskaya D. V., Pavlov T. S., Levchenko V., Negulyaev Y. A., and Staruschenko A. (2011) Cortical actin binding protein cortactin mediates ENaC activity via Arp2/3 complex. FASEB J. 25, 2688–2699 [DOI] [PubMed] [Google Scholar]
- 10. Butterworth M. B., Edinger R. S., Frizzell R. A., and Johnson J. P. (2009) Regulation of the epithelial sodium channel by membrane trafficking. Am. J. Physiol. Renal Physiol. 296, F10–F24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Butterworth M. B., Edinger R. S., Silvis M. R., Gallo L. I., Liang X., Apodaca G., Frizzell R. A., and Johnson J. P. (2012) Rab11b regulates the trafficking and recycling of the epithelial sodium channel (ENaC). Am. J. Physiol. Renal Physiol. 302, F581–F590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Staub O., Gautschi I., Ishikawa T., Breitschopf K., Ciechanover A., Schild L., and Rotin D. (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 16, 6325–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mazzochi C., Bubien J. K., Smith P. R., and Benos D. J. (2006) The carboxyl terminus of the α-subunit of the amiloride-sensitive epithelial sodium channel binds to F-actin. J. Biol. Chem. 281, 6528–6538 [DOI] [PubMed] [Google Scholar]
- 14. Copeland S. J., Berdiev B. K., Ji H. L., Lockhart J., Parker S., Fuller C. M., and Benos D. J. (2001) Regions in the carboxy terminus of α-βENaC involved in gating and functional effects of actin. Am. J. Physiol. Cell Physiol. 281, C231–C240 [DOI] [PubMed] [Google Scholar]
- 15. Cantiello H. F., Stow J. L., Prat A. G., and Ausiello D. A. (1991) Actin filaments regulate epithelial Na+ channel activity. Am. J. Physiol. 261, C882–C888 [DOI] [PubMed] [Google Scholar]
- 16. Jovov B., Tousson A., Ji H. L., Keeton D., Shlyonsky V., Ripoll P. J., Fuller C. M., and Benos D. J. (1999) Regulation of epithelial Na+ channels by actin in planar lipid bilayers and in the Xenopus oocyte expression system. J. Biol. Chem. 274, 37845–37854 [DOI] [PubMed] [Google Scholar]
- 17. Karpushev A. V., Ilatovskaya D. V., Pavlov T. S., Negulyaev Y. A., and Staruschenko A. (2010) Intact cytoskeleton is required for small G protein dependent activation of the epithelial Na+ channel. PLoS One 5, e8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith P. R., Saccomani G., Joe E. H., Angelides K. J., and Benos D. J. (1991) Amiloride-sensitive sodium channel is linked to the cytoskeleton in renal epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 88, 6971–6975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rotin D., Goldstein B. J., and Fladd C. A. (1994) Expression of the tyrosine phosphatase LAR-PTP2 is developmentally regulated in lung epithelia. Am. J. Physiol. 267, L263–L270 [DOI] [PubMed] [Google Scholar]
- 20. Kordeli E., Lambert S., and Bennett V. (1995) AnkyrinG: a new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J. Biol. Chem. 270, 2352–2359 [DOI] [PubMed] [Google Scholar]
- 21. Peters L. L., John K. M., Lu F. M., Eicher E. M., Higgins A., Yialamas M., Turtzo L. C., Otsuka A. J., and Lux S. E. (1995) Ank3 (epithelial ankyrin), a widely distributed new member of the ankyrin gene family and the major ankyrin in kidney, is expressed in alternatively spliced forms, including forms that lack the repeat domain. J. Cell Biol. 130, 313–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Doctor R. B., Chen J., Peters L. L., Lux S. E., and Mandel L. J. (1998) Distribution of epithelial ankyrin (Ank3) spliceoforms in renal proximal and distal tubules. Am. J. Physiol. 274, F129–F138 [DOI] [PubMed] [Google Scholar]
- 23. Hedstrom K. L., Ogawa Y., and Rasband M. N. (2008) AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J. Cell Biol. 183, 635–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sobotzik J. M., Sie J. M., Politi C., Del Turco D., Bennett V., Deller T., and Schultz C. (2009) AnkyrinG is required to maintain axo-dendritic polarity in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 17564–17569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brachet A., Leterrier C., Irondelle M., Fache M. P., Racine V., Sibarita J. B., Choquet D., and Dargent B. (2010) Ankyrin G restricts ion channel diffusion at the axonal initial segment before the establishment of the diffusion barrier. J. Cell Biol. 191, 383–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Makara M. A., Curran J., Little S. C., Musa H., Polina I., Smith S. A., Wright P. J., Unudurthi S. D., Snyder J., Bennett V., Hund T. J., and Mohler P. J. (2014) Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ. Res. 115, 929–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sato P. Y., Coombs W., Lin X., Nekrasova O., Green K. J., Isom L. L., Taffet S. M., and Delmar M. (2011) Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ. Res. 109, 193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pan Z., Kao T., Horvath Z., Lemos J., Sul J. Y., Cranstoun S. D., Bennett V., Scherer S. S., and Cooper E. C. (2006) A common ankyrin-G-based mechanism retains KCNQ and Nav channels at electrically active domains of the axon. J. Neurosci. 26, 2599–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lowe J. S., Palygin O., Bhasin N., Hund T. J., Boyden P. A., Shibata E., Anderson M. E., and Mohler P. J. (2008) Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J. Cell Biol. 180, 173–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kizhatil K., Yoon W., Mohler P. J., Davis L. H., Hoffman J. A., and Bennett V. (2007) Ankyrin-G and β2-spectrin collaborate in biogenesis of lateral membrane of human bronchial epithelial cells. J. Biol. Chem. 282, 2029–2037 [DOI] [PubMed] [Google Scholar]
- 31. Kizhatil K., and Bennett V. (2004) Lateral membrane biogenesis in human bronchial epithelial cells requires 190-kDa ankyrin-G. J. Biol. Chem. 279, 16706–16714 [DOI] [PubMed] [Google Scholar]
- 32. He M., Abdi K. M., and Bennett V. (2014) Ankyrin-G palmitoylation and βII-spectrin binding to phosphoinositide lipids drive lateral membrane assembly. J. Cell Biol. 206, 273–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jenkins P. M., Vasavda C., Hostettler J., Davis J. Q., Abdi K., and Bennett V. (2013) E-cadherin polarity is determined by a multifunction motif mediating lateral membrane retention through ankyrin-G and apical-lateral transcytosis through clathrin. J. Biol. Chem. 288, 14018–14031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stankewich M. C., Moeckel G. W., Ji L., Ardito T., and Morrow J. S. (2016) Isoforms of spectrin and ankyrin reflect the functional topography of the mouse kidney. PLoS One 11, e0142687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu M., and Cooper E. C. (2015) An ankyrin-G N-terminal gate and protein kinase CK2 dually regulate binding of voltage-gated sodium and KCNQ2/3 potassium channels. J. Biol. Chem. 290, 16619–16632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Montersino A., Brachet A., Ferracci G., Fache M. P., Angles d'Ortoli S., Liu W., Rueda-Boroni F., Castets F., and Dargent B. (2014) Tetrodotoxin-resistant voltage-gated sodium channel Nav 1.8 constitutively interacts with ankyrin G. J. Neurochem. 131, 33–41 [DOI] [PubMed] [Google Scholar]
- 37. Cooper E. C. (2011) Made for “anchorin”: Kv7.2/7.3 (KCNQ2/KCNQ3) channels and the modulation of neuronal excitability in vertebrate axons. Semin. Cell Dev. Biol. 22, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Edinger R. S., Coronnello C., Bodnar A. J., Labarca M., Bhalla V., LaFramboise W. A., Benos P. V., Ho J., Johnson J. P., and Butterworth M. B. (2014) Aldosterone regulates microRNAs in the cortical collecting duct to alter sodium transport. J. Am. Soc. Nephrol. 25, 2445–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bens M., Vallet V., Cluzeaud F., Pascual-Letallec L., Kahn A., Rafestin-Oblin M. E., Rossier B. C., and Vandewalle A. (1999) Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J. Am. Soc. Nephrol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 40. Edinger R. S., Bertrand C. A., Rondandino C., Apodaca G. A., Johnson J. P., and Butterworth M. B. (2012) The epithelial sodium channel (ENaC) establishes a trafficking vesicle pool responsible for its regulation. PLoS One 7, e46593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hughey R. P., Bruns J. B., Kinlough C. L., Harkleroad K. L., Tong Q., Carattino M. D., Johnson J. P., Stockand J. D., and Kleyman T. R. (2004) Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 279, 18111–18114 [DOI] [PubMed] [Google Scholar]
- 42. Butterworth M. B., Edinger R. S., Johnson J. P., and Frizzell R. A. (2005) Acute ENaC stimulation by cAMP in a kidney cell line is mediated by exocytic insertion from a recycling channel pool. J. Gen. Physiol. 125, 81–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Erlij D., De Smet P., Mesotten D., and Van Driessche W. (1999) Forskolin increases apical sodium conductance in cultured toad kidney cells (A6) by stimulating membrane insertion. Pflugers Arch. 438, 195–204 [DOI] [PubMed] [Google Scholar]
- 44. Thevananther S., Kolli A. H., and Devarajan P. (1998) Identification of a novel ankyrin isoform (AnkG190) in kidney and lung that associates with the plasma membrane and binds α-Na, K-ATPase. J. Biol. Chem. 273, 23952–23958 [DOI] [PubMed] [Google Scholar]
- 45. Shibayama T., Nakaya K., and Nakamura Y. (1993) Differential binding activity of erythrocyte ankyrin to the α-subunits of Na+,K+-ATPases from rat cerebral and axonal membrane. Cell Struct. Funct. 18, 79–85 [DOI] [PubMed] [Google Scholar]
- 46. Zhang Z., Devarajan P., Dorfman A. L., and Morrow J. S. (1998) Structure of the ankyrin-binding domain of α-Na,K-ATPase. J. Biol. Chem. 273, 18681–18684 [DOI] [PubMed] [Google Scholar]
- 47. Liu X., Spicarová Z., Rydholm S., Li J., Brismar H., and Aperia A. (2008) Ankyrin B modulates the function of Na,K-ATPase/inositol 1,4,5-trisphosphate receptor signaling microdomain. J. Biol. Chem. 283, 11461–11468 [DOI] [PubMed] [Google Scholar]
- 48. Nelson W. J., and Veshnock P. J. (1987) Ankyrin binding to (Na+ + K+) ATPase and implications for the organization of membrane domains in polarized cells. Nature 328, 533–536 [DOI] [PubMed] [Google Scholar]
- 49. Stabach P. R., Devarajan P., Stankewich M. C., Bannykh S., and Morrow J. S. (2008) Ankyrin facilitates intracellular trafficking of α1-Na+-K+-ATPase in polarized cells. Am. J. Physiol. Cell Physiol. 295, C1202–C1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carattino M. D., Edinger R. S., Grieser H. J., Wise R., Neumann D., Schlattner U., Johnson J. P., Kleyman T. R., and Hallows K. R. (2005) Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J. Biol. Chem. 280, 17608–17616 [DOI] [PubMed] [Google Scholar]
- 51. Snyder P. M., Steines J. C., and Olson D. R. (2004) Relative contribution of Nedd4 and Nedd4-2 to ENaC regulation in epithelia determined by RNA interference. J. Biol. Chem. 279, 5042–5046 [DOI] [PubMed] [Google Scholar]
- 52. Kabra R., Knight K. K., Zhou R., and Snyder P. M. (2008) Nedd4-2 induces endocytosis and degradation of proteolytically cleaved epithelial Na+ channels. J. Biol. Chem. 283, 6033–6039 [DOI] [PubMed] [Google Scholar]
- 53. Hughey R. P., Mueller G. M., Bruns J. B., Kinlough C. L., Poland P. A., Harkleroad K. L., Carattino M. D., and Kleyman T. R. (2003) Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J. Biol. Chem. 278, 37073–37082 [DOI] [PubMed] [Google Scholar]
- 54. Garvin J. L., Simon S. A., Cragoe E. J. Jr, and Mandel L. J. (1985) Phenamil: an 7irreversible inhibitor of sodium channels in the toad urinary bladder. J. Membr. Biol. 87, 45–54 [DOI] [PubMed] [Google Scholar]
- 55. Hansson J. H., Schild L., Lu Y., Wilson T. A., Gautschi I., Shimkets R., Nelson-Williams C., Rossier B. C., and Lifton R. P. (1995) A de novo missense mutation of the β subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc. Natl. Acad. Sci. U.S.A. 92, 11495–11499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bennett V., and Baines A. J. (2001) Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol. Rev. 81, 1353–1392 [DOI] [PubMed] [Google Scholar]
- 57. Lopez C., Métral S., Eladari D., Drevensek S., Gane P., Chambrey R., Bennett V., Cartron J. P., Le Van Kim C., and Colin Y. (2005) The ammonium transporter RhBG: requirement of a tyrosine-based signal and ankyrin-G for basolateral targeting and membrane anchorage in polarized kidney epithelial cells. J. Biol. Chem. 280, 8221–8228 [DOI] [PubMed] [Google Scholar]
- 58. San-Cristobal P., Lainez S., Dimke H., de Graaf M. J., Hoenderop J. G., and Bindels R. J. (2014) Ankyrin-3 is a novel binding partner of the voltage-gated potassium channel Kv1.1 implicated in renal magnesium handling. Kidney Int. 85, 94–102 [DOI] [PubMed] [Google Scholar]
- 59. Saxena S. K., Horiuchi H., and Fukuda M. (2006) Rab27a regulates epithelial sodium channel (ENaC) activity through synaptotagmin-like protein (SLP-5) and Munc13–4 effector mechanism. Biochem. Biophys. Res. Commun. 344, 651–657 [DOI] [PubMed] [Google Scholar]
- 60. Karpushev A. V., Levchenko V., Pavlov T. S., Lam V. Y., Vinnakota K. C., Vandewalle A., Wakatsuki T., and Staruschenko A. (2008) Regulation of ENaC expression at the cell surface by Rab11. Biochem. Biophys. Res. Commun. 377, 521–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Barry J., Gu Y., Jukkola P., O'Neill B., Gu H., Mohler P. J., Rajamani K. T., and Gu C. (2014) Ankyrin-G directly binds to kinesin-1 to transport voltage-gated Na+ channels into axons. Dev. Cell 28, 117–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lorenzo D. N., Badea A., Davis J., Hostettler J., He J., Zhong G., Zhuang X., and Bennett V. (2014) A PIK3C3-ankyrin-B-dynactin pathway promotes axonal growth and multiorganelle transport. J. Cell Biol. 207, 735–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Piepenhagen P. A., Peters L. L., Lux S. E., and Nelson W. J. (1995) Differential expression of Na+-K+-ATPase, ankyrin, fodrin, and E-cadherin along the kidney nephron. Am. J. Physiol. 269, C1417–C1432 [DOI] [PubMed] [Google Scholar]
- 64. Loo C. S., Chen C. W., Wang P. J., Chen P. Y., Lin S. Y., Khoo K. H., Fenton R. A., Knepper M. A., and Yu M. J. (2013) Quantitative apical membrane proteomics reveals vasopressin-induced actin dynamics in collecting duct cells. Proc. Natl. Acad. Sci. U.S.A. 110, 17119–17124 [DOI] [PMC free article] [PubMed] [Google Scholar]