

Abstract
Agnoprotein is an important regulatory protein of polyomaviruses, including JCV, BKV, and SV40. In the absence of its expression, these viruses are unable to sustain their productive life cycle. It is a highly basic phosphoprotein that localizes mostly to the perinuclear area of infected cells, although a small amount of the protein is also found in nucleus. Much has been learned about the structure and function of this important regulatory protein in recent years. It forms highly stable dimers/oligomers in vitro and in vivo through its Leu/Ile/Phe-rich domain. Structural NMR studies revealed that this domain adopts an alpha-helix conformation and plays a critical role in the stability of the protein. It associates with cellular proteins, including YB-1, p53, Ku70, FEZ1, HP1α, PP2A, AP-3, PCNA, and α-SNAP; and viral proteins, including small t antigen, large T antigen, HIV-1 Tat, and JCV VP1; and significantly contributes the viral transcription and replication. This review summarizes the recent advances in the structural and functional properties of this important regulatory protein.
Identification of agnoprotein
Evident in the earliest DNA sequence data from simian vacuolating virus-40 (SV40) in the 1970s (Dhar et al., 1977, 1978; Fiers et al., 1978) was a segment in the late proximal region of the genome between map positions 0.72 and 0.76 with a potential open reading frame predicted to encode a protein of 62 amino acids in length. This gene was dubbed agnogene since no protein was known at the time. In 1980, Jay et al. (1981) infected African green monkey cells with SV40 and labeled with [14C]-arginine and detected a protein with an apparent molecular weight of 7.9 kDa, which was absent from uninfected cells or cells in which the portion of the SV40 genome between map positions 0.72 and 0.76 was deleted. Direct sequencing of the N-terminus of the protein by Edman degradation proved that this was the gene product of the agnogene and it was named agnoprotein. In a broad sense, Gerits and Moens (2012) have recently reviewed the advances regarding the agnoprotein field from different species. In this review, however, we will focus on detailed structural and functional properties of agnoprotein from the human (JCV and BKV) and simian (SV40) polyomaviruses.
Polyomavirus genome and agnoprotein expression
Polyomaviridae are a family of encapsidated small DNA viruses with a closed circular DNA genome of about 5 kb packaged within an icosahedral capsid structure (Imperiale, 2001). Although the early region of the genome encodes regulatory proteins, large T (Swenson and Frisque, 1995; Pipas, 1998; Simmons, 2000; Sullivan and Pipas, 2002; Saribas et al., 2012), small t (Khalili et al., 2008), and T’ proteins (Bollag et al., 2000, 2006; Frisque, 2001; Prins and Frisque, 2001); the late region encodes a combination of structural (capsid) proteins, VP1, VP2, and VP3 (Safak, 2014; Saribas et al., 2014) and a regulatory agnoprotein (Rinaldo et al., 1998; Safak et al., 2001; Darbinyan et al., 2002; Khalili et al., 2005; Akan et al., 2006; Myhre et al., 2010; Unterstab et al., 2010; Sariyer et al., 2011; Gerits and Moens, 2012; Saribas et al., 2012, 2013; Coric et al., 2014; Gerits et al., 2015). Both early and late regions are transcribed in opposite directions from a common promoter/enhancer known as the regulatory region (RR), which also contains the origin of viral DNA replication (Ori) (Lynch and Frisque, 1990, 1991; Lynch et al., 1994; Saribas et al., 2014). The first polyomaviruses to be discovered were mouse polyomavirus (MPyV) (Stewart et al., 1958) and simian vacuolating virus-40 (SV40) (Sweet and Hilleman, 1960) and these served as early models to study basic cellular processes including DNA replication, transcription, transformation and cell signaling. Next, two human polyomaviruses were discovered in 1971: polyoma JC virus (JCV) (Padgett et al., 1971)—the causative agent of progressive multifocal leukoencephalopathy (PML) (Padgett et al., 1971) and polyoma BK virus (BKV)—the causative agent of polyomavirus-associated nephropathy (Gardner et al., 1971). More recently, new technical advances in the identification of low abundance microorganisms have led to the discovery of at least eight new human polyomaviruses, including one was isolated from a human respiratory tract samples by PCR amplification (Karolinska Institute polyomavirus [KIPyV]) (Allander et al., 2007); and another was isolated from a patient with symptoms of acute respiratory tract infection (Washington University polyomavirus [WUPyV]) (Gaynor et al., 2007). Follow-up studies do not provide an evidence for the association between infections with WUPyV and KIPyV polyomaviruses and respiratory tract diseases. Another polyomavirus was isolated from Merkel cell carcinoma (a rare neuroendocrine skin tumor) by pyrosequencing of the cDNA made from the host transcripts followed by bioinformatics subtraction analysis of the host sequences (Merkel cell carcinoma-associated polyomavirus [MCPyV]) (Feng et al., 2008). Another polyomavirus was isolated from Trichodysplasia spinulosa samples by rolling circle amplification of the viral genome with specific primers followed by DNA sequence analysis (Trichodysplasia spinulosa-associated polyomavirus [TSPyV]) (van der Meijden et al., 2010). The rest of the recently discovered human polyomaviruses include HPyV-6, HPyV-7, HPyV-9, HPyV-10, HPyV-12 (Moens et al., 2011), Malawi polyomavirus (MWPyV) (Siebrasse et al., 2012), and Mexico X polyomavirus (MXPyV) (Yu et al., 2012). They were isolated from either serum, stool, or skin samples of a patient but were not yet definitively linked to a particular human disease (White et al., 2013; Safak, 2014). It is interesting to note that agnoprotein is expressed only from the late proximal region of a subset of polyomaviruses, including JCV, BKV, and SV40.
Expression and Cellular Distribution of Agnoprotein
Subcellular localization of agnoprotein
Agnoprotein shows a primarily cytoplasmic distribution with high concentrations accumulating in the perinuclear region of infected cells (Fig. 1). However, it has been consistently observed that 15–20% of this protein can also be detected in the nucleus by deconvolution microscopy studies (Saribas et al., 2012). Prediction studies show that agnoprotein has a weak bipartite nuclear localization signal (Dingwall and Laskey, 1986, 1991) localized to the N-terminal region of the protein which supports these observations (Saribas et al., 2012). Consistent with this finding, employing confocal microscopy techniques, Suzuki et al. (2010) also demonstrated a heavy perinuclear localization of agnoprotein in JCV-infected cells, co-localizing with endoplasmic reticulum (ER) markers, including calreticulin and BiP. In addition, these investigators have examined the impact of several agnoprotein N-terminal amino acid substitutions in the N-terminal region on its distribution. Some of these substitutions include those from the nuclear localization signal (NLS) region of the protein. Substitution of the Arg8 and Lys9 residues to Ala did not affect the targeting of the protein to ER. That is, these variants maintained some localization to the ER.
Fig. 1.

Cellular distribution of JCV agnoprotein in infected cells. Detection of agnoprotein and VP1 by immunocytochemistry was described previously (Sariyer et al., 2011). Briefly, SVG-A cells were transfected/infected with JCV Mad-1 strain and 15th day post-infection, cells were fixed with cold acetone on glass chambers and incubated with a primary anti-Agno polyclonal (Del Valle et al., 2002) and anti-VP1 (PAb597) monoclonal antibodies followed by incubation with a FITC-conjugated goat anti-rabbit and rhodamine-conjugated goat anti-mouse secondary antibodies and examined under a fluorescence microscope.
However, substitution of the Lys22, Lys23, and Arg24 residues to Ala, Ala, and Gly, respectively, resulted in loss of such an activity, suggesting that some of these basic amino acid residues located within the NLS region of agnoprotein are important for ER targeting (Suzuki et al., 2010). Localization of agnoprotein in both cytoplasm and nucleus suggests possible roles for this protein in both compartments. Unterstab et al. (2010) investigated the co-localization of the BKV agnoprotein using EGFP fusion constructs. By employing confocal microscopy, this group demonstrated co-localization of agnoprotein with “lipid-droplets (LD)” in infected cells and amino acids 20–42 turned out to be important for this phenomenon. The same investigators also analyzed the functional consequences of BKV agnoprotein double substitution mutants, Ala25Asp and Phe39Glu in transfected cells. Results suggested that disruption of the predicted α-helix of BKV agnoprotein abrogated the targeting of the agnoprotein into the LD (Unterstab et al., 2010).
Agnoprotein Forms Dimers and Oligomers
Discovery of dimer/oligomer formation by agnoprotein
Recent analysis of the purified MBP-Agno-fusion proteins by Saribas et al. (2011) on SDS-polyacrylamide gels (SDS-PAGE) followed by Coomassie blue staining consistently demonstrated the presence of additional bands migrating higher than the expected size of monomeric agnoprotein (Safak et al., 2001; Sariyer et al., 2006). These protein complexes were originally thought to be bacterial proteins that strongly bound and thereby co-purified with agnoprotein expressed in Escherichia coli. The nature of these complexes was investigated by mass spectrometry which demonstrated that each band mostly contained the fusion protein (maltose binding protein, MBP) and agnoprotein with only minor impurities from bacterial proteins (Saribas et al., 2011). These findings strongly suggested that these additional protein bands migrating at greater than expected sizes resulted from the intrinsic homodimer and oligomer formation of agnoprotein (Fig. 2A, lane 3). Since agnoproteins from JCV, BKV, and SV40 share a high degree of homology, we expected that these proteins could also form stable dimers and oligomers. SDS-PAGE studies revealed that MBP-BKV-Agno and MBP-SV40-Agno also formed such higher order structures (Fig. 2A, lanes 4 and 5).
Fig. 2.

A: Agnoprotein from JCV, BKV, and SV40 forms highly stable dimers and oligomers in vitro. Agnoprotein was fused to MBP, expressed in E. coli, and purified as described previously (Saribas et al., 2011). MBP and MBP-Agnoprotein were analyzed by SDS-10% polyacrylamide gel electrophoresis followed by Coomassie blue staining. The positions of monomers, dimers, and oligomers are indicated by arrows. B: Agnoprotein forms stable dimers and oligomers in vivo. Whole cell extracts were prepared from SVG-A cells infected with JCV Mad-1 strain at the 21st day post-infection and analyzed by Western blotting using primary anti-agnoprotein polyclonal (Del Valle et al., 2002) antibody. The positions of monomer, dimer, and oligomers are indicated by arrows. Inf., infection.
Stable dimer/oligomer formation is not an artifact of bacterially expressed agnoprotein; in fact, these high order structures of agnoprotein were also present during infection of human cells. When whole cell extracts prepared from cells infected with JCV (21st day post-infection) were analyzed by immunoblotting, higher order complexes of agnoprotein were also detected by Western blotting, suggesting the formation of stable dimers and oligomers during the infection cycle in culture (Fig. 2B, lane 2). These findings are in agreement with the observations by Del Valle et al. (2002) and Merabova et al. (2008) who reported the detection of higher agnoprotein bands under denaturing conditions by Western blotting; however, no explanation was provided for their identity. Fluorescence resonance energy transfer (FRET) and cross-linking studies by Suzuki et al. (2010) also demonstrated the formation of agnoprotein dimers/oligomers in transfected cells in culture. Together, these findings suggest that agnoprotein may function as both monomer and/or dimers/oligomers during the viral replication cycle.
Agnoprotein dimers and oligomers are highly resistant to strong denaturing conditions
Under normal circumstances, the running conditions for reducing SDS-PAGE denature proteins. Saribas et al. (2011) have recently reported the detection of dimeric and oligomeric forms of agnoprotein on SDS-PAGE, which is in disagreement with this principle. There are a number of viral and eukaryotic proteins that form SDS-resistant dimers or oligomers that have also been reported. Nitric oxide synthetase (NOS) was, for example, demonstrated to form highly stable dimers in cells that are resistant to heat and denaturing reagents (Kolodziejski et al., 2003), supporting the idea that the active form of this enzyme is a stable dimer. Several human immunodeficiency virus (HIV) regulatory proteins, including Rev, Vpr, Nef, Vif, and tat were also reported to form stable dimers and oligomers (Frankel et al., 1988a,b; Cullen, 1998; Kwak et al., 2010). Another example to a viral protein that forms homodimers is the Ebola virus transcription factor VP30 (Hartlieb et al., 2003).
The resistance of the agnoprotein dimers/oligomers to higher concentrations of additional denaturing agents, including urea and SDS, was also tested through the time-course studies where MBP-Agno was treated with 8 M urea (final concentration) at 25°C (10–120 min) and analyzed by SDS-PAGE. Surprisingly, 8 M urea did not show a significant effect on the integrity of the dimers/oligomers. Similarly, these complexes were also treated with high concentration of SDS, (ranging from 4% to 20%) and even the highest concentrations of SDS showed a minimal effect on the integrity of the dimers and oligomers demonstrating that these complexes are highly resistant to strong denaturing agents (Saribas et al., 2011).
The high stability observed for the dimers and oligomers under different strong denaturing conditions could be either the result of covalent bond formation between the side chains of agnoprotein due to transglutaminases which are the contaminants from the environment or strong non-covalent interactions, such as salt bridges and hydrophobic interactions, between the agnoprotein monomers. These two possibilities were also tested by Saribas et al. (2011) employing a prolonged heat treatment of the protein samples by reasoning that the non-covalent interactions would be less resistant to thermal denaturation than covalent bonding. The protein samples were subjected to a prolonged heat treatment ranging from 5 to 60 min at 95°C followed by reducing SDS-PAGE analysis and found out that majority of the oligomers and nearly half of the dimers were decreased while monomeric form of agnoprotein increased substantially (Saribas et al., 2011), suggesting that the high resistance of the dimers and oligomers against strong denaturing agents is not the result of covalent bond formation between the monomers, but likely from the hydrophobic interactions and numerous strong ionic attractions. Another interesting observation about these higher order complexes is that the level of dimer and oligomers increases over time upon storage at temperatures above 0°C suggesting that the level of dimer and oligomer formation is time and temperature dependent (Saribas et al., 2011).
The Leu/Ile/Phe-rich domain of agnoprotein is essential for dimer/oligomer formation and protein stability
Deletion mutagenesis studies of agnoprotein using an MBP-Agno construct by Saribas et al. (2011) initially mapped the region responsible for dimer/oligomer formation to a region from amino acids 17 to 42 for JCV agnoprotein. Subsequent systematic mutagenesis analysis of this region was performed to delineate the amino acids responsible for stable dimer/oligomer formation. These studies showed that elimination of the amino acids from the N- and C-terminus of the Leu/Ile/Phe-rich domain significantly affected stable dimer and oligomer formation (Saribas et al., 2013). In particular, internal deletion of the Leu/Ile/Phe-rich domain completely abrogated the dimer/oligomer formation and destabilized the protein. Even the removal of the all three residues, Glu34/Phe35/Leu36, was sufficient to eliminate the oligomer formation and weaken stable dimer formation (Saribas et al., 2013) demonstrating that the Leu/Ile/Phe-rich domain of agnoprotein plays a major role in stable dimer/oligomer formation. The Leu/Ile/Phe-rich domain may also play a role in stable expression of agnoprotein (Table 1). The removal of the Leu/Ile/Phe-rich domain renders agnoprotein to an intrinsically unstable structure, thereby leading to agnoprotein degradation, which resembles a previously reported case for heat shock proteins where they undergo rapid self-degradation (Mitchell et al., 1985). The behavior of all the agnoprotein mutants used in in vitro and cell culture studies is summarized in Table 1.
TABLE 1.
Stable dimer/oligomer formation and protein expression by JCV agnoprotein mutants
| Agnoprotein WT and its mutants fused to MBP |
Dimer | Oligomer | Expression in the viral background |
|---|---|---|---|
 |
+++ | +++ | +++ |
| 1–16 | − | − | N/A |
| 1–33 | − | − | N/A |
| 1–42 | +++ | +++ | N/A |
| 17–71 | +++ | +++ | N/A |
| 25–71 | +++ | +++ | N/A |
| 28–71 | +++ | ++ | N/A |
| 34–71 | ++ | +/− | N/A |
| 35–71 | + | − | N/A |
| 36–71 | + | − | N/A |
| 37–71 | − | + | N/A |
| 43–71 | − | − | N/A |
| Δ(17–42) | − | − | − |
| Δ(30–37) | − | − | − |
| Δ(34–36) | + | − | +/− |
| I30A | ++ | ++ | ++ |
| L33A | ++ | ++ | ++ |
| I30A+L33A | ++ | ++ | ++ |
| E34A | ++ | ++ | ++ |
| E34A+D38A | ++ | ++ | ++ |
| L29A | ++ | ++ | + |
| L32A | ++ | ++ | +++ |
| L36A | ++ | ++ | + |
| L32A+L36 | ++ | ++ | + |
| L29A+L32A+L36A | ++ | ++ | − |
N/A, not available.
Specific mutations within the Leu/Ile/Phe-rich domain greatly affect viral replication and agnoprotein stability
Although agnoprotein does not have a measurable activity (i.e., enzymatic), effects of its mutations on viral replication can be assessed by the DpnI/Southern blot assays (Hirt, 1967; Saribas et al., 2013). Saribas et al. (2013) have analyzed the replication properties of agnoprotein mutants relative to WT in SVG-A by this assay. Although the replication efficiency of each mutant was indistinguishable from that of WT by the 5th day post-transfection/infection, it drastically changed by the 15th day suggesting that analyzing the first replication cycle is not fully informative to observe the full effect of any mutation on agnoprotein on the viral replication cycle. Therefore, it is always recommended to carry out DpnI analysis after the second and/or third replication cycle of JCV to observe the full effect of any mutation of agnoprotein. For example, two deletion mutants (Δ[34–36] and Δ[30–37]) substantially lost their replication efficiencies (~90% and ~70%, respectively) by the 15th day post-transfection/infection although they had similar replication efficiencies at the 5th day (Saribas et al., 2013). These replication results were correlated with those obtained from immunocytochemistry and Western blot analysis studies, where it was observed that agnoprotein expression was barely detectable for Δ(34–36) and undetectable for Δ(30–37) mutants (Saribas et al., 2013), suggesting that agnoprotein expression is critical for efficient replication of JCV as reported previously (Sariyer et al., 2011).
Structural Features of Agnoprotein
Primary and secondary structures of agnoprotein
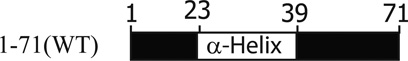
Agnoprotein is one of the critical regulatory proteins of primate polyomaviruses (JCV, BKV, and SV40). These viruses are not capable of sustaining a productive life cycle without this small protein (Saribas et al., 2013). It has a highly basic amino acid composition with multiple Arg (R) and Lys (K) residues, located at the carboxy- and amino-termini of the protein and these positively charged residues are highly conserved among the polyomaviruses. In contrast, the middle portion of the protein, ranging amino acids from 28 to 39, is composed of mostly hydrophobic residues, including Leu, Ile, and Phe and designated as Leu/Ile/Phe-rich domain. Some of these hydrophobic residues, Phe35, Glu34, Leu36, Phe39, and a charged residue Glu34 are highly conserved between JCV, BKV, and SV40 (Fig. 3) suggesting a shared functional and structural importance of these residues. It is noteworthy that all the agnoproteins aforementioned contains only three Phe residues and all are confined within the Leu/Ile/Phe-rich domain (Fig. 3). Interestingly, there is only one cysteine residue (Cys40) in JCV, BKV, and SV40 agnoproteins is located right after the Leu/Ile/Phe-rich domain. Although agnoproteins of these three viruses show high degree of amino acid sequence similarity in their N-terminal and central domains, the carboxy terminus of each protein is much more diverse. For instance, 41 of the amino acid residues of JCV agnoprotein are conserved in BKV agnoprotein and 30 of these are located in the N-terminal half of the protein (Fig. 3D). The sequence identity between JCV and BKV agnoprotein is quite high, which is 83% in amino acids 1–36, 73% in amino acids 37–50, whereas the C-terminal amino acids (51–71) show no significant identity. With respect to SV40 agnoprotein, there is only 61% sequence identity with JCV in the N-terminal region, amino acids, 1–36.
Fig. 3.

A–C: Primary structures of the JCV, BKV, and SV40 agnoproteins, respectively. The Leu/Ile/Phe-rich domain of each protein is indicated by a box. Phosphorylation sites in JCV and BKV agnoproteins are designated by the letter “P.” D: Amino acid sequence alignment of JCV, BKV, and SV40 agnoproteins.
Hydrophilic and hydrophobic regions of agnoprotein
Analyses of the agnoprotein sequences with computer programs that predict the hydrophilic and hydrophobic regions suggest that the agnoproteins of JCV, BKV, and SV40 are highly basic (Fig. 4). For instance, JCV agnoprotein has a predicted isoelectric point of 10. The hydrophilic residues are mostly located at the N- and C-termini of the protein, while the central portion exhibits a strong hydrophobic property. In particular, amino acids from 28 to 39 are highly hydrophobic and are involved in an alpha-helix formation as discussed below. Similarly, agnoproteins of BKV and SV40 also possess highly similar distributions of their hydrophilic and hydrophobic regions (Fig. 4).
Fig. 4.

Hydrophobic and hydrophilic properties of JCV, BKV, and SV40 agnoproteins. This graph was made by data obtained using the EMBOSS program (Rice et al., 2000).
NMR structure of JCV agnoprotein peptide encompassing amino acids Thr17–Gln52
Although the three dimensional structure of full-length agnoprotein is yet to be resolved, earlier modeling studies allowed us to predict the presence of two α-helices within the protein (Coric et al., 2014). Both α-helices, for example, in JCV agnoprotein are located in the N-terminus and in the central portion of the protein, from Val2 to Ala10 and Gly20 to Cys40, respectively (Combet et al., 2000; McGuffin et al., 2000; Roy et al., 2010; Unterstab et al., 2010; Saribas et al., 2013). The rest of the protein is predicted to adopt an intrinsically disordered conformation from Ser11 to Ser19 and Thr41 to Thr71. Several groups have attempted to resolve the X-ray crystal structure of JCV agnoprotein using purified proteins from baculovirus and bacterial systems with no success (personal communications). This was due to technical difficulties associated with purification of the protein and a dynamic inter-conversion of monomers to dimers or oligomers (Saribas et al., 2011). Recently, Coric et al. (2014) used a highly pure synthetic peptide of JCV agnoprotein encompassing the amino acids Thr17 to Gln52 to determine its first three dimensional (3D) structure by NMR (Fig. 5). These NMR studies demonstrated that the peptide contains an α-helix, amino acids ranging from Lys23 to Phe39, as had been predicted (Unterstab et al., 2010; Saribas et al., 2013; Coric et al., 2014) (Fig. 5A). The NMR studies also showed that surrounding regions of this α-helix adopt an intrinsically unstructured conformation. Helical wheel presentation of the α-helical region revealed four faces: an aromatic face, containing Phe31, Phe35, Ile28, and Phe39; a hydrophobic face, containing Ala25, Leu29, Leu32, and Leu36; another hydrophobic face, containing Ile30, Leu33, and Leu37; and a hydrophilic face containing Lys23, Arg27, Glu34, and Asp38 (Fig. 5B) (Coric et al., 2014).
Fig. 5.

A: NMR structure of the agnoprotein peptide was determined in the presence of 30% trifluoroethanol (TFE) (pH 3.0, temperature 313K by calculation using ARIA [Nilges, 1995]) and CNS (Brunger et al., 1998) Superimposition of the 17 energetically most favorable structures of the peptide on the backbone atoms of the domain from Lys (K) 23 to Phe (F) 39 of the averaged structure. The a-helix region is designated in red and unstructured regions from residues Thr (T) 17 to Lys (K) 22 and Cys (C) 40 to Gln (Q) 52, are designated in gray. B: Helical wheel representation of the a-helix (Lys23 to Phe39) of the agnoprotein peptide. Hydrophilic and hydrophobic faces are indicated. Hydrophobic residues occupy approximately 75% of the surface area of the wheel while hydrophilic residues form a narrow surface.
Intrinsic random structural features of N-terminus and C-terminal regions of agnoprotein
The immediate flanking sequences of the central agnoprotein α-helix exhibit intrinsically disordered conformations. In other words, both N-terminal and C-terminal regions shown here do not have a stable tertiary structure. This type of disordered conformation results from a combined high net charge and low overall hydrophobicity providing considerable flexibility for agnoprotein to interact with its various partners in different conformations, a mechanism by which it may diversify and amplify its functions (Dyson and Wright, 2005; Fink, 2005). There are several examples in the literature showing small viral proteins that often possess this type of feature. For instance, adenovirus small protein, E4-ORF3, was demonstrated to form multivalent functional matrices in infected cells by exploiting its intrinsically disordered structure (Ou et al., 2012). This then leads to the inactivation of various tumor suppressor proteins including, MRE11/Rad50/NBS1 (MRN), TIM24, p53, and promyelocytic leukemia factor (PML). Similarly, the majority of amino acid sequences located at the C-terminus and N-terminus of agnoprotein are predicted to have such intrinsically unstructured properties and these unstructured regions may play important roles in agnoprotein function in interactions with its binding-partners in infected cells and thereby contribute to the overall viral replication cycle.
NMR structure-based theoretical models for agnoprotein dimer formation
NMR studies not only revealed the monomeric agnoprotein structure but also provided important clues for possible dimer formation. In order to model the dimeric form of the protein using the NMR data (Fig. 5B), groups of amino acid residues on the each face of the alpha helix were targeted for site-directed mutagenesis and the resulting mutants were analyzed for agnoprotein structural integrity and viral replication. When Ile30 and Leu33 residues were mutated to Ala individually or combinatorically, agnoprotein dimer/oligomer formation and levels of the virally expressed agnoprotein did not change significantly. Similar results were obtained with Ala substitutions of Asp38 and Glu34. However, when Leu29 and Leu36 were singly mutated to Ala, virally expressed agnoprotein levels were significantly down-regulated during the viral replication cycle. This effect was so dramatic when three of the leucines, Leu29, Leu 32, and Leu 36, were mutated simultaneously to Ala (Leu29Ala + Leu32Ala + Leu36Ala). That is, there was no detectable level of agnoprotein produced after 15 day post-viral infection. These results indicated that Leu29 and Leu36 residues are critical for the structural integrity of agnoprotein and perhaps to dimer formation. Leu32 has a negligible effect on the stability of the protein (Coric et al., 2014). It is conceivable that the dimer structure is the stable form of agnoprotein and protects it from degradation. Based on NMR-structure-guided mutational analysis of the α-helix region of agnoprotein, a theoretical dimeric form of agnoprotein was build employing the program X-PLOR (Brunger, 1992; Brunger et al., 1998). According to this model, agnoprotein forms a dimer through the interaction of Leu29, Leu32, and Leu36 residues in an anti-parallel rather than parallel manner (Coric et al., 2014). These NMR based mutational studies also suggested that particularly the Leu29, Leu32, and Leu36 region of the α-helix can be targeted by small interfering molecules to prevent the dimer formation in order to develop therapeutics against JCV infections.
Formation of dimers and oligomers by agnoprotein through its Leu/Ile/Phe-rich domain has been previously reported (Saribas et al., 2011). There is a striking similarity between the HIV Vpr leucine zipper like domain located at its C-terminus and the Leu/Ile/Phe-rich domain of agnoprotein. Using the Vpr leucine zipper-like region as a model, a similar dimer formation by agnoprotein was also explored employing the X-PLOR program (Coric et al., 2014). This model shows that the two α-helices can associate in an antiparallel manner through the possible interactions between hydrophobic residues Leu29 and Leu36 from each monomer (Coric et al., 2014) (Fig. 6). An additional hydrophobic surface constituted by Ile30 and Leu33 is also eligible for intra- and intermolecular interactions and should not be ruled out completely; perhaps, these residues are involved in heterodimer interactions of agnoprotein with other cellular or viral proteins.
Fig. 6.

(A and B) A theoretical model of dimeric agnoprotein structure. The energetically most favorable structure of agnoprotein calculated with X-PLOR is shown with the backbone in a ribbon representation (Brunger, 1992; Brunger et al., 1998). The side chains of the aromatic and hydrophobic residues are shown in stick representations. The structure is characterized by two antiparallel a-helical domains from residues Lys23 to Phe39 with an interface composed of residues, Leu29, Leu32, and Leu36 (red). Residues Ile30, Leu33, and Leu37 (pink) are located at the opposite side of the aromatic residues Phe31, Phe35, and Phe39 (green). The part B is the 180° rotation presentaion of the part A on the y-axis. (C and D) Parts C and D represent a view from the top side of parts A and B, respectively.
Moreover, the aromatic interface, containing Phe31, Phe35, and Phe39 also appears to be available for intermolecular interactions, perhaps interaction with other proteins. The functional importance of these Phe residues has been recently analyzed by alanine scanning mutagenesis demonstrating that they exhibit combinatorial effect on viral DNA replication rather than forming dimers or oligomers (Saribas et al., 2012). It is also possible that the three hydrophobic surfaces in the α-helix region may form multiple potential conformational variants that are present within the infected cells. It is known that agnoprotein is distributed to nucleus, cytoplasm, and perhaps into the cellular membranes, suggesting that agnoprotein exists in different conformations in different compartments of the infected cells to allow a functional flexibility to the protein. The only Cys residue, Cys40 was originally thought to be involved in dimerization by a disulfide bridge. Hidaka et al. (2015) reported that Cys40 can form a disulfide bridge between two synthetic agnoprotein peptides spanning from aa 22 to 40 and, therefore, is responsible for dimerization/oligomerization of agnoprotein. In fact, stable agnoprotein dimer/oligomers consistent form under reducing conditions suggesting that the findings by Hidaka et al. (2015) may have resulted from the use of a short peptide in an in vitro system.
Post-Translational Modifications on Agnoprotein
Phosphorylation of agnoprotein
The agnoproteins of BKV (Rinaldo et al., 1998; Johannessen et al., 2008), JCV (Okada et al., 2002; Sariyer et al., 2006) and SV40 (Jackson and Chalkley, 1981) are all phosphoproteins. For BKV and JCV, phospho-acceptor sites have been mapped to Ser-7, Ser-11, and Thr-21 (Sariyer et al., 2006; Johannessen et al., 2008). Ser7 and Ser11 are also conserved in SV40, while Thr21 is replaced by Ser21 in SV40 agnoprotein. BKV, JCV, and SV40 contain a conserved Ser or Thr residues at position 45 but the in vivo functionality of this potential phospho-acceptor site is unknown. Sariyer et al. (2006) demonstrated that single and double substitutions of Ser7, Ser11, and Thr21 with Ala in JCV agnoprotein generated mutant viruses that failed to propagate. Johannessen et al. (2008) replaced Ser11 with either non-phosphorylatable Ala or phospho-mimicking aspartic acid (Asp) in BKV agnoprotein and this reduced the ability of these mutants to propagate compared to wild-type virus. Studies with non-phosphorylatable agnoprotein mutants demonstrated that phosphorylation also affects the stability of agnoprotein. Expression levels of single Thr21Ala, double Ser7Ala/Ser11Ala, and triple Ser7Ala/Ser11Ala/Thr21Ala mutant proteins were higher than those of wild-type JCV agnoprotein during the first round of replication. However, their levels reduced significantly during the second round of replication (Sariyer et al., 2006). Similarly, BKV Ser11Ala agnoprotein was less stable than wild-type or the phospho-mimicking Ser11Asp mutant protein (Sariyer et al., 2006; Johannessen et al., 2008;).
Phosphorylation of a substrate can stimulate its ubiquitination and subsequent degradation by the proteasome (Hunter et al., 2007). It is not known whether phosphorylation of agnoprotein leads to its ubiquitination and subsequent degradation, but in silico prediction algorithms of ubiquitin-conjugated sites show that agnoprotein of different polyomaviruses contain putative ubiquitin acceptor residues (Radivojac et al., 2010). Phosphorylation may also influence the subcellular location of agnoprotein (Okada et al., 2001). Although putative sumoylation, acetylation, amidation, and methylation sites are present in agnoprotein, except for phosphorylation no other post-translational modifications of agnoprotein have been demonstrated (Huang et al., 2005; Kiemer et al., 2005; Plewczynski et al., 2005; Li et al., 2006; Xue et al., 2006; Shao et al., 2009; Zhao et al., 2014), and whether these modifications occur in vivo remains to be elucidated.
Peculiar behavior of Leu29 and Phe mutants of agnoprotein
Consistent with the partial localization of agnoprotein into the nucleus (Saribas et al., 2012), recent mutational analysis of the dimerization domain by Coric et al. (2014) revealed that a single mutation of Leu29 to Ala rendered agnoprotein exclusively a nuclear protein, supporting the idea that agnoprotein is trafficking back and forth between nucleus and cytoplasm. Moreover, Saribas et al. (2012) also made a peculiar observation earlier when analyzing Phe mutants of agnoprotein by immunocytochemistry. That is, mutation of Phe31, Phe35, and Phe39 to Ala, respectively, in the viral background increased the volume of the mutant virus-infected cells observed microscopically. The precise mechanism of this phenomenon is currently unknown. However, recent findings by Otlu et al. (2014) demonstrate that agnoprotein is readily released from the infected cells. Further analysis of these three Phe mutants in release assays by our laboratory demonstrate that each may play a significant role in this process, because high proportion of each Phe-mutant agnoprotein appears to be trapped in cells during the release process. This observation, in part, may explain why the volume of the cells expressing Phe mutants is increased compared to WT (data not shown).
Nucleic Acid Binding Features of Agnoprotein
Is JCV agnoprotein a DNA binding protein?
Viroporins are general described as a family of small and hydrophobic integral membrane proteins that forms channels and play important roles during the various aspect of the viral life (Scott and Griffin, 2015). DNA binding studies employing band shift assays by Saribas et al. (2012) have demonstrated that agnoprotein enhances the DNA binding activity of large T antigen (LT-Ag) to Ori without directly interacting with DNA, suggesting that agnoprotein is involved in viral DNA replication through interaction with LT-Ag. However, agnoprotein does not seem to form an apparent stable interaction with LT-Ag/viral DNA complex because the migration pattern of DNA–protein complexes was not altered upon addition of agnoprotein into the binding mixture. This finding is consistent with a previous report by Safak et al. (2001) which demonstrated that agnoprotein interacts with the helicase domain of LT-Ag in the absence of DNA. Both findings suggest the following possibility: interaction of agnoprotein with LT-Ag off the DNA is sufficient to induce a conformational change on LT-Ag so that LT-Ag binds to its target sequences on Ori more efficiently. This suggests, in turn, that either (i) agnoprotein is not a part of an LT-Ag/DNA complex or (ii) there is such a weak interaction between agnoprotein and the LT-Ag/DNA complex that it precludes its detection as part of a ternary complex.
These findings by Saribas et al. (2012) showing no evidence of for agnoprotein–DNA interaction contrasts with a report by Gilbert et al. (1981) in which SV40 agnoprotein was proposed to be a DNA binding protein based on the premise that this protein contains various positively charged basic residues, including Arg and Lys, and, therefore, it might have a nucleic acid binding activity. The experimental settings employed by Gilbert et al. were somewhat different from those employed by Saribas et al. (2012) which may account for the discrepant results from the two groups. Saribas et al. used a purified recombinant protein (GST-Agno) and a short-labeled synthetic oligonucleotide corresponding to the large T antigen binding site I (BS I) and inverted repeat region (IR) of origin of DNA replication (Ori) of JCV in the binding assays and concluded that purified agnoprotein does not directly interact with DNA. In the study by Gilbert et al., however, metabolically labeled whole-cell extracts prepared from African Green Monkey Kidney (AGMK) cells infected with SV40 were run through columns packed with either a single-stranded or a double-stranded calf thymus DNA-cellulose and the columns were then subsequently washed with a low salt and high salt buffers. The various fractions were then analyzed by SDS-PAGE followed by autoradiography in order to detect the labeled agnoprotein in the fractions. Labeled agnoprotein was only detected in fractions eluted with high salt but not with low salt buffer (Gilbert et al., 1981). It is important to note that, many other labeled proteins, in addition to agnoprotein, were also detected in the eluted fractions, thus making the conclusions somewhat complicated. These results also suggest a strong possibility that agnoprotein did not necessarily bind to DNA directly but did so indirectly interacting with cellular protein(s) that were retained in the DNA columns due to their own intrinsic DNA binding activities. Therefore, the results reported by Gilbert et al. appear to be inconclusive with respect to the DNA binding activity of SV40 agnoprotein by itself.
Role of Agnoprotein in Viral Life Cycle
Regulation of viral DNA replication and transcription
The precise role of agnoprotein in the viral life cycle is unclear. However, deletion of the entire gene (dl2304) or 2 bp insertion into the agnoprotein coding gene in the SV40 genome by Hou-Jong et al. (1987) revealed smaller plaque sizes in infected monolayer cells compared to wild-type (WT). A similar study was also performed by Akan et al. (2006) in which the entire agnoprotein coding region was deleted in the JC viral background and the effect of these mutations was analyzed. It was observed that both the level of JC viral gene expression and its replication were dramatically reduced in the mutant virus as compared to WT. This finding is consistent with those for the SV40 agnoprotein deletion mutant as stated above (Hou-Jong et al., 1987). In addition, when the translation initiation codon (ATG) was mutated to CAG, no agnoprotein was produced, accompanied by significant inhibition of viral transcription and replication during the first round of viral infection, but not to the extent that was observed when the entire agnoprotein gene was deleted. However, in later rounds of viral replication, the effects of the point mutations approached those of the full agnogene deletion mutant (Akan et al., 2006). Further analysis of the agnoprotein coding region by DNA footprinting, followed by band-shift assays using nuclear extracts from JCV infected cells, demonstrated that certain cis-acting DNA elements within the agnoprotein coding gene served as binding sites for infection-induced factors and contributed to the viral replication cycle (Akan et al., 2006). Moreover, analysis by Ellis and Koralnik (2015) of a specific agnoprotein coding region deletion mutant (Mad-1 376–396) isolated from a PML patient’s brain samples exhibited substantial defects in viral replication, as had been observed by Akan et al. (2006) suggesting that specific regions in the agnoprotein coding region contribute to viral replication, perhaps at the DNA level, in a cis-acting manner. Furthermore, in support of these findings, Myhre et al. (2010) reported that several clinical variants of BK virus with specific agnoprotein gene deletions are also severely defective in gene expression and viral DNA replication but can be rescued by agnoprotein gene expression in cis- and trans-complementation experiments, suggesting the importance of agnoprotein for the BKV replication cycle as well.
Over the years, JCV agnoprotein has been shown to interact with other viral proteins (large T- (Safak et al., 2001) and small t- (Sariyer et al., 2008) antigen, capsid protein VP1 (Suzuki et al., 2012) and host proteins, including Y-box protein binding protein (YB-1) (Safak et al., 2002), p53 (Darbinyan et al., 2002), FEZ1 (Suzuki et al., 2005), and adaptor protein complex 3 (Suzuki et al., 2013). Particularly, its functional interaction with large T-antigen and YB-1 by transfection assays by Safak et al. (2001, 2002) demonstrated that it downregulates large T-antigen mediated viral DNA replication and YB-1 mediated viral transcription from the viral early and late promoters. Gerits et al. (2015) also demonstrated the interaction of BKV agnoprotein with proliferating cell nuclear antigen (PCNA) and its inhibitory effect on PCNA-dependent DNA synthesis in vitro, suggesting that agnoprotein may play a role in switching off viral DNA replication late in the replication cycle to allow for efficient assembly of replicated genomes into virions. Studies with siRNA directed against agnoprotein transcripts also demonstrated the importance of the agnoprotein in viral transcription and replication, in which investigators observed reduced levels of viral transcription and replication (Orba et al., 2004; Radhakrishnan et al., 2004; Matoba et al., 2008).
Involvement of agnoprotein in virion biogenesis and release
The effect of agnoprotein on the formation of “viral like particles (VLPs)” was analyzed in studies by Shishido-Hara et al. using a construct in which the entire JCV late coding region, excluding agnogene, was cloned under a heterologous promoter (cytomegalovirus, CMV). This construct expressed all viral capsid proteins, VP1, VP2, and VP3 but not agnoprotein. Results showed that the VLPs were polymorphic, rather than having a normal uniform shape, suggesting a role for agnoprotein in efficient viral progeny formation (Shishido-Hara, 2015; Shishido-Hara et al., 2004, 2012, 2014). Similar observations were made by Suzuki et al. (2010) regarding virion maturation in the absence of agnoprotein, in which irregularly shaped virions were formed. These investigators also demonstrated that the interaction of agnoprotein with JCV VP1 enhanced VP1 pentamer formation, further suggesting a role for agnoprotein in virion assembly. Agnoprotein appears to play a minimal role in release of the viral particles. A study by Sariyer et al. (2011) demonstrated that JCV and SV40 agnoprotein null mutants, where translation initiation codon (ATG) was mutated to abrogate its expression were able to release virions efficiently, however, the released virions were found to be mostly deficient in DNA content (Sariyer et al., 2011), which is consistent with the other studies regarding the role of agnoprotein in virion biogenesis (Shishido-Hara et al., 2004, 2012, 2014; Suzuki et al., 2010; Shishido-Hara, 2015).
Is agnoprotein a viroporin?
Viroporins are in general small and hydrophobic integral transmembrane proteins that play important roles in various aspects of many RNA and DNA viruses (Scott and Griffin, 2015). One of the interesting characteristics of viroporins is their ability to form pores in cell membranes by oligomerization. However, these pores lack the highly regulated gating behaviors of many cellular channels (Scott and Griffin, 2015). Such viroporins have been reported in the literature, including HIV-1 Vpu, influenza virus M2 protein (Nieva et al., 2012) and HPV16 E5 protein (Hu and Ceresa, 2009; Krawczyk et al., 2010). JCV agnoprotein has also been recently reported to have viroporin-like properties by Suzuki et al. (2010) (Scott and Griffin, 2015), such as being an integral part of cell membrane resulting in permeabilization of it and thereby aiding the release of virions from the infected cells (Suzuki et al., 2010). In previous similar virion release studies by Sariyer et al. (2011) JCV and SV40 agnoprotein null mutants in which the translation initiation codons (ATG) was mutated to CAG so that no agnoprotein was expressed from the virus were also used to investigate the contribution of agnoprotein to the viral release (Sariyer et al., 2011). These studies initially demonstrated that there is a substantial downregulation of viral transcription and replication suggesting a possibility that the virus particles were trapped inside the infected cells and are therefore not released efficiently. This then impaired the propagation of the virus particles and subsequently the viral life cycle. Analysis of virion release from the infected cells using these JCV and SV40 agnoprotein null mutants by immunoprecipitation followed by Western and Southern blot analyses revealed that viral particles were efficiently released from the infected cells in the absence of agnoprotein but the DNA content of the viral particles was severely diminished. This suggested a role for agnoprotein in encapsidation of the viral genome into the virions rather than playing a major role in their release from the infected cells. These findings argue that a viroporin activity of agnoprotein reported by Suzuki et al. (2010) may not necessarily be the main factor in virion release (Suzuki et al., 2010). Two recent publications also argue against the membrane integration activity agnoprotein. In one of the reports, it was demonstrated that agnoprotein is released from infected and transfected cells into the culture medium (Otlu et al., 2014) suggesting that if agnoprotein is an integral component of the cell membrane, it is highly unlikely that it will be readily secreted into the cell culture medium unless one of the various forms of agnoprotein (monomeric, dimeric or oligomeric (Saribas et al., 2011) are favorably suitable for this process. In a second report, Johannessen et al. (2011) demonstrated that BKV agnoprotein interacts with α-soluble N-ethylmaleimide-sensitive fusion attachment protein (α-SNAP) (Johannessen et al., 2011), which is a ubiquitous and indispensable component of the membrane fusion machinery and is a part of a special membrane system such as Golgi apparatus, suggesting that agnoprotein may interact with cell surface molecules rather than inserting itself into the cell membranes. Again, there is also the possibility that one of three forms of agnoprotein may interact with α-SNAP and the others may have viroporin-like activity.
Impact of Agnoprotein on Cellular Processes
As described above, agnoprotein is an essential participant in the viral life cycle during productive infections. It is also becoming clear that agnoprotein has a role in modulating the cellular life cycle that is independent of the virus-specific effects occurring during infection. Thus, much as large T-antigen has viral-specific functions, such as binding to the viral origin of replication, and cellular functions, such as binding p53 and pRb and driving the cell cycle and transformation (White and Khalili, 2004, 2006), agnoprotein also has a set of specific cellular binding partners and cellular processes which it perturbs. These interactions are discussed below and summarized in Table 2.
TABLE 2.
Cellular binding partners of agnoprotein and associated functions
| Virus | Agnoprotein binding partner | Binding domain within agnoprotein | Functional event | References |
|---|---|---|---|---|
| JCV | JCV T-Ag | 1–36 | Inhibition of JCV transcription and replication | Safak et al. (2001) |
| JCV | JCV VP1 | ND | Virion assembly | Suzuki et al. (2012) |
| JCV | HIV-1 Tat | 18–54 | Inhibition of Tat-mediated HIV-1 promoter induction | Kaniowska et al. (2006) |
| JCV | p53 | 1–36 | Suppression of cell cycle progression | Darbinyan et al. (2002) |
| JCV | Ku70 | 1–36 | Double-strand break DNA repair | Darbinyan et al. (2004) |
| JCV | PP2A | 18–36 | Dephosphorylation of agnoprotein | Sariyer et al. (2008) |
| JCV | YB-1 | 1–36 | Inhibition of YB-1-mediated transcription | Safak et al. (2002) |
| JCV | FEZ1 | ND | Microtubule extension | Suzuki et al. (2005) |
| JCV | HP1a | 1–18 | Nuclear membrane permeability | Okada et al. (2005) |
| BKV | α-SNAP | 1–15 | Vesicle transport | Johannessen et al. (2011) |
| BKV | PCNA | ND | Inhibition of DNA replication | Gerits et al. (2015) |
| JCV | BP-3 | ND | Inhibition of PB3-mediated vesicular trafficking | Suzuki et al. (2013) |
Agnoprotein and cell cycle progression
One approach to investigate the function of agnoprotein is to express it ectopically. In this way, the effect of agnoprotein can be studied in the absence of the other viral proteins. When JCV agnoprotein was stably expressed in mammalian cells, it significantly interfered with the cell cycle progression (Darbinyan et al., 2002). FACS analysis studies showed that agnopositive cells mostly accumulate at the G2/M phase of the cell cycle (Darbinyan et al., 2002). This was associated with a decline in the H1 kinase activity of cyclins A and B, which correlated with the induction of expression of the cyclin inhibitor p21/WAF-1. The promoter for p21/WAF-1 is positively regulated by p53 and this positive regulation was found to be enhanced using a p21/WAF-1 promoter reporter with p53−/− Saos-2 cells transfected with various combinations of expression plasmids for agnoprotein and p53 (Darbinyan et al., 2002). This function of agnoprotein appears to be mediated by direct interaction with p53 since immunoprecipitation/Western blotting and GST pull-down experiments demonstrated the N-terminal 36 amino acids of agnoprotein bind to p53 (Darbinyan et al., 2002).
Agnoprotein and DNA repair
Darbinyan et al. (2004) has recently showed that agnopositive cells are more sensitive to treatment with cisplatin than controls as measured by viability and clonogenicity assays performed after a single exposure to cisplatin. Metaphase spreads prepared from these cells showed near tetraploidy while cisplatin-treated agnonegative cells showed some degree of chromosomal breakage and rejoining. However, cisplatin-treated agnopositive cells a remarkable degree of chromosomal breakage was evident with most chromosomes fragmented into many pieces (Darbinyan et al., 2004). In addition, in cisplatin-treated agnopositive cells exhibited enhanced micronuclei formation, which is another measure of chromosomal damage. FACS analysis showed that while cisplatin-treated agnonegative cells accumulated in S phase, agnopositive cells did not, but rather they become aneuploid. Assay of nuclear extracts in vitro for their ability to ligate linearized plasmid substrates showed that agnoprotein expression was associated with impaired nonhomologous end-joining (NHEJ) double-strand-break DNA repair activity (Darbinyan et al., 2004). Also observed was the subcellular relocalization of the Ku70 NHEJ DNA repair protein to the perinuclear space, where agnoprotein was also localized. Protein–protein interaction studies showed an interaction of agnoprotein with Ku70. The ability of agnoprotein to inhibit NHEJ repair activity when added to cellular extracts was mediated by aa 1–36 region of the protein. Since Ku70 is a component of DNA-dependent protein kinase that is involved in DNA repair and for DNA damage-induced cell cycle signaling, agnoprotein interaction with Ku70 may be important in the JCV life cycle (Darbinyan et al., 2004).
Agnoprotein and oligodendrocyte differentiation and apoptosis
Although JCV is able to productively infect both astrocytes and oligodendrocytes in the CNS, it is the infection of oligodendrocytes, which function to produce and maintain myelin that is likely responsible for the generation of the regions of demyelination that are the pathological hallmark of PML. Although primary cultures of oligodendrocytes are difficult to obtain in significant numbers, a number of cell lines are available as alternatives. CG-4 is a bipotential cell line of rat CNS glial precursors that proliferate well in the presence of conditioned medium from the B104 neuroblastoma cell line and undergo differentiation into oligodendrocytes after withdrawal of mitogen (removal of the conditioned medium) or into astrocytes after replacement of the mitogen with 20% fetal calf serum (Louis et al., 1992). When agnoprotein was expressed in CG-4 cells by retroviral transduction, it was found that the cells could still differentiate into oligodendrocytes upon mitogen withdrawal but they showed lower morphological complexity and less arborization than control cells (Merabova et al., 2008). The induction of differentiation in the agnoprotein-expressing cells was also associated with the appearance of subdiploid cells in FACS analysis, a reduction in viability in MTT assays, DNA damage in COMET assays and caspase 3 activity. These findings indicate possible involvement of agnoprotein in biological events leading to the oligodendrocyte damage and loss that are involved in the pathological demyelination of PML lesions (Merabova et al., 2008). Interestingly, the effects of agnoprotein to inhibit CG4 oligodendroglial cells differentiation are similar to the effects of JCV infection on human primary oligodendrocyte precursor cells, in which inhibition of differentiation into oligodendrocytes is observed (Darbinyan et al., 2013).
Agnoprotein and cytokine secretion by oligodendrocytes
In addition to their role in myelination, oligodendrocytes produce a number of neurotrophic factors that have indispensable roles in the protection of axonal function and promotion of neuronal survival. To look at the effect of agnoprotein on the extracellular factors involved in communication between oligodendrocytes and neurons, the CG-4-Agno and CG-4 cells described in the previous section again provide a suitable culture system (Merabova et al., 2008). Culturing rat cortical neurons with conditioned medium (CM) from rat CG4-Agno resulted in significantly reduced arborization and loss of neuronal processes evident in cells labeled with antibody to class III β-tubulin compared to CG4 controls (Merabova et al., 2012). CXCL5/LIX is a rat chemokine that is closely related to the human CXCL5/ENA78 and CXCL6/GCP-2 chemokines, and is essential for neuronal cell survival. In CM from agnoprotein-producing CG-4 cells, it was found that the level of CXCL5/LIX was decreased compared to control cells and this reduced expression of CXCL5/LIX in the CM from CG4-Agno positive cells triggered a cascade of signaling events in cortical neurons including the MAPK and GSK3 pathways (Merabova et al., 2012). In turn, this signaling is involved in mechanisms of neuronal apoptosis in response to the depletion of CXCL5/LIX signaling. These data suggest that agnoprotein induces dysregulation of the chemokine production by oligodendrocytes contributing to the injuries to neurons and axons that are found in the pathogenesis of PML lesions (Merabova et al., 2012). Interestingly, the effects of agnoprotein on cytokine secretion by CG4 oligodendroglial cells are similar to the effects of JCV infection on human primary oligodendrocyte precursor cells, where substantial dysregulation of several chemokines, including CCL5/RANTES, GRO, CXCL1/GROα, CXCL16, CXCL8/IL-8, CXCL5/ENA-78, and CXCL10/IP-10 are observed (Darbinyan et al., 2013).
Agnoprotein and interference with the host secretion process
A powerful approach for investigating protein–protein interactions is the yeast two-hybrid assay. Using this approach with BKV agnoprotein as bait, α-soluble N-ethylmaleimide-sensitive fusion attachment protein (α-SNAP) was identified as a potential target (Johannessen et al., 2011). Further studies showed a partial subcellular colocalization of BKV agnoprotein and α-SNAP and that they could be co-immunoprecipitated together from cell extracts. Peptide mapping studies and GST pull-down experiments showed that the amino-terminal 37 amino acids of BKV agnoprotein mediate its interaction with α-SNAP. Functional studies with a VSVG-EGFP reporter construct demonstrated that BKV agnoprotein inhibits vesicular movement from the endoplasmic reticulum (ER) to the plasma membrane suggesting that it has a regulatory role in the cellular secretion process (Johannessen et al., 2011).
Conclusions and Future Directions
In this review, we have summarized the recent advances in structure–function relationship of agnoproteins of polyomaviruses, including JCV, BKV, and SV40. Agnoprotein is a small regulatory protein and plays important roles during the viral life cycle. These DNA viruses require agnoprotein for the successful completion of their life cycle. Recent structural studies by NMR provided a partial 3D structure of a JCV agnoprotein using a synthetic peptide encompassing amino acids Thr17 through Gln52 and revealed that agnoprotein contains a α-helix domain at the central portion of the protein, Lys23-Phe39. This region is rich in hydrophobic residues, including Leu, Ile, and Phe. Structure-based mutational analysis of the region demonstrated that α-helix region plays an essential role in protein stability and dimer/oligomer formation. Particularly, residues Leu29 and Leu36 are highly critical for the stable expression of the protein. Theoretical modeling studies support these findings where these two residues interact with one another when this protein forms a dimer in an antiparallel manner. The structure-based mutational analysis of the region also provided with an opportunity to target the α-helix region by small molecules to disrupt the dimer/oligomer formation for drug discovery purposes against JCV infections.
DNA binding studies with pure recombinant agnoprotein demonstrate that this protein does not interact with DNA directly. However, RNA chromatin immunoprecipitation (RNA-Chip) studies provide evidence that that it rather interacts with RNA (data not shown). In addition, α-helix region contains a predicted nuclear export signal (NES) similar to HIV-1 Rev (data not shown) (la Cour et al., 2003) and interacts with the chromosomal region maintenance protein-1 (CRM-1). CRM-1 is one of the regulatory host proteins involved in nucleo-cytoplasmic export of transcripts (Harris and Hope, 2000; Daugherty et al., 2010; Schmid et al., 2012). Future studies are required to further elucidate these observations. Our future studies also include the identification and characterization of new cellular partners of agnoprotein with respect to their relevance to the viral replication cycle.
Another interesting characteristic of agnoprotein is its ability to be released from the infected cells (Otlu et al., 2014). This recent finding naturally leads to several unanswered questions as follows: what is the function of this released protein in the extracellular matrix? Does it enter the uninfected neighboring cells, including astrocytes, neurons, oligodendrocytes, and microglia, if so, what pathway? If enters, how does it affect the health of those cells? Alternatively, it may interact with the cell surface molecules/receptors and trigger various signaling pathways internally through which it exploits the cellular pathways to protect the incoming virus or allow the virus replicate more efficiently in those cells. Exploring answers to these relevant questions will open up new avenues to better understand the polyomavirus life cycle and provide with ample opportunities to develop effective therapeutic approaches toward JCV and BKV related diseases.
Acknowledgments
We thank past and present members of the Department of Neuroscience for their insightful discussion and sharing of ideas and reagents. This work was made possible by grants awarded to M.S. (RO1NS090949) by the NIH and by the Temple University Drug Discovery Initiative (161398).
Footnotes
Conflicts of interest: Authors declare no conflict of interest.
Literature Cited
- Akan I, Sariyer IK, Biffi R, Palermo V, Woolridge S, White MK, Amini S, Khalili K, Safak M. Human polyomavirus JCV late leader peptide region contains important regulatory elements. Virology. 2006;349:66–78. doi: 10.1016/j.virol.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Allander T, Andreasson K, Gupta S, Bjerkner A, Bogdanovic G, Persson MA, Dalianis T, Ramqvist T, Andersson B. Identification of a third human polyomavirus. J Virol. 2007;81:4130–4136. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag B, Kilpatrick LH, Tyagarajan SK, Tevethia MJ, Frisque RJ. JC virus T′135, T′136 and T′165 proteins interact with cellular p107 and p130 in vivo and influence viral transformation potential. J Neurovirol. 2006;12:428–442. doi: 10.1080/13550280601009553. [DOI] [PubMed] [Google Scholar]
- Bollag B, Prins C, Snyder EL, Frisque RJ. Purified JC virus T and T’ proteins differentially interact with the retinoblastoma family of tumor suppressor proteins. Virology. 2000;274:165–178. doi: 10.1006/viro.2000.0451. [DOI] [PubMed] [Google Scholar]
- Brunger AT. A system for X-Ray crystallography and NMR. New Haven, CT: Yale University Press; 1992. [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Combet C, Blanchet C, Geourjon C, Deleage G. NPS@: Network protein sequence analysis. Trends Biochem Sci. 2000;25:147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- Coric P, Saribas AS, Abou-Gharbia M, Childers W, White MK, Bouaziz S, Safak M. Nuclear magnetic resonance structure revealed that the human polyomavirus JC virus agnoprotein contains an alpha-helix encompassing the Leu/Ile/Phe-rich domain. J Virol. 2014;88:6556–6575. doi: 10.1128/JVI.00146-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen B. HIV-1 auxiliary proteins: Making connections in dying cell. Cell. 1998;93:685–692. doi: 10.1016/s0092-8674(00)81431-2. [DOI] [PubMed] [Google Scholar]
- Darbinyan A, Darbinian N, Safak M, Radhakrishnan S, Giordano A, Khalili K. Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene. 2002;21:5574–5581. doi: 10.1038/sj.onc.1205744. [DOI] [PubMed] [Google Scholar]
- Darbinyan A, Kaminski R, White MK, Darbinian-Sarkissian N, Khalili K. Polyomavirus JC infection inhibits differentiation of oligodendrocyte progenitor cells. J Neurosci Res. 2013;91:116–127. doi: 10.1002/jnr.23135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbinyan A, Siddiqui KM, Slonina D, Darbinian N, Amini S, White MK, Khalili K. Role of JC virus agnoprotein in DNA repair. J Virol. 2004;78:8593–8600. doi: 10.1128/JVI.78.16.8593-8600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Liu B, Frankel AD. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat Struct Mol Biol. 2010;17:1337–1342. doi: 10.1038/nsmb.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Valle L, Gordon J, Enam S, Delbue S, Croul S, Abraham S, Radhakrishnan S, Assimakopoulou M, Katsetos CD, Khalili K. Expression of human neurotropic polyomavirus JCV late gene product agnoprotein in human medulloblastoma. J Natl Cancer Inst. 2002;94:267–273. doi: 10.1093/jnci/94.4.267. [DOI] [PubMed] [Google Scholar]
- Dhar R, Reddy VB, Weissman SM. Nucleotide sequence of the DNA encoding the 5′-terminal sequences of simian virus 40 late mRNA. J Biol Chem. 1978;253:612–620. [PubMed] [Google Scholar]
- Dhar R, Subramanian KN, Pan J, Weissman SM. Structure of a large segment of the genome of simian virus 40 that does not encode known proteins. Proc Natl Acad Sci USA. 1977;74:827–831. doi: 10.1073/pnas.74.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingwall C, Laskey RA. Protein import into the cell nucleus. Annu Rev Cell Biol. 1986;2:367–390. doi: 10.1146/annurev.cb.02.110186.002055. [DOI] [PubMed] [Google Scholar]
- Dingwall C, Laskey RA. Nuclear targeting sequences—A consensus? Trends Biochem Sci. 1991;16:478–481. doi: 10.1016/0968-0004(91)90184-w. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Ellis LC, Koralnik IJ. JC virus nucleotides 376–396 are critical for VP1 capsid protein expression. J Neurovirol. 2015;21:671–678. doi: 10.1007/s13365-014-0278-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiers W, Contreras R, Haegemann G, Rogiers R, Van de Voorde A, Van Heuverswyn H, Van Herreweghe J, Volckaert G, Ysebaert M. Complete nucleotide sequence of SV40 DNA. Nature. 1978;273:113–120. doi: 10.1038/273113a0. [DOI] [PubMed] [Google Scholar]
- Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Frankel AD, Bredt DS, Pabo CO. Tat protein from human immunodeficiency virus forms a metal-linked dimer. Science. 1988a;240:70–73. doi: 10.1126/science.2832944. [DOI] [PubMed] [Google Scholar]
- Frankel AD, Chen L, Cotter RJ, Pabo CO. Dimerization of the tat protein from human immunodeficiency virus: A cysteine-rich peptide mimics the normal metal-linked dimer interface. Proc Natl Acad Sci USA. 1988b;85:6297–6300. doi: 10.1073/pnas.85.17.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisque RJ. Structure and function of JC virus T′ proteins. J Neurovirol. 2001;7:293–297. doi: 10.1080/13550280152537120. [DOI] [PubMed] [Google Scholar]
- Gardner SD, Feild AM, Colleman DV, Hulme B. New human papovavirus (BK) isolated from urine after renal transplantation. Lancet. 1971;1:1253–1257. doi: 10.1016/s0140-6736(71)91776-4. [DOI] [PubMed] [Google Scholar]
- Gaynor AM, Nissen MD, Whiley DM, Mackay IM, Lambert SB, Wu G, Brennan DC, Storch GA, Sloots TP, Wang D. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerits N, Johannessen M, Tummler C, Walquist M, Kostenko S, Snapkov I, van Loon B, Ferrari E, Hubscher U, Moens U. Agnoprotein of polyomavirus BK interacts with proliferating cell nuclear antigen and inhibits DNA replication. Virol J. 2015;12:7. doi: 10.1186/s12985-014-0220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerits N, Moens U. Agnoprotein of mammalian polyomaviruses. Virology. 2012;432:316–326. doi: 10.1016/j.virol.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J, Nomura S, Anderson CW, George K. Identification of the SV40 agnoproduct: A DNA binding protein. Nature. 1981;291:346–349. doi: 10.1038/291346a0. [DOI] [PubMed] [Google Scholar]
- Harris ME, Hope TJ. RNA export: Insights from viral models. Essays Biochem. 2000;36:115–127. doi: 10.1042/bse0360115. [DOI] [PubMed] [Google Scholar]
- Hartlieb B, Modrof J, Muhlberger E, Klenk HD, Becker S. Oligomerization of Ebola virus VP30 is essential for viral transcription and can be inhibited by a synthetic peptide. J Biol Chem. 2003;278:41830–41836. doi: 10.1074/jbc.M307036200. [DOI] [PubMed] [Google Scholar]
- Hidaka K, Hojo K, Fujioka S, Nukuzuma S, Tsuda Y. Oligomerization of neutral peptides derived from the JC virus agnoprotein through a cysteine residue. Amino Acids. 2015;47:2205–2213. doi: 10.1007/s00726-015-2004-3. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Hou-Jong MH, Larsen SH, Roman A. Role of the agnoprotein in regulation of simian virus 40 replication and maturation pathways. J Virol. 1987;61:937–939. doi: 10.1128/jvi.61.3.937-939.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu LL, Ceresa BP. Characterization of the plasma membrane localization and orientation of HPV16 E5 for cell-cell fusion. Virology. 2009;393:135–143. doi: 10.1016/j.virol.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HD, Lee TY, Tzeng SW, Horng JT. KinasePhos: A web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 2005;33:W226–W229. doi: 10.1093/nar/gki471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JM, Lesort M, Johnson GV. Ubiquitin-proteasome system alterations in a striatal cell model of Huntington’s disease. J Neurosci Res. 2007;85:1774–1788. doi: 10.1002/jnr.21287. [DOI] [PubMed] [Google Scholar]
- Imperiale M, Major EO. Polyomaviruses. In: Knipe MK, Howley PM, editors. Fields virology. 5th. Philadelphia: 2001. pp. 2263–2298. [Google Scholar]
- Jackson V, Chalkley R. Use of whole-cell fixation to visualize replicating and maturing simian virus 40: Identification of new viral gene product. Proc Natl Acad Sci USA. 1981;78:6081–6085. doi: 10.1073/pnas.78.10.6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay G, Nomura S, Anderson CW, Khoury G. Identification of the SV40 agnogene product: A DNA binding protein. Nature. 1981;291:346–349. doi: 10.1038/291346a0. [DOI] [PubMed] [Google Scholar]
- Johannessen M, Myhre MR, Dragset M, Tummler C, Moens U. Phosphorylation of human polyomavirus BK agnoprotein at Ser-11 is mediated by PKC and has an important regulative function. Virology. 2008;379:97–109. doi: 10.1016/j.virol.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Johannessen M, Walquist M, Gerits N, Dragset M, Spang A, Moens U. BKV agnoprotein interacts with alpha-soluble N-ethylmaleimide-sensitive fusion attachment protein, and negatively influences transport of VSVG-EGFP. PLoS ONE. 2011;6:e24489. doi: 10.1371/journal.pone.0024489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniowska D, Kaminski R, Amini S, Radhakrishnan S, Rappaport J, Johnson E, Khalili K, Del Valle L, Darbinyan A. Cross-interaction between JC virus agnoprotein and human immunodeficiency virus type 1 (HIV-1) Tat modulates transcription of the HIV-1 long terminal repeat in glial cells. J Virol. 2006;80:9288–9299. doi: 10.1128/JVI.02138-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili K, Sariyer IK, Safak M. Small tumor antigen of polyomaviruses: Role in viral life cycle and cell transformation. J Cell Physiol. 2008;215:309–319. doi: 10.1002/jcp.21326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili K, White MK, Sawa H, Nagashima K, Safak M. The agnoprotein of polyomaviruses: A multifunctional auxiliary protein. J Cell Physiol. 2005;204:1–7. doi: 10.1002/jcp.20266. [DOI] [PubMed] [Google Scholar]
- Kiemer L, Bendtsen JD, Blom N. NetAcet: Prediction of N-terminal acetylation sites. Bioinformatics. 2005;21:1269–1270. doi: 10.1093/bioinformatics/bti130. [DOI] [PubMed] [Google Scholar]
- Kolodziejski PJ, Rashid MB, Eissa NT. Intracellular formation of “undisruptable” dimers of inducible nitric oxide synthase. Proc Natl Acad Sci USA. 2003;100:14263–14268. doi: 10.1073/pnas.2435290100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk E, Suprynowicz FA, Sudarshan SR, Schlegel R. Membrane orientation of the human papillomavirus type 16 E5 oncoprotein. J Virol. 2010;84:1696–1703. doi: 10.1128/JVI.01968-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak YT, Raney A, Kuo LS, Denial SJ, Temple BR, Garcia JV, Foster JL. Self-association of the lentivirus protein, Nef. Retrovirology. 2010;7:77. doi: 10.1186/1742-4690-7-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Cour T, Gupta R, Rapacki K, Skriver K, Poulsen FM, Brunak S. NESbase version 1.0: A database of nuclear export signals. Nucleic Acids Res. 2003;31:393–396. doi: 10.1093/nar/gkg101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Xue Y, Jin C, Wang M, Yao X. Prediction of nepsilon-acetylation on internal lysines implemented in Bayesian discriminant method. Biochem Biophys Res Commun. 2006;350:818–824. doi: 10.1016/j.bbrc.2006.08.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis JC, Magal E, Muir D, Manthorpe M, Varon S. CG-4, a new bipotential glial cell line from rat brain, is capable of differentiating in vitro into either mature oligodendrocytes or type-2 astrocytes. J Neurosci Res. 1992;31:193–204. doi: 10.1002/jnr.490310125. [DOI] [PubMed] [Google Scholar]
- Lynch KJ, Frisque RJ. Identification of critical elements within the JC virus DNA replication origin. J Virol. 1990;64:5812–5822. doi: 10.1128/jvi.64.12.5812-5822.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KJ, Frisque RJ. Factors contributing to the restricted DNA replicating activity of JC virus. Virology. 1991;180:306–317. doi: 10.1016/0042-6822(91)90035-a. [DOI] [PubMed] [Google Scholar]
- Lynch KJ, Haggerty S, Frisque RJ. DNA replication of chimeric JC virus-simian virus 40 genomes. Virology. 1994;204:819–822. doi: 10.1006/viro.1994.1600. [DOI] [PubMed] [Google Scholar]
- Matoba T, Orba Y, Suzuki T, Makino Y, Shichinohe H, Kuroda S, Ochiya T, Itoh H, Tanaka S, Nagashima K, Sawa H. An siRNA against JC virus (JCV) agnoprotein inhibits JCV infection in JCV-producing cells inoculated in nude mice. Neuropathology. 2008;28:286–294. doi: 10.1111/j.1440-1789.2007.00878.x. [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Merabova N, Kaminski R, Krynska B, Amini S, Khalili K, Darbinyan A. JCV agnoprotein-induced reduction in CXCL5/LIX secretion by oligodendrocytes is associated with activation of apoptotic signaling in neurons. J Cell Physiol. 2012;227:3119–3127. doi: 10.1002/jcp.23065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merabova N, Kaniowska D, Kaminski R, Deshmane SL, White MK, Amini S, Darbinyan A, Khalili K. JC virus agnoprotein inhibits in vitro differentiation of oligodendrocytes and promotes apoptosis. J Virol. 2008;82:1558–1569. doi: 10.1128/JVI.01680-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell HK, Petersen NS, Buzin CH. Self-degradation of heat shock proteins. Proc Natl Acad Sci USA. 1985;82:4969–4973. doi: 10.1073/pnas.82.15.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens U, Ludvigsen M, Van Ghelue M. Human polyomaviruses in skin diseases. Patholog Res Int. 2011;2011:123491. doi: 10.4061/2011/123491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhre MR, Olsen GH, Gosert R, Hirsch HH, Rinaldo CH. Clinical polyomavirus BK variants with agnogene deletion are non-functional but rescued by trans-complementation. Virology. 2010;398:12–20. doi: 10.1016/j.virol.2009.11.029. [DOI] [PubMed] [Google Scholar]
- Nieva JL, Madan V, Carrasco L. Viroporins: Structure and biological functions. Nat Rev Microbiol. 2012;10:563–574. doi: 10.1038/nrmicro2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilges M. Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J Mol Biol. 1995;245:645–660. doi: 10.1006/jmbi.1994.0053. [DOI] [PubMed] [Google Scholar]
- Okada Y, Endo S, Takahashi H, Sawa H, Umemura T, Nagashima K. Distribution and function of JCV agnoprotein. J Neurovirol. 2001;7:302–306. doi: 10.1080/13550280152537148. [DOI] [PubMed] [Google Scholar]
- Okada Y, Sawa H, Endo S, Orba Y, Umemura T, Nishihara H, Stan AC, Tanaka S, Takahashi H, Nagashima K. Expression of JC virus agnoprotein in progressive multifocal leukoencephalopathy brain. Acta Neuropathol. 2002;104:130–136. doi: 10.1007/s00401-002-0526-8. [DOI] [PubMed] [Google Scholar]
- Okada Y, Suzuki T, Sunden Y, Orba Y, Kose S, Imamoto N, Takahashi H, Tanaka S, Hall WW, Nagashima K, Sawa H. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: Role in nuclear egress of viral particles. EMBO Rep. 2005;6:452–457. doi: 10.1038/sj.embor.7400406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orba Y, Sawa H, Iwata H, Tanaka S, Nagashima K. Inhibition of virus production in JC virus-infected cells by postinfection RNA interference. J Virol. 2004;78:7270–7273. doi: 10.1128/JVI.78.13.7270-7273.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otlu O, De Simone FI, Otalora YL, Khalili K, Sariyer IK. The agnoprotein of polyomavirus JC is released by infected cells: Evidence for its cellular uptake by uninfected neighboring cells. Virology. 2014;468–470:88–95. doi: 10.1016/j.virol.2014.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou HD, Kwiatkowski W, Deerinck TJ, Noske A, Blain KY, Land HS, Soria C, Powers CJ, May AP, Shu X, Tsien RY, Fitzpatrick JA, Long JA, Ellisman MH, Choe S, O’Shea CC. A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell. 2012;151:304–319. doi: 10.1016/j.cell.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett BL, Zu Rhein GM, Walker DL, Echroade R, Dessel B. Cultivation of papova-like virus from human brain with progressive multifocal leukoencephalopathy. Lancet. 1971;1:1257–1260. doi: 10.1016/s0140-6736(71)91777-6. [DOI] [PubMed] [Google Scholar]
- Pipas JM. Molecular chaperone function of the SV40 large T antigen. Dev Biol Stand. 1998;94:313–319. [PubMed] [Google Scholar]
- Plewczynski D, Tkacz A, Wyrwicz LS, Rychlewski L. AutoMotif server: Prediction of single residue post-translational modifications in proteins. Bioinformatics. 2005;21:2525–2527. doi: 10.1093/bioinformatics/bti333. [DOI] [PubMed] [Google Scholar]
- Prins C, Frisque RJ. JC virus T’ proteins encoded by alternatively spliced early mRNAs enhance T antigen-mediated viral DNA replication in human cells. J Neurovirol. 2001;7:250–264. doi: 10.1080/13550280152403290. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan S, Gordon J, Del Valle L, Cui J, Khalili K. Intracellular approach for blocking JC virus gene expression by using RNA interference during viral infection. J Virol. 2004;78:7264–7269. doi: 10.1128/JVI.78.13.7264-7269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, Iakoucheva LM. Identification, analysis, and prediction of protein ubiquitination sites. Proteins. 2010;78:365–380. doi: 10.1002/prot.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rinaldo CH, Traavik T, Hey A. The agnogene of the human polyomavirus BK is expressed. J Virol. 1998;72:6233–6236. doi: 10.1128/jvi.72.7.6233-6236.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M. Polyomaviruses and Papillomaviruses. Reference Module in Biomedical Sciences. 2014 [Google Scholar]
- Safak M, Barrucco R, Darbinyan A, Okada Y, Nagashima K, Khalili K. Interaction of JC virus agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J Virol. 2001;75:1476–1486. doi: 10.1128/JVI.75.3.1476-1486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Sadowska B, Barrucco R, Khalili K. Functional interaction between JC virus late regulatory agnoprotein and cellular Y-box binding transcription factor, YB-1. J Virol. 2002;76:3828–3838. doi: 10.1128/JVI.76.8.3828-3838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Abou-Gharbia M, Childers W, Sariyer IK, White MK, Safak M. Essential roles of Leu/Ile/Phe-rich domain of JC virus agnoprotein in dimer/oligomer formation, protein stability and splicing of viral transcripts. Virology. 2013;443:161–176. doi: 10.1016/j.virol.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Arachea BT, White MK, Viola RE, Safak M. Human polyomavirus JC small regulatory agnoprotein forms highly stable dimers and oligomers: Implications for their roles in agnoprotein function. Virology. 2011;420:51–65. doi: 10.1016/j.virol.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Mun S, Johnson J, El-Hajmoussa M, White MK, Safak M. Human polyoma JC virus minor capsid proteins, VP2 and VP3, enhance large T antigen binding to the origin of viral DNA replication: Evidence for their involvement in regulation of the viral DNA replication. Virology. 2014;449:1–16. doi: 10.1016/j.virol.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, White MK, Safak M. JC virus agnoprotein enhances large T antigen binding to the origin of viral DNA replication: Evidence for its involvement in viral DNA replication. Virology. 2012;433:12–26. doi: 10.1016/j.virol.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Akan I, Palermo V, Gordon J, Khalili K, Safak M. Phosphorylation mutants of JC virus agnoprotein are unable to sustain the viral infection cycle. J Virol. 2006;80:3893–3903. doi: 10.1128/JVI.80.8.3893-3903.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Khalili K, Safak M. Dephosphorylation of JC virus agnoprotein by protein phosphatase 2A: Inhibition by small t antigen. Virology. 2008;375:464–479. doi: 10.1016/j.virol.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Saribas AS, White MK, Safak M. Infection by agnoprotein-negative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol J. 2011;8:255. doi: 10.1186/1743-422X-8-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Gonzalez RA, Dobner T. CRM1-dependent transport supports cytoplasmic accumulation of adenoviral early transcripts. J Virol. 2012;86:2282–2292. doi: 10.1128/JVI.06275-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott C, Griffin S. Viroporins: Structure, function and potential as antiviral targets. J Gen Virol. 2015;96:2000–2027. doi: 10.1099/vir.0.000201. [DOI] [PubMed] [Google Scholar]
- Shao J, Xu D, Tsai SN, Wang Y, Ngai SM. Computational identification of protein methylation sites through bi-profile Bayes feature extraction. PLoS ONE. 2009;4:e4920. doi: 10.1371/journal.pone.0004920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido-Hara Y. Progressive multifocal leukoencephalopathy: Dot-shaped inclusions and virus-host interactions. Neuropathology. 2015;35:487–496. doi: 10.1111/neup.12203. [DOI] [PubMed] [Google Scholar]
- Shishido-Hara Y, Ichinose S, Higuchi K, Hara Y, Yasui K. Major and minor capsid proteins of human polyomavirus JC cooperatively accumulate to nuclear domain 10 for assembly into virions. J Virol. 2004;78:9890–9903. doi: 10.1128/JVI.78.18.9890-9903.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido-Hara Y, Ichinose S, Uchihara T. JC virus intranuclear inclusions associated with PML-NBs: Analysis by electron microscopy and structured illumination microscopy. Am J Pathol. 2012;180:1095–1106. doi: 10.1016/j.ajpath.2011.11.036. [DOI] [PubMed] [Google Scholar]
- Shishido-Hara Y, Yazawa T, Nagane M, Higuchi K, Abe-Suzuki S, Kurata M, Kitagawa M, Kamma H, Uchihara T. JC virus inclusions in progressive multifocal leukoencephalopathy: Scaffolding promyelocytic leukemia nuclear bodies grow with cell cycle transition through an S-to-G2-like state in enlarging oligodendrocyte nuclei. J Neuropathol Exp Neurol. 2014;73:442–453. doi: 10.1097/NEN.0000000000000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebrasse EA, Reyes A, Lim ES, Zhao G, Mkakosya RS, Manary MJ, Gordon JI, Wang D. Identification of MW polyomavirus, a novel polyomavirus in human stool. J Virol. 2012;86:10321–10326. doi: 10.1128/JVI.01210-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DT. SV40 large T antigen functions in DNA replication and transformation. Adv Virus Res. 2000;55:75–134. doi: 10.1016/s0065-3527(00)55002-7. [DOI] [PubMed] [Google Scholar]
- Stewart SE, Eddy BE, Borgese N. Neoplasms in mice inoculated with a tumor agent carried in tissue culture. J Natl Cancer Inst. 1958;20:1223–1243. doi: 10.1093/jnci/20.6.1223. [DOI] [PubMed] [Google Scholar]
- Sullivan CS, Pipas JM. T antigens of simian virus 40: Molecular chaperones for viral replication and tumorigenesis. Microbiol Mol Biol Rev. 2002;66:179–202. doi: 10.1128/MMBR.66.2.179-202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Okada Y, Semba S, Orba Y, Yamanouchi S, Endo S, Tanaka S, Fujita T, Kuroda S, Nagashima K, Sawa H. Identification of FEZ1 as a protein that interacts with JC virus agnoprotein and microtubules: Role of agnoprotein-induced dissociation of FEZ1 from microtubules in viral propagation. J Biol Chem. 2005;280:24948–24956. doi: 10.1074/jbc.M411499200. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Orba Y, Makino Y, Okada Y, Sunden Y, Hasegawa H, Hall WW, Sawa H. Viroporin activity of the JC polyomavirus is regulated by interactions with the adaptor protein complex 3. Proc Natl Acad Sci USA. 2013;110:18668–18673. doi: 10.1073/pnas.1311457110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Orba Y, Okada Y, Sunden Y, Kimura T, Tanaka S, Nagashima K, Hall WW, Sawa H. The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Pathog. 2010;6:e1000801. doi: 10.1371/journal.ppat.1000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Semba S, Sunden Y, Orba Y, Kobayashi S, Nagashima K, Kimura T, Hasegawa H, Sawa H. Role of JC virus agnoprotein in virion formation. Microbiol Immunol. 2012;56:639–646. doi: 10.1111/j.1348-0421.2012.00484.x. [DOI] [PubMed] [Google Scholar]
- Sweet BH, Hilleman MR. The vacuolating virus, S.V.40. Proc Soc Exp Biol Med. 1960;105:420–427. doi: 10.3181/00379727-105-26128. [DOI] [PubMed] [Google Scholar]
- Swenson JJ, Frisque RJ. Biochemical characterization and localization of JC virus large T antigen phosphorylation domains. Virology. 1995;212:295–308. doi: 10.1006/viro.1995.1487. [DOI] [PubMed] [Google Scholar]
- Unterstab G, Gosert R, Leuenberger D, Lorentz P, Rinaldo CH, Hirsch HH. The polyomavirus BK agnoprotein co-localizes with lipid droplets. Virology. 2010;399:322–331. doi: 10.1016/j.virol.2010.01.011. [DOI] [PubMed] [Google Scholar]
- van der Meijden E, Janssens RW, Lauber C, Bouwes Bavinck JN, Gorbalenya AE, Feltkamp MC. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog. 2010;6:e1001024. doi: 10.1371/journal.ppat.1001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MK, Gordon J, Khalili K. The rapidly expanding family of human polyomaviruses: Recent developments in understanding their life cycle and role in human pathology. PLoS Pathog. 2013;9:e1003206. doi: 10.1371/journal.ppat.1003206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MK, Khalili K. Polyomaviruses and human cancer: Molecular mechanisms underlying patterns of tumorigenesis. Virology. 2004;324:1–16. doi: 10.1016/j.virol.2004.03.025. [DOI] [PubMed] [Google Scholar]
- White MK, Khalili K. Interaction of retinoblastoma protein family members with large T-antigen of primate polyomaviruses. Oncogene. 2006;25:5286–5293. doi: 10.1038/sj.onc.1209618. [DOI] [PubMed] [Google Scholar]
- Xue Y, Zhou F, Fu C, Xu Y, Yao X. SUMOsp: A web server for sumoylation site prediction. Nucleic Acids Res. 2006;34:W254–W257. doi: 10.1093/nar/gkl207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Greninger AL, Isa P, Phan TG, Martinez MA, de la Luz Sanchez M, Contreras JF, Santos-Preciado JI, Parsonnet J, Miller S, DeRisi JL, Delwart E, Arias CF, Chiu CY. Discovery of a novel polyomavirus in acute diarrheal samples from children. PLoS ONE. 2012;7:e49449. doi: 10.1371/journal.pone.0049449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Xie Y, Zheng Y, Jiang S, Liu W, Mu W, Liu Z, Zhao Y, Xue Y, Ren J. GPSSUMO: A tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res. 2014;42:W325–W330. doi: 10.1093/nar/gku383. [DOI] [PMC free article] [PubMed] [Google Scholar]